

Abstract
Chronic lymphocytic leukemia deletion gene 7 (Clld7) is a candidate tumor suppressor on chromosome 13q14. Clld7 encodes an evolutionarily conserved protein that contains an RCC1 domain plus broad complex, tramtrack, bric-a-brac (BTB) and POZ domains. In this study, we investigated the biological functions of Clld7 protein in inducible osteosarcoma cell lines. Clld7 induction inhibited cell growth, decreased cell viability, and increased gamma-H2AX staining under conditions of caspase inhibition, indicating activation of the DNA damage/repair pathway. Real-time PCR analysis in tumor cells and normal human epithelial cells revealed Clld7 target genes that regulate DNA repair responses. Furthermore, depletion of Clld7 in normal human epithelial cells conferred resistance to apoptosis triggered by DNA damage. Taken together, the biological actions of Clld7 are consistent with those of a tumor suppressor.
Keywords: tumor suppressor, DNA damage/repair pathway, 13q14, primary human keratinocyte, apoptosis
Introduction
Chromosome 13q14 deletions frequently occur in several human malignancies, including B-cell chronic lymphocytic leukemia (1, 2), non-Hodgkin’s lymphoma (3, 4), acute lymphoblastic leukemia (5), lung cancer (6, 7), prostate cancer (8, 9), head and neck cancer (10), and esophageal cancer (11, 12), suggesting the presence of one or more tumor suppressor gene(s) in this region. The chronic lymphocytic leukemia deletion gene 7 (Clld7) (also called RCBTB1, CLLL7, E4.5 (13), and GLP (14)) is a candidate tumor suppressor that maps to chromosome 13q14 (15). The CLLD7 gene encodes a 531-amino-acid protein that contains two functional domains: a regulator of chromosome condensation (RCC1) domain at the N-terminus and a broad complex, tramtrack, and bric-a-brac (BTB) domain at the C-terminus. The prototype RCC1 protein functions as a guanine nucleotide exchange factor for the Ras-related nuclear protein (Ran) GTPase (16). The C-terminal BTB domain mediates protein-protein interactions, and serves as an adaptor for cullin 3 based ubiquitin ligases (17–19).
Despite the fact that Clld7 is a candidate tumor suppressor, there is limited information on the biological activities of this protein. The sole functional study with Clld7 to date is a report linking Clld7 overexpression to cellular hypertrophy in cultured rat vascular smooth muscle and renal proximal tubule cells (14).
In this study, we report that Clld7 expression is decreased in colon cancer, cervical cancer, and lymphoma cell lines. Clld7 overexpression in U2OS human osteosarcoma cells reduced colony formation and significantly decreased cell viability. Moreover, upon expression of Clld7, the DNA damage/repair pathway was activated as evidenced by increased γ-H2AX staining and increased transcription of DNA damage related genes such as GADD34, GADD45A, and GADD153. In addition, depletion of Clld7 in primary human epithelial cells caused a marked decrease in apoptosis in response to treatment with the chemotherapy agent cisplatin. These findings suggest that Clld7 may regulate DNA damage/repair pathways and are consistent with a potential role of Clld7 as a tumor suppressor.
Materials and Methods
Cell lines and plasmids
Human osteosarcoma U2OS, cervical carcinoma CaSki, SiHa, HeLa, C33A, colon cancer Caco-2, Colo205, HCT116, and RKO cells and human leukemia and lymphoma K562, GP-10, Jurkat, and U937 lines were obtained from ATCC cells cultured as recommended. The human Burkitt’s lymphoma cell lines, BJAB and BL41, were provided by Dr. Elliot Kieff (Brigham and Women’s Hospital, Boston, MA) and cultured in RPMI 1640 medium (Invitrogen), 10% calf serum, penicillin (50 units/ml), and streptomycin (50 μg/ml). U2OS tet on cells (Becton Dickinson) were cultured in DMEM, 10% tetracycline free fetal bovine serum (Invitrogen) and 200 μg/ml G418. Clld7 expressing cell lines were established by transfecting U2OS tet on cells with pTRE2hygro (Becton Dickinson) or pTRE2hygro containing C terminally hemagglutinin (HA)-tagged Clld7. After 14 days of selection with hygromycin, individual colonies were picked and expanded. To induce the expression of Clld7, cells were treated with 2 μg/ml doxycycline (Dox) for 3 days unless indicated otherwise.
Primary human foreskin and cervical keratinocytes were prepared as described (20, 21) and cultured in Keratinocyte Serum Free Medium (K-SFM, Invitrogen) containing human epidermal growth factor 1–53 (EGF 1–53), bovine pituitary extract (BPE), penicillin (100 U/ml), streptomycin (100 μg/ml), gentamicin (10 μg/ml), and amphotericin B (0.5 μg/ml).
Colony formation assay
U2OS cells were seeded into six-well plates at a density of 8×104 cells/well. Cells were transfected using Fugene 6 (Roche) with 1μg Clld7-pcDNA 3.1 plasmid, which contains C-terminally HA-tagged Clld7 cDNA. Transfection of pcDNA 3.1(Invitrogen) was used as a control. Twenty-four hours after transfection, cells were selected with 600 μg/ml G418. At 10 days post selection, surviving cells were stained with sulforhodamine B (Sigma), and quantified in a plate reader (22).
