

Cep152 interacts with the cryptic Polo-box of Plk4 and is required for Plk4-induced centriole overduplication.
Abstract
Both gain and loss of function studies have identified the Polo-like kinase Plk4/Sak as a crucial regulator of centriole biogenesis, but the mechanisms governing centrosome duplication are incompletely understood. In this study, we show that the pericentriolar material protein, Cep152, interacts with the distinctive cryptic Polo-box of Plk4 via its N-terminal domain and is required for Plk4-induced centriole overduplication. Reduction of endogenous Cep152 levels results in a failure in centriole duplication, loss of centrioles, and formation of monopolar mitotic spindles. Interfering with Cep152 function prevents recruitment of Plk4 to the centrosome and promotes loss of CPAP, a protein required for the control of centriole length in Plk4-regulated centriole biogenesis. Our results suggest that Cep152 recruits Plk4 and CPAP to the centrosome to ensure a faithful centrosome duplication process.
Introduction
The centrosome is the primary microtubule-organizing center of an animal cell that consists of two centrioles surrounded by pericentriolar material (PCM; Bettencourt-Dias and Glover, 2007). Centrosomes duplicate once per cell cycle, which involves the growing of procentrioles (daughter centrioles) orthogonally to each of the two parental centrioles (Nigg, 2007; Strnad and Gönczy, 2008). In early mitosis, the two centrosomes separate and participate in mitotic spindle pole formation (Hinchcliffe and Sluder, 2001). Interestingly, there is a correlation between excess centrosomes, aneuploidy, and cancer (Nigg, 2006; Ganem et al., 2009). Extra centrosomes generate chromosomal instability by exacerbating erroneous attachments of chromosomes to spindle microtubules (Ganem et al., 2009), which may contribute to cancer progression. Thus, understanding the regulatory mechanisms governing centrosome duplication may provide insights into both normal cell behavior and tumorigenesis.
Centriole formation is triggered by a conserved kinase, Plk4 (SAK; Bettencourt-Dias et al., 2005; Habedanck et al., 2005). Activation of Plk4 in human cells induces a cascade, including hsSas6 (Leidel et al., 2005), CPAP (Kohlmaier et al., 2009; Tang et al., 2009), Cep135 (Ohta et al., 2002), γ-tubulin, and CP110 (Kleylein-Sohn et al., 2007) that are required at different stages of procentriole formation. Plk4 also induces de novo centriole formation and amplification of centrioles, leading to tumorigenesis in flies (Peel et al., 2007; Basto et al., 2008). Plk4+/− mice develop spontaneous liver and lung tumors, suggesting that reduced Plk4 gene dosage increases the probability of mitotic errors and cancer development (Ko et al., 2005). Recent data suggest that restricting centriole duplication to once per cell cycle is regulated by the F-box protein Slimb, which mediates proteolytic degradation of SAK in Drosophila melanogaster (Cunha-Ferreira et al., 2009; Rogers et al., 2009). In human cells, an autoregulatory feedback loop places Plk4 stability under direct control of its own activity and may form an important mechanism to limit normal centriole duplication to once per cell cycle (Holland et al., 2010). Although Plk4 function is crucial for the regulation of centriole formation, the underlying mechanisms remain scarce.
