

Abstract

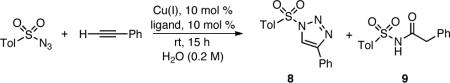
An efficient room temperature method for the synthesis of 1-sulfonyl-1,2,3-triazoles from in situ generated copper(I) acetylides and sulfonyl azides is described. Copper(I) thiophene-2-carboxylate (CuTC) catalyst produces the title compounds under both non-basic anhydrous and aqueous conditions in good yields.
As latent diazo compounds, 1-sulfonyl-1,2,3-triazoles serve as convenient progenitors of reactive azavinyl carbenes.1 These electron-deficient heterocycles stand out as an important exception in the family of generally stable and unreactive 1,2,3-triazoles: the weakened N1–N2 bond in 1-sulfonyl derivatives facilitates their ring-chain isomerism, which leads to the formation of diazoimines, and subsequent decomposition to the transition metal stabilized carbenes.
Copper(I) catalyzed azide-alkyne cycloaddition (CuAAC)2 could well be the most direct and practical route to 1-sulfonyl-1,2,3-triazoles. High fidelity, efficiency, and compatibility with a broad range of functional groups and conditions have made it a widely utilized method for the synthesis of small molecules, bioconjugates, and complex molecular architectures.3 However, the outcome of the CuAAC reaction of electron-deficient azides is affected by a wide variety of factors. Mirroring the well-known Dimroth rearrangement,4,5,6 reaction medium,7 temperature,8 triazole ring substituents,9,10 and ligand choice11,12 all influence the outcome of the reaction (Scheme 1). Judicious choice of reaction conditions currently enables isolation of either N-acyl sulfonamides 77 or 1-sulfonyl triazoles 4. Although several recent reports have described synthesis of 1-sulfonyl triazoles,8,12 none offer the level of generality and simplicity required for wide adoption. In search of a practical, simple, and robust synthesis of sulfonyl triazoles, we examined effects of various ligands on the outcome of the CuAAC reaction of sulfonyl azides. This communication reports our findings.
Scheme 1.