Real-time PCR
Total RNA from cells was extracted using the RNeasy Mini Kit (Qiagen). Genomic DNA was eliminated by on-column digestion during RNA extraction using RNase-Free DNase Set (Qiagen). Clld7 expression in was quantified by real-time PCR using the QuantiTect SYBR Green kit (Qiagen) using an ABI Prism 7300 instrument (Applied Biosystems); RNA from normal colon epithelial cells (Invitrogen) and human peripheral blood lymphocytes (Invitrogen) were used as controls. Each sample was tested in triplicate. The average cycle number at threshold [Ct] was normalized against GAPDH. The expression level of Clld7 was calculated based on the 2−ΔΔCt method. Water was used as a negative control. PCR primers are summarized in Table S1.
The human DNA damage RT2 Profiler PCR Array and RT2 Real-Time SYBR Green/ROX PCR Mix were purchased from SA Biosciences (Frederick, MD). PCR array analysis was performed and analyzed according to the manufacturer’s instructions (http://www.sabiosciences.com/pcr/arrayanalysis.php).
Fluorescence-activated cell sorting (FACS)
For cell cycle analysis, U2OS tet on lines transfected with either empty vector or expressing Clld7 (H661) were seeded into a six-well plate at the density of 5 × 104 cells/well. Cells were harvested after induction with 2 μg/ml Dox for three days, washed with phosphate buffered saline (PBS) and fixed in 75% ethanol. DNA was stained with propidium iodide (50 μg/ml) and analyzed with a FACSCalibur system using CellQuest software (Becton Dickinson).
For quantification of γ-H2AX activation in H661 cells, cells were treated with 2 μg/ml Dox for two days, fixed and processed using the γ-H2AX phosphorylation assay kit for flow cytometry (Millipore, #17-344, Billerica, MA). Fixed Cells were incubated with either FITC labeled γ-H2AX antibody or mouse IgG as control at 4°C overnight followed by propidium iodide staining and analyzed using a FACSCalibur instrument (Becton Dickinson). Data were analyzed using FlowJo software (Tree Star, San Carlos, CA).
Cell proliferation analysis
AlamarBlue (Biosource) was used to assess cell viability/proliferation. H661 cells and control cells were seeded into a 96-well plate at a density of 2,000 cells/well. AlamarBlue was added to the culture medium for 10 hours and the fluorescence signal of the dye was captured in a Wallac VICTOR3 1420 plate reader (Perkin Elmer) and analyzed using the Wallac 1420 program.
To determine the effect of caspase inhibitors on cell viability, H661 and control cells were seeded into a 96-well plate at a density of 2,000 cells/well. Twelve hours after induction with 2μg/ml Dox, pan-caspase inhibitors QVD-Oph (20 μM) or z-VAD (50 μM) were added to cells for 48 hours before AlamarBlue staining or Western blot analysis. To test whether Clld7 induced growth inhibition is p53-dependent, H661 cells were transfected with either p53 siRNA (Thermo Fisher Scientific, L-003329-00-0005) or non-targeting siRNA control (Thermo Fisher Scientific, D-001810-10-05) using Lipofectamine 2000 (Invitrogen). At 16 hours after transfection, cells were seeded into 96-well plates at a density of 5,000 cells/well. Cells were induced for 2 days or 3 days before AlamarBlue assays.
Immunological Methods
Immunofluorescence experiments were performed as previously described (23). Theγ-H2AX antibody (Millipore, #05-636) and an Alexa Fluor 488 labeled secondary antibody (Invitrogen, #A11001) was used for γ-H2AX detection. Images were taken with a laser scanning Zeiss Axioskop PCM2000 confocal fluorescence microscope. Cells with >5γ-H2AX foci were counted as positive. Phase contrast pictures were taken using a Zeiss AxioCam Mrm inverted microscope and processed with AxionVision Rel 4.4 software. For Western blotting, cells were lysed in 0.5% Nonidet P-40 (NP40), 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), and protease inhibitor cocktail (Roche) and cleared by centrifugation at 4°C at 16,000 g for 5 min. Protein concentrations were determined by the Bradford method (Bio-Rad). 150 μg aliquots were analyzed by SDS-PAGE and transferred to PVDF membranes (Immobilon-P, Millipore). Membranes were blocked for one hour in 5% nonfat dry milk in TNET buffer (200 mM Tris-HCl, 1 M NaCl, 50 mM EDTA, 0.1% Tween 20, pH 7.5) and probed with primary antibodies for one hour. Antibodies used were α-actin (Chemicon, #MAB1501), α-tubulin (Cell Signalling, #2144), HA (Santa Cruz, #sc-805), CDK7 (Cell Signaling, #2916),γ-H2AX (Millipore, #05-636), GADD45A (Santa Cruz, #sc-797), p53 (Millipore, #CBL404) and Phospho-Ser15-p53 (Cell Signaling, #9286). Secondary antibodies were horseradish peroxidase-linked anti-mouse or anti-rabbit IgG (GE Healthcare). Detection was done with enhanced chemiluminescence (Perkin Elmer Life Sciences, Inc.) followed by digital acquisition and quantification on a Kodak 4000R Image Station (Kodak) using Kodak Imaging Software (version 4.0).
See supplemental materials and methods for immunoprecipitation.
RNAi transfection
Human foreskin keratinocytes were transfected with 200 pmol of Clld7 specific siRNAs, or a control siRNA duplex (Thermo Fisher Scientific) using the Human Keratinocyte Nucleofector Kit (Lonza Company).
For the apoptosis resistance experiment, siRNA transfected HFKs were seeded on coverslips. Three days after transfection, cells were treated with 10 μg/ml cisplatin (Sigma) for 23 hours. After treatment, cells were fixed and nuclei were stained as described above. At least 1,000 cells for each condition were counted and cells showing nuclear fragmentation were defined as apoptotic cells.