Results and discussion
To identify proteins that bind to Plk4, we prepared centrosome-enriched fractions from KE37 cells by sucrose gradients followed by biochemical pull-down assays with extracts derived from KI-extracted centrioles and recombinant double-tagged Plk4 (N-terminal zz tag and C-terminal His tag) as bait. Mass spectrometrical analysis of eluted binding partners identified Cep152, a so far poorly characterized protein. Cep152 is the human orthologue (Blachon et al., 2008) of the Drosophila Asterless protein, a centriolar component required for centriole duplication (Varmark et al., 2007), and has been previously identified in a proteomic screen for centrosomal proteins in human cells (Andersen et al., 2003). To verify binding between Plk4 and Cep152, we first performed pull-down assays. Fig. 1 A shows an in vitro interaction between maltose-binding protein (MBP)–Plk4 and in vitro–translated [35S]Cep152. These results suggest that the binding of Plk4 to Cep152 is direct. To further characterize the interaction between both proteins, we have generated rabbit polyclonal antibodies against Cep152. Ab1140 was selected for Western blotting (Fig. S1 A), and Ab26 was selected for immunofluorescence (Fig. S3 D). Ab26 did not detect endogenous Cep152 in total cell extracts but recognizes overexpressed Cep152 (Fig. S1 B) and in centrosome-enriched fractions (Fig. S1 C). To analyze the interaction in vivo, we generated rabbit polyclonal and mouse monoclonal Plk4 antibodies that detect endogenous Plk4 (Fig. S1 D) and asked whether complexes between Plk4 and Cep152 could be detected in vivo. As seen in Fig. 1 B, endogenous Plk4 was present in Cep152 immunoprecipitates. In addition, interactions between ectopically produced Myc-Plk4 and GFP-Cep152 that were coexpressed in 293T cells could also be detected in vivo (Fig. 1 C). These results demonstrate that Plk4 and Cep152 stably associate, confirming our initial findings based on mass spectrometry. In contrast to other Polo-like kinase family members, the Polo-box (PB) domain of Plk4 exhibits only a single PB, and the structural basis for its interaction with its binding partners is not fully understood (Leung et al., 2002). In addition, Plk4 contains a cryptic PB that is required for its localization to the centrosome (Habedanck et al., 2005). To study the interaction in more detail, we mapped the binding sites between Plk4 and Cep152. We found that a fragment comprising the cryptic PB of Plk4 (aa 581–879) is required for binding to Cep152, whereas an interaction with the PB itself could not be detected (Fig. 1 D). These results indicate that the interaction mechanism of Plk4 with its binding partners is distinct from Plk1, which requires both PBs for phospho-dependent substrate targeting (Elia et al., 2003b).
Figure 1.

Cep152 interacts with Plk4 in vitro and in vivo. (A) To analyze in vitro binding between Cep152 and Plk4, either MBP tag alone or MBP-tagged recombinant Plk4 immobilized on amylose beads was used in a binding assay with in vitro–translated [35S]Cep152. Binding of [35S]Cep152 to Plk4 was detected by autoradiography. Equal pull-down of MBP and MBP-Plk4 was shown by Coomassie staining. PD, pull-down. (B) Endogenous Cep152 was immunoprecipitated from U2OS cell extracts using Cep152 (Ab1140). Coprecipitated endogenous Plk4 was detected with a mouse anti-Plk4 antibody by Western blotting. Immunoprecipitation control, random rabbit IgGs. (C) Coimmunoprecipitation of Myc-Plk4 and GFP-Cep152 after coexpression in 293T cells. Reciprocal immunoprecipitation Western blots were performed with anti-Myc and anti-GFP antibodies. Coprecipitated proteins were detected by Western blotting against the corresponding tag. (D, left) Different Flag-Plk4 fragments were coexpressed with GFP-Cep152 in 293T cells. Anti-Flag immunoprecipitates were analyzed in immunoblots for coprecipitated GFP-Cep152 using GFP antibodies. (right) Scheme of expressed Plk4 fragments.
To analyze the centrosomal localization of Cep152, we performed colocalization experiments with centrin-2, a centriolar marker, and found that the major fraction of Cep152 localized around the centriole, and only a minor part colocalized with centrioles (Fig. 2 A). Immunoelectron microscopy was used to obtain more definitive insights into the localization of Cep152. We observed that endogenous Cep152 localizes to the PCM cloud embedding the outer wall at the proximal ends of the centriole but that it was not found in the centriolar lumen (Fig. 2 B). Moreover, endogenous Cep152 partially colocalized with Plk4. We detected a symmetrical localization of Plk4 to centrioles, whereas Cep152 staining was more asymmetrical, exhibiting a stronger signal around one of the two centrioles (Fig. 2 C). Upon analysis of Cep152 in an interphase stage and during different stages of mitosis, we found that Cep152 was always localized to centrosomes (Fig. S2 A). Analysis of the expression pattern of Cep152 in U2OS cells reveals that Cep152 protein levels are low in mitosis but gradually increase during late G1 until S/G2 (Fig. S2, B and C). Despite the lower expression of Cep152 during mitosis, the protein still localizes to the mitotic centrosome but to a lesser extent (Fig. S2 D).