Proposed Intermediates in the Copper(I)-Catalyzed Reaction of Alkynes and Sulfonyl Azides.
In general, ligands that exhibit most dramatic effect on the course of the reaction contain nitrogen heterocycles, (e.g. tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA)13 or 2,6-lutidine).8,11 However, as a “borderline soft” Lewis acid, copper(I) also partners well with sulfur ligands, for example methionine in copper transport proteins and copper-containing enzymes.14 Herein, we report that copper(I) thiophene-2-carboxylate (Liebeskind's reagent)15 is a convenient, bench-stable catalyst for the synthesis of 1-sulfonyl-1,2,3-triazoles effective at ambient temperature both under anhydrous conditions and as a heterogeneous suspension in water.
A variety of sulfur σ-donating ligands (Table 1) were screened under heterogeneous aqueous conditions for their ability to effect selective formation of the 1-sulfonyl triazole 8 over N-acyl sulfonamide 9 (presumably formed via a highly reactive N-sulfonyl ketenimine intermediate such as 6). All results were compared to the CuSO4/sodium ascorbate system (entry 1), which generates and maintains copper in its +1 oxidation state in situ.
Table 1.
1-Sulfonyl Triazole Synthesis: Effect of Copper(I) Source and Sulfur Ligand
| entry | coppert(I) source | ligand | PhCCH consumed (%)a | ratio 8:9a |
|---|---|---|---|---|
| 1 | CuSO4/NaAscb | – | 89c | 6:1 |
| 2 | CuBr | t-Bu2S | 40 | 10:1 |
| 3 | CuBr | BnSPh | 43 | 12:1 |
| 4 | CuBr | thiophene | 37 | 14:1 |
| 5 | CuBr | thiolane | 85 | 2:1 |
| 6 | CuBr | Bn2S | 44 | 3:1 |
| 7 | CuBr | Met | 23 | 1:1 |
| 8 | Cu2O | t-Bu2S | 13 | 1:1 |
| 9 | CuSO4/NaAsc | t-Bu2S | 85c | 13:1 |
| 10 | CuPF6•(MeCN)4 | t-Bu2S | 86c | 13:1 |
| 11 | CuSCN | – | 17 | 2:1 |
| 12 | CuBr•SMe2 | – | 32 | 2:1 |
| 13 | CuTC d | – | 95 c | >100:1 |
As determined by LC/MS analysis, see Supporting Information for details.
NaAsc = sodium ascorbate.
TsN3 undetected by LC/MS.
CuTC = copper(I) thiophene-2-carboxylate. Tol = tolyl.
Relative to the control reaction (entry 1), tert-butyl sulfide, benzyl phenyl sulfide, and thiophene (entries 2–4) in conjunction with CuBr showed improved selectivity for triazole 8 at the expense of conversion. In contrast, thiolane (entry 5) exhibited high conversion with diminished selectivity. Addition of tert-butyl sulfide to the CuSO4/sodium ascorbate system (entry 9) resulted in an improved ratio and maintained high conversion. Attempts to achieve high conversion and selectivity by screening different copper(I) sources in the presence of tert-butyl sulfide were modestly successful (entries 9 and 10). Finally, screening other sulfur containing copper(I) complexes (entries 11–13) led to the identification of copper(I)-thiophene-2-carboxylate (CuTC, entry 13), which dramatically influenced the course of the reaction. Conversion remained high, as seen in entries 1, 9, and 10, yet no N-acyl sulfonamide 9 was detected by LC/MS— resulting in exclusive selectivity under heterogeneous aqueous conditions.
Examination of a panel of solvents (see Supporting Information) for the CuTC-catalyzed 1-sulfonyl triazole synthesis identified water and toluene as optimal solvents, with both leading to complete reactions in 2 h. Interestingly, traditional CuAAC mixed co-solvent conditions (2:1 tert-butyl alcohol:H2O) resulted in both poorer conversions and selectivity. Acetonitrile, a strongly coordinating solvent for copper(I), suppressed product formation entirely, while dichloromethane and chloroform led to complete conversion after 5 h. At lower catalyst loadings (2 mol %), complete conversion and selectivity were achieved in 15 h, attesting to the stability of the catalyst.
A variety of 1-sulfonyl triazoles were synthesized (Scheme 2) using water as the solvent. Despite cooling in an ice water bath, an observed exotherm, most pronounced in the presence of excess sulfonyl azide, prompted a change to toluene as the solvent of choice.
Scheme 2.
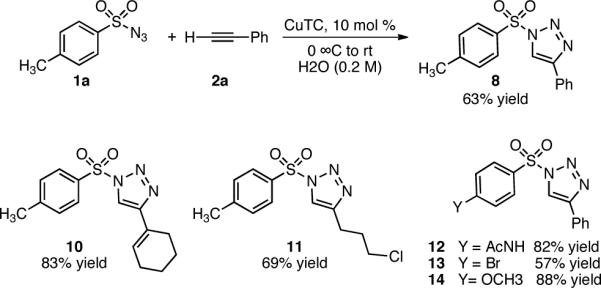
Synthesis of 1-Sulfonyl -1,2,3-Triazoles in Watera
a1.0 mmol scale: CuTC (10 mol %), H2O (0.2 M), alkyne (1–1.3 equiv), azide (1 equiv), 0 °C to rt, 2–18 h.
The general procedure16 for the synthesis of 1-sulfonyl triazoles results in good isolated yields over a range of alkynes with tosyl azide (Table 2). Both aryl alkynes (entry 1) and aliphatic alkynes (entries 2–10) are competent substrates, as well as the strongly electron-donating ethyl ethynyl ether (entry 11). The products were isolated via extraction into ethyl acetate and removal of residual copper by treatment with Cuprisorb resin.17
Table 2.
N-Sulfonyl Triazole Synthesis: Scope with Respect to Alkynea

| entry | alkyne | product | time (h) | yield (%)b |
|---|---|---|---|---|
| 1c |

|
8 | 3.5 | 89 |
| 2d |
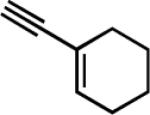
|
10 | 7 | 64 |
| 3c |
|
11 | 1 | 75 |
| 4 |
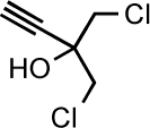
|
15 | 2.5 | 75 |
| 5 |

|
16 | 3.5 | 65 |
| 6 |

|
17 | 4 | 75 |
| 7 |

|
18 | 2 | 83 |
| 8e |
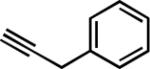
|
19 | 3 | 75 |
| 9f |
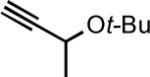
|
20 | 1 | 82 |
| 10e |
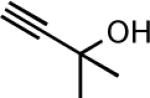
|
21 | 2.5 | 71 |
| 11g |

|
22 | 7 | 75 |
1.0 mmol scale unless otherwise noted; CuTC (10 mol %), toluene (0.2 M), alkyne (1 equiv), tosyl azide (1 equiv), rt.
Isolated yield.
5.0 mmol scale.
1.1 equiv alkyne.
1.3 equiv alkyne.
2.4 equiv alkyne.
1.2 equiv alkyne.
In addition, the general procedure can be applied to a variety of alkyl- and arylsulfonyl azides and alkynes (Table 3).
Table 3.
N-Sulfonyl Triazole Synthesis: Scope with Respect to Sulfonyl Azidea