Statistics
Student unpaired t test or two-way ANOVA analysis was used.
Results
Clld7 copy number is reduced and expression is decreased in human tumor cell lines
Deletions in the region of chromosome 13q14, where Clld7 is localized, have been detected in various lymphoid neoplasms (5) as well as in solid human tumors (6, 8). To confirm and extend these observations, we performed online data mining to evaluate Clld7 copy numbers in different tumor cell lines provided by the Wellcome Trust Cancer Genome Project (http://www.sanger.ac.uk). Consistent with a previous study we found no evidence for amplification or point mutation of Clld7 among the 776 human tumor cell lines in the database. Homozygous deletions of Clld7, however, were detected in two tumor cell lines, the SF126 astrocytoma cell line and the acute myeloid leukemia derived KMOE-2 cell line. Evidence for loss of heterozygosity (LOH) of the Clld7 gene was detected in 316 tumor cell lines, representing 29 tumor types. For example, 4 out of 12 cervical cancer samples show evidence for LOH within the Clld7 gene locus. LOH of Clld7 is almost as frequent as that of a well-established tumor suppressor, Rb1, in this collection of cell lines (Table S2).
Furthermore, we evaluated Clld7 expression in 14 tumor cell lines by quantitative real-time reverse transcription (QRT) PCR. RNAs from corresponding normal cells were used as controls. Three out of four cervical cancer cell lines expressed Clld7 at ~50% lower levels than normal human cervical keratinocytes (Figure 1). Moreover, three colon cancer cell lines showed >50% decreased expression of Clld7 as compared to normal colon epithelial cells. In lymphoma and leukemia cell lines, expression of Clld7 was reduced in five out of six cell lines examined. Hence, Clld7 expression is reduced in a variety of human tumor cell lines.
Figure 1.

Real-time PCR analysis of Clld7 expression in tumor lines. Clld7 expression in each tumor cell line (black bars) is indicated by its ratio to that of the corresponding normal tissues (white bars). GAPDH was used as the reference.
Clld7 has growth inhibitory activity in osteosarcoma cells
We next aimed to test the effect of ectopic Clld7 expression in cells with reduced endogenous Clld7 expression. The human osteosarcoma cell line U2OS, which expresses relatively low levels of Clld7, was chosen as a model (Figure S1A). U2OS cells were transfected with either a pcDNA3 based expression vector for C-terminally HA-tagged Clld7 or pcDNA 3 vector as a control followed by G418 selection. While we obtained numerous drug resistant colonies of control vector transfected U2OS cells, only very few, small colonies were obtained when cells were transfected with the Clld7 expression vector (Figure 2A, left panel). Quantification of live cells revealed that Clld7 expression caused a statistically significant inhibition of cell growth (100 ±0.067% for control versus 23.4±0.24% in Clld7 expressing cells; p<0.001, Figure 2A, right panel). To further investigate this growth suppressive activity of Clld7, we generated clonal U2OS cell lines with tetracycline-inducible expression of C-terminally HA-tagged Clld7 (HA-Clld7). Single-cell clones were selected and expanded. Clone H661, which shows robust induction of HA-Clld7 upon doxycycline treatment was chosen for further experiments. Consistent with our ectopic expression experiments, doxycyclin mediated induction of Clld7 (Figure 2B), caused growth suppression in U2OS cells (Figure 2C). To quantitate Clld7 mediated growth suppression we used AlamarBlue, a dye that probes mitochondrial fitness. These experiments showed that there was an ~40% percent decrease of cell proliferation/viability within 3 days of doxycycline treatment (Figure 2D). Similar growth inhibitory effects of Clld7 expression were observed with various inducible expression levels of Clld7 (Figure S1B) and in three additional clones (data not shown). To determine whether the growth inhibitory effect induced by Clld7 expression is p53 dependent, we depleted p53 by transfecting H661 cells with a pool of p53 specific siRNAs or a non-targeting control siRNA. Western blot analysis documented efficient p53 knockdown (Figure 2D, lower right panel) in H661 cells. Induction of Clld7 expression in p53 depleted H661 cell caused a decrease in cell proliferation/viability similar to control siRNA transfected cells (Figure 2D lower left panel). This indicates that Clld7 inhibits cell proliferation/viability through p53-independent pathway.
Figure 2.