Figure 2.
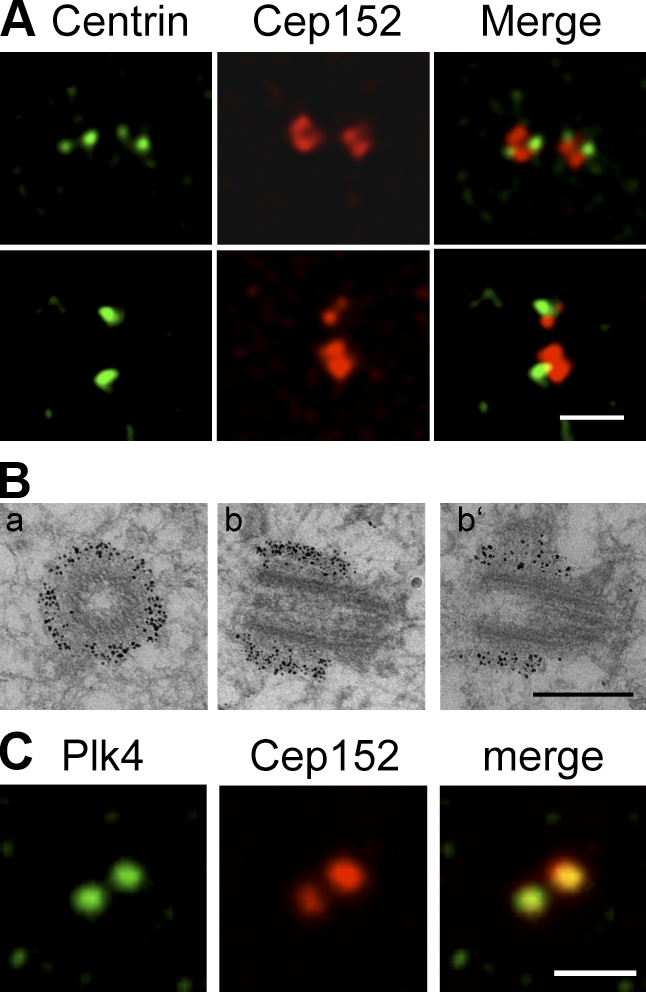
Cep152 localizes to the PCM. (A) Immunofluorescence images showing that a minor fraction of endogenous Cep152 (red) colocalizes with the centriolar marker centrin-2 (green) in U2OS cells. (top) Cell with four centrioles. (bottom) Cell with two centrioles. (B) Immunogold EM of U2OS cells shows that Cep152 localizes to the PCM cloud surrounding the outer wall at the proximal ends of the centriole. Cep152 localization on transversial sections (a) and longitudinal sections of one centriole (b and b’) are shown. (C) Costaining of U2OS cells with antibodies against Cep152 (red) and Plk4 (green). Bars: (A and C) 2 µm; (B) 0.5 µm.
To determine whether centriole reduplication in S phase–arrested U2OS cells required the presence of Cep152, the protein was down-regulated by specific siRNAs (O1 and O2). As Cdk2 activity is required for centriole duplication (Hinchcliffe et al., 1999; Lacey et al., 1999; Matsumoto et al., 1999; Meraldi et al., 1999), Cdk2 siRNAs were used as a positive control. As summarized in Fig. 3 A, ablation of Cep152 function interfered with centriole reduplication. Next, we analyzed whether down-regulation of Cep152 also plays a role in the unperturbed centrosome cycle in U2OS cells. Because a reduction in centriole numbers was expected to produce the most striking phenotypes during cell division, we focused our analysis on mitotic cells in the siRNA-treated populations. 72 h after Cep152 depletion, >20–25% of the mitotic cells displayed monopolar spindles in comparison with 4% in control cells. (Fig. 3 B). Cep152 contains eight predicted coiled-coil regions (Fig. S3 A). Mapping of the region in Cep152 that binds to Plk4 reveals that a fragment comprising the first three coiled-coil regions (aa 1–512) is sufficient for binding to Plk4 (Fig. S3 A). Interestingly, although Cep152 binds to Plk4 via its N-terminal region (fragment Cep152 1–512), the region that confers centrosomal localization of Cep152 is distinct and located at its C terminus (Fig. S3 B). We also found that overexpression of the Cep152 N-terminal fragment exhibits a dominant-negative effect and therefore interferes with centriole duplication (Fig. 3 C). Interestingly, we observed that expression of the dominant-negative Cep152 1–512 fragment prevents Plk4 localization at the centrosome (Fig. 3 D). These data implicate that an interaction between these proteins might be essential for centrosomal recruitment of Plk4.