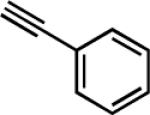
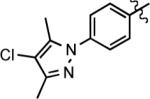
| entry | R1 | alkyne | product | time (h) | yield (%)b |
|---|---|---|---|---|---|
| 1c | 4-BrC6H4 |

|
13 | 3 | 86 |
| 2 | 4-BrC6H4 |

|
23 | 4.5 | 78 |
| 3d | 4-H3COC6H4 |
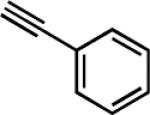
|
14 | 6.5 | 94 |
| 4 | 4-H3COC6H4 |

|
24 | 7 | 79 |
| 5 | Bn |

|
25 | 3.5 | 87 |
| 6e | Me |
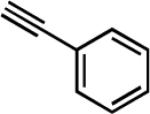
|
26 | 12 | 88 |
| 7e | i-Pr |
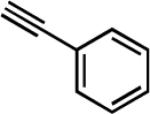
|
27 | 12 | 75 |
| 8 | n-C8H17 |

|
28 | 3.5 | 92 |
| 9f |
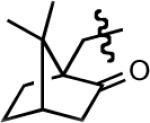
|
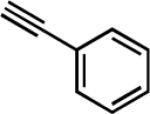
|
29 | 8.5 | 90 |
| 10g |