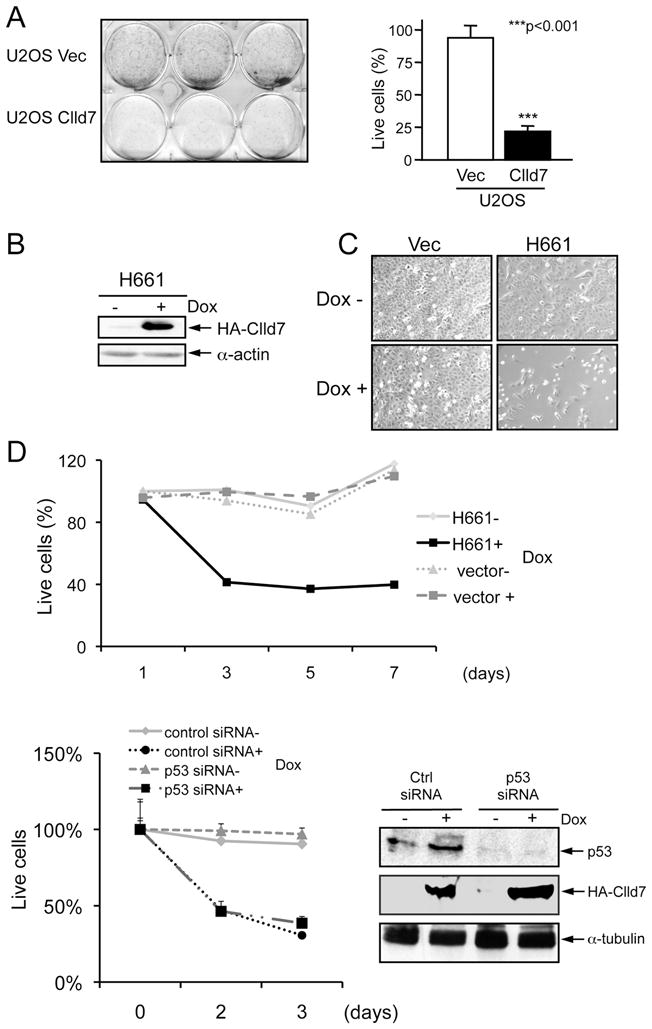
Suppression of cell proliferation by Clld7 in U2OS cells. A. Overexpression of Clld7 decreases colony formation in U2OS cells. After 10 days of G418 selection, live cells were stained with sulforhodamine B (SRB). In the left panel, wells in the upper row are cells transfected with vector; wells in the lower row are cells transfected with Clld7. Right panel is a bar graph depicting the percentage of live cells in the experiment shown in the left panel. Results represent averages and standard deviations from triplicate wells. B. Inducible expression of Clld7 in H661, a clonal U2OS tet on line expressing C-terminal HA epitope tagged Clld7, as assessed by Western blotting. α-actin was used as a loading control. C. Decreased H661 cell density after 3 days of Clld7 expression by doxycycline (Dox) treatment (2 μg/ml). Treatments with and without Dox are indicated by + and −. D. Cell viability of H661 and U2OS tet on-vector cells as assessed by AlamarBlue staining. Upper panel: H661 cells were treated with Dox for the indicated times. Lower Panel: H661 cells were transfected with p53 specific siRNA or a non-targeting control siRNA followed by Dox treatment for the indicated times. Efficiency of p53 depletion and Clld7 expression were confirmed by Western blot (right panel). α-tubulin was used as loading control.
Decreased cell viability as a consequence of Clld7 expression is partially due to apoptosis
We next determined the effect of Clld7 expression on the cell cycle profile of U2OS cells. Flow cytometric analyses revealed a significant increase of cells with sub-G1 DNA content from 1% to 32% at 72 hours of Clld7 expression, representing increased DNA fragmentation upon Clld7 expression (Figure 3A; lower panels, 3B). In addition, the percentages of cells in G0/G1 and G2/M dropped from 48% to 30% and 28% to 18%, respectively. In contrast, doxycyline treated control cells showed no increase in the percentage of cells with sub-G1 DNA content and minimal changes in their G0/G1 and G2/M populations (57% to 54% and 22% to 23%, respectively) (Figure 3A; upper panels, 3B).
Figure 3.

Clld7 overexpression decreases cell viability through induction of apoptosis. A. Cell cycle profile determined by FACS in H661 cells. Representative data from three independent experiments are shown. Percentage of sub-G1 phase is graphed in B. C. Pan-caspase inhibitor QVD (20 μM) abrogates Dox mediated Clld7 expression in H661 cells. D. Cell viability in C is determined by AlamarBlue staining. Caspase inhibitors z-VAD (50 μM) and QVD (20 μM) were added at 12 hours after Dox treatment for 48 hours. Cell viability in H661 cells without Dox and caspase inhibitor treatment is defined as 100%.
To determine whether the observed increase in the sub G1 population upon doxycyline treatment represents caspase-dependent apoptosis, we treated cells with the pan-caspase inhibitors, QVD-Oph or z-VAD. As expected, caspase inhibition partially abrogated the decrease in cell density upon Clld7 expression (Figure 3C). Quantification by AlamarBlue staining showed that QVD-Oph and z-VAD significantly increased H661 cell survival cells from 26±1.1% to 64%±7.13 and 45±3.34% (p<0.001), respectively (Figure 3D).
Clld7 activates the DNA damage/repair pathway
To investigate the molecular mechanism that may cause apoptosis in response to Clld7 expression, we first evaluated whether the DNA damage/repair pathway may be activated due to expression of Clld7. We performed immunofluorescence staining for serine 139 phosphorylation of histone H2AX (γ-H2AX), a marker for DNA double-strand breaks. We observedγ-H2AX foci in H661 cells but not in control cells upon Dox treatment (Figure 4A, left panel). The incidence of γ-H2AX foci (>5 foci/cell) was significantly higher in Dox treated H661 cells than in untreated H661 cells (15.3±8% compared to 1.4±1.3%, p<0.05, Figure 4A, middle panel), and γ-H2AX foci co-localized with phosphorylated Chk2 (Figure S2). Increased expression of γ-H2AX in Dox treated H661 cells was confirmed by immunoblot experiments (Figure 4A, right panel). Quantification by FACS showed thatγ-H2AX positive cells increased from 2.4% to 12.5% after Clld7 induction by Dox in H661 cells. Theγ-H2AX positive cells are distributed throughout the different phases of cell cycle and are not specifically increased in apoptotic cells with sub G1 content (Figure 4B).
Figure 4.