Figure 3.

Cep152 is required for centriole duplication. (A) U2OS cells were transfected with siRNAs against Cdk2 (positive control), Cep152 (O1 and O2), GL2 (negative control), and arrested in S phase by aphidicolin treatment. 70 h later, cells with more than four centrioles were counted. Red, Cep152; green, centrin-2; blue, DNA. (B) U2OS cells were transfected with either GL2 or Cep152 siRNAs (O1 or O2). Spindle poles were depicted with centrin staining (green, centrin-2) and mitotic spindles with α-tubulin antibodies (red) or DNA (blue). 72 h after transfection, cells with monopolar or bipolar mitotic spindles were counted. (C) U2OS cells were transfected with either GFP or GFP-Cep152 1–512 (green). Centrioles were visualized with centrin-2 staining (red). 48 h and 72 h after transfection, cells with less than two centrioles were counted. (left) Representative pictures of the observed phenotypes. (D) Cep152 full length (FL), Cep152 fragments (1–512 and 508–end), or GFP were overexpressed in U2OS cells. 48 h after transfection, cells with or without centriolar Plk4 staining (blue) were counted. GT335 (red; Bobinnec et al., 1998), an antibody to modified tubulin, was used as a marker for centrosomes. Insets show enlargements of centrosomes as merged image and individual channels. Error bars show the SDs of at least three independent experiments. Bars, 5 µm.
To address whether Cep152 is required for Plk4-induced centrosome overduplication, we generated an HA-Plk4–overexpressing HeLa cell line under the control of the tetracycline-inducible promoter. Plk4 was induced through addition of doxycycline, and this resulted in a multiplication of γ-tubulin–positive dots (Fig. 4 A). Simultaneous down-regulation of Cep152 by siRNAs clearly interfered with Plk4-induced centrosome reduplication by abrogating the reduplication phenotype (Fig. 4 A). The ability of Plk4 to induce the formation of multiple centrioles is regulated during the cell cycle. Cells that are synchronized and held at the G1/S transition respond to Plk4 induction by the formation of flower-like centriolar structures (Fig. S3 C; Kleylein-Sohn et al., 2007). Down-regulation of Cep152 levels by siRNA leads to a reduction of flowers, as shown by centrin or CP110 stainings. This reveals that the formation of Plk4-induced flower-like structures is dependent on Cep152 (Fig. S3 C). Next, we asked whether the centrosomal localization of Plk4 may be dependent on Cep152 and vice versa. Using Plk4-specific antibodies, we found that Plk4 was still detectable at the centrosome during the gradual knockdown of Cep152 (Fig. S3 D). These data suggest that the maintenance of Plk4 at the centrosome is not dependent on functional Cep152. Plk4 siRNA expression shows that centrosomal binding of Cep152 was also independent of Plk4 (Fig. S3 D). To find out whether the recruitment of newly synthesized Plk4 to the centrosome was impaired by the absence of Cep152, we again made use of the Plk4-inducible HeLa cell line (Fig. 4 B) and found that the recruitment of newly synthesized Plk4 to the centrosome was severely impaired when Cep152 was absent (Fig. 4 B), although overall levels of induced Plk4 remained unchanged (Fig. 4 B). To address the involvement of Cep152 in the dynamics of Plk4 recruitment to the centrosome, FRAP was performed with U2OS cells transiently expressing GFP-Plk4 for a short time. Cells were treated with GL2 or Cep152 siRNAs 60 h before the plasmid DNA transfections. As shown in Fig. 4 C, for both GL2- and Cep152-siRNA treated cells, Plk4 recovery reached a plateau after ∼70 s with a total intensity of ∼20% of the prebleaching value. These data indicate the presence of rapidly exchanging (∼20%) and static pools (∼80%) of Plk4 on the centrosome. Interestingly, the rate of fluorescence recovery on the centrosome was partially impaired in cells down-regulated for Cep152. The mean t1/2 of Cep152 siRNA-treated cells was calculated to be 17.4 s, whereas the mean t1/2 for GL2-transfected cells was 11.2 s. This may imply that Plk4 exchanges between centrosomal and noncentrosomal pools with slower kinetics in the absence (or partial loss) of Cep152. Thus, Cep152 might function as a protein that recruits Plk4 to the centrosome, thus facilitating centrosome duplication.