|
|
30 | 3 | 82 |
| 11g |
|

|
31 | 3 | 83 |
1.0 mmol scale unless otherwise noted; CuTC (10 mol %), solvent (0.2 M), alkyne (1 equiv), sulfonyl azide (1 equiv), rt.
Isolated yield.
5.0 mmol scale.
1.1 equiv alkyne.
1.3 equiv alkyne.
2.4 equiv alkyne.
1.2 equiv alkyne.
Further experiments were designed in order to gain insights into the reactivity of CuTC catalyst. A comparison of three different copper(I) carboxylates (Table 4) revealed decreased conversion and selectivity in aqueous suspensions (entries 2, 3, and 4). Although selectivity remained high for CuOAc (entry 2) and CuOTf (entry 3) in toluene, conversions were low even after 15 h, requiring days to reach completion at room temperature. In stark contrast, the CuTC-catalyzed reaction (entry 1) reached completion both in toluene and under heterogeneous aqueous conditions in 2 h. The furan analog of CuTC (CuFC, entry 4) benefits from the same intramolecular arrangement of σ-donors as CuTC, and as such performed comparably to CuTC in toluene, despite a small decrease in selectivity under aqueous conditions. Interestingly, the addition of thiophene to CuOAc (entry 5) resulted in little improvement in toluene compared to CuOAc alone (entry 2) and lowered both the selectivity and conversion in an aqueous suspension to that of thiophene and CuBr (Table 1, entry 4).
Table 4.
N-Sulfonyl Triazole Synthesis: Scope with Respect to Copper(I) Carboxylate
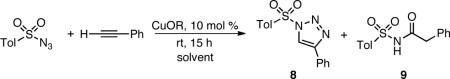
| entry | CuOR | ratio 7:8a (PhCCH consumed (%a)) |
||
|---|---|---|---|---|
| toluene | water | |||
| 1 | CuTC | >100:1 | (98)b,c | >100:1 (95)b,c |
| 2 | CuOAc | >100:1 | (40) | 26:1 (88)b |
| 3 | CuOTf | >100:1 | (17) | 5:1 (27) |
| 4 | CuFCd | >100:1 | (>99) | 31:1 (93)b |
| 5 | CuOAce | >100:1 | (55) | 13:1 (32) |
As determined by LC/MS, see Supporting Information for details.
TsN3 undetected by LC/MS.
2 h.
CuFC = copper(I)-furan-2-carboxylate.
With the addition of thiophene (10 mol %).
CuAAC catalysis is complex and involves multiple dynamic equilibria between copper acetylide species of different coordination geometry, aggregation state, and ligand environment.18 Controlling these equilibria is of paramount importance for channeling the catalysis in the desired direction. Reactions of sulfonyl azides are further complicated by the instability of (1,2,3-triazol-5-yl)copper species 3. Therefore, a successful catalyst should (a) stabilize this intermediate, thereby disfavoring its irreversible conversion to the ketenimine 6 (a thermodynamic consideration) and/or (b) facilitate the protiolysis step, thus favoring formation of the desired triazole product (a kinetic consideration). We contend that CuTC excels at both. It stabilizes the triazolyl intermediate 3 by increasing the electron density of its π-system, thus disfavoring the departure of dintrogen. If this irreversible formation of ketenimine 6 does not occur, formation of 5-H triazole product 4 would be favored.
The carboxylate group of CuTC may also act as a base in toluene, forming 2-thiophene carboxylic acid upon the deprotonation of the alkyne. Assuming that the acid remains coordinated to copper throughout the catalytic cycle, the protiolysis step (3 to 4 in Scheme 1) becomes intramolecular and therefore facile and fast.
That copper(I) acetylides are bona fide intermediates in the path to 1-sulfonyl triazoles finds strong support from the failure of internal alkynes (e.g., diphenylacetylene) to react. Notably, other copper(I) complexes, such as CuBr•SMe2 required the addition of 2,6-lutidine in order to observe conversion in toluene.19 This observation supports our hypothesis that carboxylate of CuTC acts as a base in toluene, and may do so in water. However, its role as a σ-donor cannot currently be ruled out, nor can a contribution from the varying solubility of the various copper(I) sources and ligands.
The data presented in Table 4 indicates that the intramolecular presentation of sulfur, the five-membered aromatic heterocycle, and a carboxylate, are all necessary to achieve both selectivity and rapid conversion.
The disclosed method offers an efficient and broad-in-scope means of accessing 1-sulfonyl-1,2,3-triazoles. The procedure is experimentally simple and allows rapid access to these important intermediates, paving the way for future studies of their utility as well as for probing the mechanism of the CuAAC reaction. These studies are underway and will be reported elsewhere.
Supplementary Material
Acknowledgment
The authors thank Dr. Suresh M. Pitram, Dr. Jason E. Hein, and Mr. Sen Wai Kwok (TSRI) for valuable advice and stimulating discussions. Financial support from the National Institutes of Health, National Institute of General Medical Sciences (GM087620), Eli Lilly (JR), and the Skaggs Institute for Chemical Biology is gratefully acknowledged.
Footnotes