Clld7 induced γ-H2AX foci in U2OS cells. A. Left panel: representative immunofluorescence images showing accumulation of γ-H2AX foci (green) upon inducible expression of Clld7 (H661+). Empty vector transfected cells are shown as a control (Vec− and Vec+). Inserted pictures are zoomed-in images. Middle panel: quantification of γ-H2AX positive cells (>5 foci/cell) in H661+/− and Vec+/−. ~300 cells were counted for each group. Averages and standard deviations are from three independent experiments. Right panel: Western blot analysis of γ-H2AX in H661+/− and Vec+/− cells. B. Live cells stained with γ-H2AX are quantified in H661 upon Dox mediated Clld7 induction by FACS. H661 cells +/− Dox treatment were stained with FITC labeled γ-H2AX antibodies or FITC labeled mouse IgG as a control. Cells were co-stained with Propidium iodide to determine DNA content. C. Immunofluorescence images of γ-H2AX foci (green) (left panel) and quantification of γ-H2AX staining (middle panel) in H661 cells with or without Dox and caspase inhibitor QVD (20 μM) treatment. Approximately 300 cells were counted for each group. Averages and standard deviation are from three independent experiments. Right panel, Western blot analysis of γ-H2AX and inducible expression of HA-Clld7 in H661 cells with and without QVD treatment. D. Flow cytometry analysis of cells used in C after cells were stained with Propidium iodide.
To ensure that γ-H2AX foci did not result from nucleosomal DNA fragmentation during apoptosis, we treated H661 cells with the pan-caspase inhibitor QVD. These experiments revealed that both the number of γ-H2AX foci per cell and the size of γ-H2AX foci in H661 cells upon Clld7 induction by Dox were not dramatically altered upon QVD treatment (Figure 4C), whereas QVD efficiently inhibited nucleosomal DNA fragmentation (Figure 4D).
In combination these data indicate that γ-H2AX focus formation upon inducible expression of Clld7 likely represent a primary effect of Clld7 expression and is not caused by DNA fragmentation during apoptosis.
To determine whether Clld7 may play a role in the DNA damage/repair pathway, we performed a pathway-focused qRT-PCR array analysis (SA Biosciences) in H661 cells with and without Dox treatment. Using 2.5-fold modulation of gene expression as a cutoff, we found that 17 of 84 genes were up-regulated (Figure 5A) and four genes were down-regulated upon Clld7 expression (Figure S2A, B). 13 of the 17 up-regulated genes have well-established roles in DNA replication, recombination, and repair pathways, which are vital to maintain genome integrity (24). To validate these results, we performed qRT-PCR experiments. BTG2 showed a 2.5-fold increase, ERCC2, CCNH, CDK7, GADD34, and GADD135 displayed 4-fold increases and GADD45A showed an ~15-fold increase in expression in H661 cells after Clld7 induction (Figure 5B). Increased expression of GADD45A in H661 upon Dox treatment was also confirmed by Western blotting (Figure 5C). These results suggest that Clld7 activates the DNA damage/repair pathway at least in part through transcriptional up-regulation of DNA damage/repair pathway genes.
Figure 5.

Overexpression of Clld7 deregulates expression of genes in the DNA damage/repair pathway. A. PCR-array analysis documents 17 genes with increased expression upon inducible expression of Clld7 in H661 cells. B. Real-time PCR documents increased expression of 7 genes in H661+ cells as compared to H661- cells. Vector transfected cells were used as control. C. Increased GADD45A protein expression upon Dox-induced expression of Clld7 in H661 cells as assessed by Western blot. α–tubulin was used as a loading control.
Clld7 depletion in primary human epithelial cells confers resistance to apoptosis in response to DNA damaging chemotherapy agents
Given that overexpression of Clld7 decreased cell viability and increased formation of γ-H2AX foci in a human tumor cell line, we set out to determine whether Clld7 may play a similar role in normal human epithelial cells. We depleted expression of Clld7 in primary human foreskin keratinocytes (HFKs) by siRNA. Transfection of scrambled siRNA was used as a control. Clld7 mRNA was reduced by ~70% as assessed qRT-PCR (Figure 6A). Next we assessed whether Clld7 depletion in HKFs affects expression of DNA damage/repair pathway genes. QRT- PCR analysis revealed an ~50% decrease in GADD34 and GADD45 expression in HFKs transfected with Clld7 specific siRNAs as compared to control siRNA transfected HFKs (Figure 6A). These experiments suggest that Clld7 is involved in controlling expression of DNA damage/repair pathway genes in normal human epithelial cells.
Figure 6.

Depletion of Clld7 in primary human keratinocytes increases resistance to cisplatin-induced apoptosis. A. Decreased expression of GADD45A and GADD34 after Clld7 depletion. Human primary foreskin keratinocytes (HFKs) were transfected with a Clld7 specific siRNA pool, an individual Clld7 specific siRNA, or control siRNA. Five days after transfection, RNA was extracted for real-time PCR detection. B. HFKs transfected with a Clld7 specific siRNA pool or a control siRNA were treated with the chemotherapy agent cisplatin (10μg/ml) for 23 hours and stained with Hochest33258. A minimum 1,000 cells were counted blindly in each group from three independent populations of HFKs. C. Representative images of HFKs from B. D. HFKs are transfected with non-targeting control and Clld7 siRNA followed by cisplatin treatment (10 μg/ml) for 23 hours. Cell lysates were analyzed by western blot for the expression of Serine 15-phosphorylated p53 and total p53. α–actin was used as a loading control.