Figure 4.

Cep152 recruits Plk4 to the centrosome. (A) Centriole overduplication in HeLa Tet-on cells was forced by induction of Plk4 expression through addition of doxycycline. Simultaneously, cells were transfected with either GL2 or one of two different Cep152 siRNAs (O1 or O2). Centrosomes were stained with γ-tubulin (green) antibodies or Cep152 (red). 72 h after transfection, cells with less than two centrosomes were counted. (B) Hela Tet-on cells were transfected with GL2 or Cep152 siRNAs (O1 and O2) treated with aphidicolin for 24 h. Plk4 expression was induced in the last 20 h of aphidicolin treatment. Cells were scored for centrosomal HA-Plk4 signal. Induced HA-Plk4 signal (green) locates to the centrosome (red, γ-tubulin). Blue, DNA. (right) Insets display enlargements of the selected regions in the indicated channels (arrowheads). The last set of insets includes a triple merge. Corresponding immunoblots from siRNA-treated and induced cells were analyzed for Plk4 levels using antibodies against HA and Plk4 in comparison with uninduced, control siRNA-treated samples. (A and B) Error bars indicate SDs (n = 3). (C) Dynamics of Plk4 at centrosomes in response to Cep152 RNAi. FRAP was performed on U2OS cells treated with either GL2 or Cep152 siRNAs (O2) for 60 h followed by GFP-Plk4 transfections for 14 h. GFP-Plk4–positive, unsplit centrosomes in the same plane of focus were selected for photobleaching and subsequent imaging. 80 × 80–pixel squares surrounding the centrosome were bleached (bleach time 2.5 s), and the recovery of GFP fluorescence on centrosomes was imaged over time. (left) Arrows mark photobleached regions on the centrosome. (right) Relative expression levels of GFP-Plk4 in GL2- and O2-transfected cells were determined. (bottom) Mean fluorescence recovery profiles of GFP-Plk4 on the centrosome were depicted in GL2- and O2-treated cells (n = 12). Mean t1/2 of experiments is shown ± SD. Student’s t test was performed with GL2 or Cep152 siRNA-treated cells for recovery of GFP-Plk4 on the centrosome with P < 0.0001. Bars: (A) 5 µm; (B) 20 µm; (C) 3 µm.