Supporting Information Available Experimental procedures and full spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(a) Horneff T, Chuprakov S, Chernyak N, Gevorgyan V, Fokin VV. J. Am. Chem. Soc. 2008;130:14972. doi: 10.1021/ja805079v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chuprakov S, Kwok SW, Zhang L, Lercher L, Fokin VV. J. Am. Chem. Soc. 2009;131:18034. doi: 10.1021/ja908075u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Miura T, Yamauchi M, Murakami M. Chem. Commun. 2009:1470. doi: 10.1039/b819162j. [DOI] [PubMed] [Google Scholar]; (d) Grimster NP, Zhang L, Fokin VV. J. Am. Chem. Soc. 2010;132:2510. doi: 10.1021/ja910187s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Tornøe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]; (b) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem., Int. Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- (3).(a) Wu P, Fokin VV. Aldrichimica Acta. 2007;40:7. [Google Scholar]; (b) Moses JE. Chem. Soc. Rev. 2007;36:1249. doi: 10.1039/b613014n. [DOI] [PubMed] [Google Scholar]; for a recent collection of reviews, see Chem. Soc. Rev. 2010;39(4)
- (4).(a) Dimroth O. Justus Liebigs Ann. Chem. 1909;364:183. [Google Scholar]; (b) El Ashry ESH, El Kilany Y, Rashed N, Assafir H. Adv. Heterocycl. Chem. 1999;75:79. [Google Scholar]
- (5).Gilchrist TL, Gymer GE. Adv. Heterocycl. Chem. 1974;16:33. [Google Scholar]
- (6).L'Abbé G. J. Heterocycl. Chem. 1984;21:627. [Google Scholar]
- (7).(a) Cho SH, Yoo EJ, Bae I, Chang S. J. Am. Chem. Soc. 2005;127:16046. doi: 10.1021/ja056399e. [DOI] [PubMed] [Google Scholar]; (b) Cassidy MP, Raushel J, Fokin VV. Angew. Chem., Int. Ed. 2006;45:3154. doi: 10.1002/anie.200503805. [DOI] [PubMed] [Google Scholar]
- (8).Yoo EJ, Ahlquist M, Kim SH, Bae I, Fokin VV, Sharpless KB, Chang S. Angew. Chem., Int. Ed. 2007;46:1730. doi: 10.1002/anie.200604241. [DOI] [PubMed] [Google Scholar]
- (9).Harmon RE, Stanley F, Jr., Gupta SK, Johnson J. J. Org. Chem. 1970;35:3444. [Google Scholar]
- (10).Himbert G, Regitz M. Liebigs Ann. Chem. 1973;9:1505. [Google Scholar]
- (11).Whiting M, Fokin VV. Angew. Chem., Int. Ed. 2006;45:3157. doi: 10.1002/anie.200503936. [DOI] [PubMed] [Google Scholar]
- (12).(a) Wang F, Fu H, Jiang Y, Zhao Y. Adv. Synth. Catal. 2008;350:1830. [Google Scholar]; (b) Cano I, Nicasio MC, Pérez PJ. Org. Biomol. Chem. 2010;8:536. doi: 10.1039/b912835b. [DOI] [PubMed] [Google Scholar]
- (13).Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org. Lett. 2004;6:2853. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- (14).Kaim W, Rall J. Angew. Chem. Int. Ed. 1996;35:43. [Google Scholar]
- (15).Allred GD, Liebeskind LS. J. Am. Chem. Soc. 1996;118:2748. [Google Scholar]
- (16).CAUTION! The reaction is exothermic, and can be accompanied by dinitrogen release. It should not be performed in a closed vessel, and adequate cooling should always be available.Procedure A: A 125-mL Erlenmyer flask was charged with CuTC (0.191 g, 1.0 mmol) and dry toluene (50 mL). Subsequently, phenylacetylene (1.10 mL, 1.02 g, 10.0 mmol) then tosyl azide (1.55 mL, 10.0 mmol) were added, and the reaction mixture was allowed to stir for 2 h. It was then diluted with saturated aq NH4Cl (50 mL) and extracted into EtOAc (3 × 50 mL). The combined organics were washed with brine, dried (Na2SO4) and concentrated in vacuo. To remove copper the concentrate was redissolved in CHCl3 and charged with Cuprisorb resin. The mixture was stirred, filtered through celite and concentrated in vacuo. Pulverizing the crude material in cold cyclohexane and collection by filtration afforded 8 (2.67 g, 89% yield) as a white powder. Procedure B: A scintillation vial was charged with copper(I) thiophene-2-carboxylate (CuTC, 0.019 g, 0.1 mmol, 0.1 equiv with respect to alkyne) and water (5 mL) and cooled in an ice-water bath. Subsequently, phenylacetylene (0.110 mL, 1.0 mmol, 1 equiv) then tosyl azide (0.155 mL, 1.0 mmol, 1 equiv) were added and the reaction mixture allowed to warm to room temperature for 2 h. The reaction mixture was diluted with saturated aq NH4Cl (5 mL) and extracted into EtOAc (2 × 5 mL). The combined organics were dried (Na2SO4) and filtered through celite. The eluent was concentrated in vacuo. To remove copper, the concentrate was redissolved in CHCl3 and charged with cuprisorb resin. The mixture was stirred, filtered through celite and concentrated in vacuo. Pulverizing the crude material in cold cyclohexane and collection by filtration afforded 8 (0.188 g, 63% yield) as an off-white powder: mp 105.2–107.0 °C (lit.1 107–108 °C); Rf = 0.44 (silica gel, hexanes:EtOAc 7:3); max(HATR)/cm−1 3142, 1386 (SO2), 1197 1170 (SO2) 988; 1H NMR (600 MHz, CDCl3) 8.31 (s, 1H) 8.03 (d, J = 8.4 Hz, 2H) 7.82 (d, J = 8.2 Hz, 2H) 7.43 (t, J = 7.6 Hz, 2H) 7.37 (m, 3H) 2.44 (s, 3H); 13C NMR (150 MHz, CDCl3) 147.3, 133.0, 130.4, 129.1, 129.0, 128.8, 128.7, 126.0, 118.9, 21.8; Anal. calcd for C15H13N3O2S: C, 60.18; H, 4.38; N, 14.04; found: C, 59.84; H, 4.47; N, 13.87.
- (17). Local dealer information is available at http://www.seachem.com.
- (18).Hein JE, Fokin VV. Chem. Soc. Rev. 2010;39:1302. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Despite attempts to exclude moisture, the formation of N-acylsulfonamide was always observed under these conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.