To analyze the effect of Clld7 depletion on apoptosis induced by DNA damage in primary human epithelial cells, we treated HFKs transfected with Clld7 specific siRNAs or control siRNA with the DNA damaging chemotherapy agent cisplatin at 3 days post-siRNA transfection. The apoptotic rate of HFKs treated with cisplatin dropped from 63±11% in control siRNA transfected HFKs to 35±8.6% in HFKs transfected with Clld7 siRNA (p<0.05, Figure 6B). Representative pictures of apoptotic cell nuclei stained with Hoechst 33258 are shown in Figure 6C. Similar results were obtained with three independent HFK populations.
To determine whether Clld7 downregulation in HFKs may inhibit p53 activation, we assessed p53 activation in response to cisplatin treatment in HFKs with siRNA mediated Clld7 depletion. These experiments revealed that Clld7 depletion did not compromise p53 Ser15 phosphorylation or p53 stabilization in HFKs (Figure 6D). Hence Clld7 may not directly control the capacity of primary cells to sense DNA damage and activate p53.
Discussion
Chromosome 13q14 deletions are frequently detected in a variety of human tumors suggesting the presence of tumor suppressors in this region. Several genes cloned from 13q14 including ARL11 (25), RGC32 (26), and RFP2 (27, 28) have been reported to inhibit tumor growth. Here we investigated whether Clld7 may also be a 13q14 tumor suppressor candidate. By mining publically available data database, we found that at least one copy of Clld7 is lost in ~50% (319/776) of tumor cell lines that have been examined (Table S2). Our analysis of this data set, however, does not provide evidence for Clld7 point mutations in these cell lines. By qRT PCR, expression of Clld7 was shown to be low in multiple tumor cell lines (Figure 1). Since analysis of CpG islands on chromosome 13q14.3 revealed no evidence for significant methylation in B-CLL patients (29), promoter methylation may not significantly contribute to the reduced expression of tumor suppressors in the vicinity of 13q14.
To determine whether Clld7 may be a growth suppressor, we expressed the protein in the U2OS human osteosarcoma line that has relatively low endogenous Clld7 expression. We found that cell growth and survival were greatly inhibited (Figure 2C, D,) and that this inhibition was independent of p53 tumor suppressor activity (Figure 2D). Similar results were also obtained with clones with lower level Clld7 expression of (data not shown) suggesting that the observed growth inhibitory effects were not due to high-level Clld7 expression.
We noted an increased incidence of nuclear foci of γ-H2AX upon Clld7 expression (Figure 4A). Nuclear γ-H2AX foci decorate double strand DNA breaks and thus serve as a marker for activation of the DNA damage/repair pathway (30). Activation of the DNA damage/repair pathway was detected in precancerous lesions by two independent groups in lung cancer (31) as well as bladder tumor specimens (32). It is purported that the activation of the DNA damage/repair pathway functions as a cell intrinsic tumor suppressor pathway in early potentially precancerous lesions (33). Since caspase inhibition inhibits cell death but does not affect the incidence of γ-H2AX nuclear foci (Figure 4C), the appearance of these foci does not represent a consequence of nucleosomal DNA fragmentation during apoptosis (Figure 4D). Therefore, we hypothesize that Clld7 may exert its tumor suppressive activity by regulating DNA damage responses, which, in turn, may lead to growth inhibition and/or apoptosis. To molecularly characterize the DNA damage response triggered by Clld7 expression, we investigated mRNA expression of genes that have been implicated in DNA damage/repair. These experiments showed that 17 of 84 genes were transcriptionally up-regulated by Clld7, including GADD45A, GADD153, and GADD34 (Figure 5A and B). To rule out that the observed activation of the DNA damage pathway represents a consequence of high-level Clld7 expression, we also performed loss-of-function experiments where we depleted Clld7 in primary human epithelial cells. These experiments revealed that Clld7 depletion caused decreased expression of GADD45A and GADD34 (Figure 6A).
Given these results we investigated whether, through targeting the DNA/damage repair pathway, Clld7 may alter cell susceptibility to DNA damaging chemotherapy agents. Indeed, Clld7 depletion in primary human keratinocytes confers resistance to cisplatin and dampens the apoptotic response through a p53 independent pathway (Figure 6B, C, D). This finding is consistent with a previous report that GADD45A depletion inhibited apoptosis upon genotoxic insults (34). Similarly, GADD34 depletion also inhibited apoptosis by cisplatin in human mesothelioma cell lines (35). Hence we hypothesize that Clld7 depletion bestows resistance to apoptotic stress through down-regulating GADD45A and GADD34 expression in primary cells. Given that, presumably due to allelic loss, Clld7 is expressed at low levels in many tumor cell lines, our results suggest that the Clld7 expression level may be predictor of the sensitivity of tumor cells to chemotherapy.
The molecular basis of transcriptional regulation of DNA damage response genes by Clld7 is unknown. The BTB domain may bind to DNA and reprogram gene transcription as most other conserved BTB proteins (36, 37). However, consistent with a previous report (14), we found no evidence for nuclear localization of Clld7 (data not shown). Nevertheless, we observed that ectopic expression of a Clld7 fragment that consists of the BTB domain more potently suppressed U2OS cell proliferation than the isolated RCC1 domain of Clld7 (data not shown), suggesting that the BTB domain may play an important role in the growth suppressive activity of Clld7. As reported previously, BTB proteins function as substrate specific adaptors for Cullin 3 ubiquitin ligase complexes (17, 18, 38). Consistent with this notion and in agreement with a recent publication (39), co-immunoprecipitation experiments showed that Clld 7 can associate with cullin 3 (supplemental Figure 4). The identification of substrate(s) of the Clld7-Cul3 complex may provide insights into the mechanism by which Clld7 regulates DNA damage/repair processes.