The canonical pathway of centriole biogenesis has been previously described in exquisite detail at the ultrastructural level (Strnad and Gönczy, 2008). Several human centrosomal proteins indispensable for Plk4-dependent procentriole formation have been identified and include hsSas6 (Leidel et al., 2005), CPAP (Kohlmaier et al., 2009), (Tang et al., 2009), CP110 (Kleylein-Sohn et al., 2007), and Cep135 (Ohta et al., 2002). To identify the role of Cep152 in this pathway, Cep152 was down-regulated by siRNA, and the centrosomal localization of these proteins was analyzed. We find that the localization of hsSas6, CP110, or Cep135 is not impaired by Cep152 siRNA treatment (Fig. 5 A). However, we cannot rule out that hsSas6, CP110, or Cep135 stably associated with centrioles before Cep152 depletion, similar to our observation with Plk4 localization. Interestingly, we observed that the SAS-4–related protein CPAP, which is required for centrosome duplication and controls centriole length (Kohlmaier et al., 2009; Schmidt et al., 2009; Tang et al., 2009), did not localize to centrioles upon Cep152 down-regulation (Fig. 5 A). Therefore, the localization of Cep152 at the centrosome is crucial for centrosomal recruitment or maintenance of CPAP. To further confirm the link between Cep152 and CPAP, we analyzed the localization of these proteins within the flower-like structure upon overexpression of Plk4 in colocalizations with the centriolar marker centrin-2 and Plk4 itself. Intriguingly, Cep152 exhibited a similar localization that was described for CPAP, namely a staining around parental centrioles and in between the nascent procentrioles (Fig. 5 B). Next, we asked whether Cep152 could interact with CPAP and found complexes between GFP-Cep152 and Flag-CPAP after coexpression in 293T cells in vivo (Fig. 5 C). Mapping of the region in Cep152 that binds to CPAP led to the finding that residues 513–1,074 of Cep152 are necessary for the interaction with CPAP (Fig. 5 D). Thus, Cep152 seems to form a scaffold with distinct regions for Plk4 and CPAP interactions and for centrosomal localization.
Figure 5.

CPAP localization at the centrosome is dependent on Cep152. (A) U2OS cells were transfected with either GL2 or Cep152 siRNAs. Colocalizations of CPAP, hSas6, Cep135, and CP110 (red) together with γ-tubulin (green) were determined by immunofluorescence. Insets show enlargements of the merged image and individual channels. (right) Protein levels of the indicated proteins were determined by Western blotting. The graph shows a quantification of the percentage of cells with centrosomal CPAP localization. Error bars indicate SDs (n = 3). (B, left) Cep152 and CPAP costainings (red) within the flower-like centrin-2 structures (green) were depicted. (B, right) Cep152 and CPAP colocalizations (red) were performed together with HA-Plk4 (green) using anti-HA antibodies. (C) Flag-CPAP and GFP or GFP-Cep152 constructs were coexpressed in 293T cells. GFP and GFP-Cep152 were immunoprecipitated 48 h after expression. Coimmunoprecipitated Flag-CPAP was detected by Western blotting against the Flag tag. (D) Different GFP-Cep152 fragments (Fig. S3 A) were coexpressed with Flag-CPAP in 293T cells. Anti-GFP immunoprecipitates were analyzed by Western blotting for coprecipitated Flag-CPAP with anti-Flag antibodies. Bars: (A) 5 µm; (B, left) 1 µm; (B, right) 0.5 µm.
In summary, our data identify the centriolar protein Cep152, a protein involved in centriole duplication, as a novel Plk4-binding protein and the first binding partner to interact with the cryptic PB of Plk4. Cep152 partially colocalizes with Plk4 around the centrioles and is required for Plk4-induced centriole overduplication. Therefore, Cep152 is a critical component of the Plk4-dependent centriole assembly pathway, as Plk4 cannot trigger centriole duplication in the absence of Cep152 (Fig. 4 A). Cep152 localization to the centrosome is also crucial for recruitment of CPAP to the centrosome. It is conceivable that the interaction between Plk4 and Cep152 might facilitate the recruitment of other components, such as CPAP, to trigger centriole duplication. Interestingly, a recent study by Chang et al. (2010) suggests that CPAP might be a physiological substrate of Plk4. Together, Cep152 seems to regulate the recruitment of Plk4 to the centrosome and the maintenance of CPAP at this structure.
The 3D structure of Plk4 suggests a binding mechanism different from Plk1 (Leung et al., 2002). The Plk1 PB binds to target proteins after their phosphorylation (Elia et al., 2003a,b). However, the interaction of Plk4 and Cep152 occurs through its single cryptic PB in the absence of phosphorylation, although we currently cannot exclude that a phosphorylation event might regulate this interaction. In conclusion, our findings provide important mechanistic insights into the process of procentriole assembly, implicating Cep152 as a critical component in Plk4-induced procentriole formation.
Materials and methods
Culturing of cells, generation of stable cell lines, transfections, and cell synchronization
HeLa cells, HeLa Tet-on cells, and U2OS cells were grown in DME (Sigma-Aldrich) containing 1 g/liter glucose, 10% fetal calf serum, and 2 mM glutamine. 293T and KE37 cells were grown in DME containing 4.5 g/liter glucose, 10% fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. Inducible HeLa Tet-on cells stably expressing HA-Plk4 were generated by cotransfection of cells that carry Tet-on transactivators (Takara Bio Inc.) with pTREtight-HA-Plk4 and pPuro vector (Takara Bio Inc.) containing a marker for puromycin resistance. For induction of HA-Plk4 expression, media were supplemented with 2 µg/ml doxycycline. Cells were analyzed 20–48 h after induction. 293T cells were transfected with Ca2+ phosphate according to standard protocols. HeLa, HeLa Tet-on, and U2OS cells were transfected with Lipofectamine 2000 (Invitrogen) for siRNA transfections or with Polyfect (QIAGEN) for plasmid DNA transfections. For overduplication assays, U2OS cells were arrested in S phase by supplementation of media with 1.9 µg/ml aphidicolin. Cells were analyzed 70 h after aphidicolin addition. U2OS cells were arrested in mitosis by supplementation of media with 100 ng/ml nocodazole for 17 h. For dT-Block, U2OS cells were treated with 4 mM thymidine for 19 h, released for 12 h, and again blocked for 15 h.
For observing Plk4-dependent flower-like procentriole formation, HeLa Tet-on HA-Plk4 wild-type cells were treated with siRNAs for 70 h and supplemented with 2 µg/ml aphidicolin for 24 h. Plk4 expression was induced for the last 20 h of aphidicolin treatment.
Antibodies and Western blotting
Rabbit anti-Plk4 and mouse anti-Plk4 antibodies were raised against a synthetic peptide spanning residues 564–579 of Plk4. Rabbit anti-Cep152 (Ab26) antibody was raised against a synthetic peptide spanning residues 26–39 of Cep152. A second rabbit anti-Cep152 (Ab1140) antibody was raised against the Cep152 fragment spanning aa 1,140–1,308 that was expressed as a GST fusion protein in Escherichia coli and used for immunization. All antibodies were affinity purified using corresponding peptides or protein fragments immobilized on CNBr-activated Sepharose.
Rabbit anti-GFP antibody (NB600-303) was purchased from Novus Biologicals. Mouse anti–c-Myc (9E10), mouse anti-Plk1 (36–298), and mouse anti–cyclin E (H12) were purchased from Santa Cruz Biotechnology, Inc. Mouse anti-FlagM2 (F3165) and mouse anti–α-tubulin were obtained from Sigma-Aldrich, mouse anti-actin (JLA20) was obtained from EMD, and mouse anti-HA was obtained from Babco. Peroxidase-conjugated donkey anti–rabbit and goat anti–mouse antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc.
Recombinant protein expression
All recombinant proteins were expressed in BL21-Rosetta in the presence of 2% glucose and 1.5% ethanol. Protein expression was induced with 0.5 mM isopropylthio-β-d-galactoside for 15 h at 18°C. Plk4 carrying a C-terminal His tag and an N-terminal zz tag (Jäkel and Görlich, 1998) was natively purified by single-step affinity chromatography using Ni-NTA Sepharose (QIAGEN) according to the instructions of the manufacturer, and MBP-Plk4 was natively purified via amylose beads (New England Biolabs, Inc.). In all cases, lysis buffer was 50 mM Tris, pH 7.5, 250 mM NaCl, 2 mM MgCl2, 5% glycerol, and 0.25% NP-40. All proteins were frozen in liquid nitrogen and stored at −80°C.