In summary, we provide data that are consistent with a role of Clld7 as a 13q14 tumor suppressor: first, the expression level of Clld7 in several cancer cell lines was significantly reduced; second, Clld7 overexpression decreased cell viability through activating the DNA damage/repair pathway; third, depletion of Clld7 in human primary keratinocytes decreased DNA damage/repair gene expression and conferred resistance to the apoptosis-inducing drug cisplatin.
Supplementary Material
Acknowledgments
Supported by PHS grant R01 DE015302 and CA081135 (K.M.).
We thank Jun Lu for help with FACS analysis, Christopher Crum and Amy Baldwin for human cervical keratinocytes, Margaret McLaughlin-Drubin and Xiaodong Sun for helpful comments on the manuscript, Wade Harper for the myc-cullin 3 plasmid and Siribang-on Piboonniyom for the Clld7 expression vector.
References
- 1.Ripolles L, Ortega M, Ortuno F, et al. Genetic abnormalities and clinical outcome in chronic lymphocytic leukemia. Cancer Genet Cytogenet. 2006;171:57–64. doi: 10.1016/j.cancergencyto.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 2.Cotter FE, Auer RL. Genetic alteration associated with chronic lymphocytic leukemia. Cytogenet Genome Res. 2007;118:310–9. doi: 10.1159/000108315. [DOI] [PubMed] [Google Scholar]
- 3.Wada M, Okamura T, Okada M, et al. Delineation of the frequently deleted region on chromosome arm 13q in B-cell non-Hodgkin’s lymphoma. Int J Hematol. 2000;71:159–66. [PubMed] [Google Scholar]
- 4.Wada M, Okamura T, Okada M, et al. Frequent chromosome arm 13q deletion in aggressive non-Hodgkin’s lymphoma. Leukemia. 1999;13:792–8. doi: 10.1038/sj.leu.2401395. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Hermanson M, Grander D, et al. 13q deletions in lymphoid malignancies. Blood. 1995;86:1911–5. [PubMed] [Google Scholar]
- 6.Kwong FM, Wong PS, Lung ML. Genetic alterations detected on chromosomes 13 and 14 in Chinese non-small cell lung carcinomas. Cancer Lett. 2003;192:189–98. doi: 10.1016/s0304-3835(02)00698-5. [DOI] [PubMed] [Google Scholar]
- 7.Wistuba, Behrens C, Milchgrub S, et al. Sequential molecular abnormalities are involved in the multistage development of squamous cell lung carcinoma. Oncogene. 1999;18:643–50. doi: 10.1038/sj.onc.1202349. [DOI] [PubMed] [Google Scholar]
- 8.Latil A, Chene L, Mangin P, Fournier G, Berthon P, Cussenot O. Extensive analysis of the 13q14 region in human prostate tumors: DNA analysis and quantitative expression of genes lying in the interval of deletion. Prostate. 2003;57:39–50. doi: 10.1002/pros.10272. [DOI] [PubMed] [Google Scholar]
- 9.Dong JT, Boyd JC, Frierson HF., Jr Loss of heterozygosity at 13q14 and 13q21 in high grade, high stage prostate cancer. Prostate. 2001;49:166–71. doi: 10.1002/pros.1131. [DOI] [PubMed] [Google Scholar]
- 10.Gupta VK, Schmidt AP, Pashia ME, Sunwoo JB, Scholnick SB. Multiple regions of deletion on chromosome arm 13q in head-and-neck squamous-cell carcinoma. Int J Cancer. 1999;84:453–7. doi: 10.1002/(sici)1097-0215(19991022)84:5<453::aid-ijc1>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 11.Pack SD, Karkera JD, Zhuang Z, et al. Molecular cytogenetic fingerprinting of esophageal squamous cell carcinoma by comparative genomic hybridization reveals a consistent pattern of chromosomal alterations. Genes Chromosomes Cancer. 1999;25:160–8. doi: 10.1002/(sici)1098-2264(199906)25:2<160::aid-gcc12>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 12.Hu N, Roth MJ, Polymeropolous M, et al. Identification of novel regions of allelic loss from a genomewide scan of esophageal squamous-cell carcinoma in a high-risk Chinese population. Genes Chromosomes Cancer. 2000;27:217–28. doi: 10.1002/(sici)1098-2264(200003)27:3<217::aid-gcc1>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 13.Solomou EE, Sfikakis PP, Kotsi P, et al. 13q deletion in chronic lymphocytic leukemia: characterization of E4.5, a novel chromosome condensation regulator-like guanine nucleotide exchange factor. Leuk Lymphoma. 2003;44:1579–85. doi: 10.3109/10428190309178782. [DOI] [PubMed] [Google Scholar]
- 14.Guo DF, Tardif V, Ghelima K, et al. A novel angiotensin II type 1 receptor-associated protein induces cellular hypertrophy in rat vascular smooth muscle and renal proximal tubular cells. J Biol Chem. 2004;279:21109–20. doi: 10.1074/jbc.M401544200. [DOI] [PubMed] [Google Scholar]
- 15.Mabuchi H, Fujii H, Calin G, et al. Cloning and characterization of CLLD6, CLLD7, and CLLD8, novel candidate genes for leukemogenesis at chromosome 13q14, a region commonly deleted in B-cell chronic lymphocytic leukemia. Cancer Res. 2001;61:2870–7. [PubMed] [Google Scholar]
- 16.Hadjebi O, Casas-Terradellas E, Garcia-Gonzalo FR, Rosa JL. The RCC1 superfamily: from genes, to function, to disease. Biochim Biophys Acta. 2008;1783:1467–79. doi: 10.1016/j.bbamcr.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 17.Geyer R, Wee S, Anderson S, Yates J, Wolf DA. BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Mol Cell. 2003;12:783–90. doi: 10.1016/s1097-2765(03)00341-1. [DOI] [PubMed] [Google Scholar]
- 18.Xu L, Wei Y, Reboul J, et al. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature. 2003;425:316–21. doi: 10.1038/nature01985. [DOI] [PubMed] [Google Scholar]
- 19.Pintard L, Willis JH, Willems A, et al. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature. 2003;425:311–6. doi: 10.1038/nature01959. [DOI] [PubMed] [Google Scholar]
- 20.Woodworth CD, Bowden PE, Doniger J, et al. Characterization of normal human exocervical epithelial cells immortalized in vitro by papillomavirus types 16 and 18 DNA. Cancer Res. 1988;48:4620–8. [PubMed] [Google Scholar]
- 21.Jones DL, Alani RM, Munger K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 1997;11:2101–11. doi: 10.1101/gad.11.16.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun X, Frierson HF, Chen C, et al. Frequent somatic mutations of the transcription factor ATBF1 in human prostate cancer. Nat Genet. 2005;37:407–12. doi: 10.1038/ng1528. [DOI] [PubMed] [Google Scholar]
- 23.Zhou X, Munger K. Expression of the human papillomavirus type 16 E7 oncoprotein induces an autophagy-related process and sensitizes normal human keratinocytes to cell death in response to growth factor deprivation. Virology. 2009;385:192–7. doi: 10.1016/j.virol.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanawalt PC. Paradigms for the three rs: DNA replication, recombination, and repair. Mol Cell. 2007;28:702–7. doi: 10.1016/j.molcel.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Yendamuri S, Trapasso F, Calin GA. ARLTS1 - a novel tumor suppressor gene. Cancer Lett. 2008;264:11–20. doi: 10.1016/j.canlet.2008.02.021. [DOI] [PubMed] [Google Scholar]
- 26.Vlaicu SI, Cudrici C, Ito T, et al. Role of response gene to complement 32 in diseases. Arch Immunol Ther Exp (Warsz) 2008;56:115–22. doi: 10.1007/s00005-008-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baranova A, Hammarsund M, Ivanov D, et al. Distinct organization of the candidate tumor suppressor gene RFP2 in human and mouse: multiple mRNA isoforms in both species- and human-specific antisense transcript RFP2OS. Gene. 2003;321:103–12. doi: 10.1016/j.gene.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 28.van Everdink WJ, Baranova A, Lummen C, et al. RFP2, c13ORF1, and FAM10A4 are the most likely tumor suppressor gene candidates for B-cell chronic lymphocytic leukemia. Cancer Genet Cytogenet. 2003;146:48–57. doi: 10.1016/s0165-4608(03)00126-2. [DOI] [PubMed] [Google Scholar]
- 29.Mertens D, Wolf S, Schroeter P, et al. Down-regulation of candidate tumor suppressor genes within chromosome band 13q14.3 is independent of the DNA methylation pattern in B-cell chronic lymphocytic leukemia. Blood. 2002;99:4116–21. doi: 10.1182/blood.v99.11.4116. [DOI] [PubMed] [Google Scholar]
- 30.Srivastava N, Gochhait S, de Boer P, Bamezai RN. Role of H2AX in DNA damage response and human cancers. Mutat Res. 2009;681:180–8. doi: 10.1016/j.mrrev.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 31.Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 32.Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 33.Venkitaraman AR. Medicine: aborting the birth of cancer. Nature. 2005;434:829–30. doi: 10.1038/434829a. [DOI] [PubMed] [Google Scholar]
- 34.Li LS, Morales JC, Hwang A, Wagner MW, Boothman DA. DNA mismatch repair-dependent activation of c-Abl/p73alpha/GADD45alpha-mediated apoptosis. J Biol Chem. 2008;283:21394–403. doi: 10.1074/jbc.M709954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adusumilli PS, Chan MK, Chun YS, et al. Cisplatin-induced GADD34 upregulation potentiates oncolytic viral therapy in the treatment of malignant pleural mesothelioma. Cancer Biol Ther. 2006;5:48–53. doi: 10.4161/cbt.5.1.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang W, Mi J, Li N, et al. Identification and characterization of DPZF, a novel human BTB/POZ zinc finger protein sharing homology to BCL-6. Biochem Biophys Res Commun. 2001;282:1067–73. doi: 10.1006/bbrc.2001.4689. [DOI] [PubMed] [Google Scholar]
- 37.Kelly KF, Daniel JM. POZ for effect--POZ-ZF transcription factors in cancer and development. Trends Cell Biol. 2006;16:578–87. doi: 10.1016/j.tcb.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J. 2004;23:1681–7. doi: 10.1038/sj.emboj.7600186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plafker KS, Singer JD, Plafker SM. The ubiquitin conjugating enzyme, UbcM2, engages in novel interactions with components of cullin-3 based E3 ligases. Biochemistry. 2009;48:3527–37. doi: 10.1021/bi801971m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.