Molecular cloning
Plk4 wild type and K41R were PCR amplified from pX-HA-Plk4 (provided by D. Spengler, German Cancer Research Center, Heidelberg, Germany) and cloned into the PciI and BamHI sites of pQE80zz (provided by D. Görlich, Max Planck Institute, Göttingen, Germany), into the BamHI and HindIII sites of pMAL-c2 (New England Biolabs, Inc.), into the BamHI and XhoI sites of pCMV-3Tag-1 (Agilent Technologies), and into the BamHI and XhoI sites of pCMV-3Tag-2 (Agilent Technologies). All Plk4 fragments were amplified by PCR and cloned into the HindIII and XhoI sites of pCMV-3Tag-1. Cep152 was amplified by PCR from pCR-XL-Topo-Cep152 (GenBank/EMBL/DDBJ accession no. NM_014985) and cloned into the KpnI and XhoI sites of pEGFP-C1 (Takara Bio Inc.) and into the EcoRV and XhoI sites of pET-30c (EMD). All Cep152 fragments were cloned either into the XhoI and KpnI sites or into the XhoI and SmaI sites of pEGFP-C1. CPAP was amplified from pEGFP-CPAP (provided by P. Gönczy, École Polytechnique Fédèrale de Lausanne, Lausanne, Switzerland) by PCR and cloned into the BamHI and SalI sites of pCMV-3Tag-2 vector.
Interaction experiments
Centrosomes were isolated from KE37 cells by discontinuous gradient ultracentrifugation as described previously (Moudjou and Bornens, 1998). In brief, cell pellet was washed with TBS and 0.1× TBS/8% sucrose. Cells were resuspended with 0.1× TBS/8% sucrose and mixed with 0.5% NP-40 lysis buffer. The suspension was shaken slowly for 30 min at 4°C and spun at 2,500 g for 10 min. Hepes buffer and DNase were added to the supernatant to final concentrations of 10 mM and 1 µg/ml, respectively. After incubation for 30 min at 4°C, the mixture was gently underlaid with 60% sucrose solution and spun at 10,000 g for 30 min. The obtained centrosomal suspension was loaded onto a discontinuous sucrose gradient (70, 50, and 40% sucrose solutions from the bottom) and spun at 120,000 g for 1 h. Fractions were collected from the top, diluted with Pipes buffer (10 mM Pipes), and spun at 20,400 g for 15 min.
Purified centrioles from ∼4.5 × 109 KE37 cells were resuspended in 2 ml of binding buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.25% NP-40, 10 mM β-glycerophosphate, and 10 mM NaF) containing 2 M KI and incubated on ice for 45 min. After centrifugation for 10 min at 13,000 rpm, the supernatant was dialyzed stepwise against binding buffer containing 1 M KI, binding buffer containing 0.5 M KI, and binding buffer without KI. Precipitated proteins were pelleted by centrifugation for 10 min at 13,000 rpm. For in vitro pull-down assay, half of the supernatant was added to either immobilized zz tag or immobilized zz-Plk4 (100 µg of each protein was immobilized on 100 µl IgG Sepharose) in binding buffer. The reactions were incubated at 4°C for 3 h and washed three times with binding buffer and once with binding buffer + 50 mM NaCl. Bound proteins were eluted with binding buffer + 850 mM NaCl, TCA precipitated, and analyzed by SDS-PAGE followed by silver staining and mass spectrometry.
For in vitro pull-down assay with in vitro–translated Cep152, 10 µg MBP or MBP-Cep152 was immobilized on 10 µl amylose beads and incubated with 20 µl of an in vitro translation reaction (50 µl total volume; TNT-Coupled Reticulocyte Lysate system; Promega) in NP-40 buffer (40 mM Tris, pH 7.5, 150 mM NaCl, 0.5% NP-40, 5 mM EDTA, 10 mM β-glycerophosphate, and 5 mM NaF). After incubation for 2 h at 4°C, beads were washed three times with NP-40 buffer and boiled in 2× SDS buffer. Eluted material was separated by SDS-PAGE and subjected to autoradiography. As input, 5% of the in vitro translation was loaded.
For coimmunoprecipitations, 293T cells were transfected with the corresponding constructs. Cells were lysed in NP-40 buffer for 30 min on ice. After centrifugation for 10 min at 13,000 rpm, 2 µg anti-Flag, anti-Myc, or anti-GFP antibody was added to the supernatant. Proteins were immunoprecipitated for 2 h at 4°C and collected by addition of 10 µl protein G–Sepharose. After extensive washing, the beads were boiled in 2× SDS buffer and analyzed by SDS-PAGE and Western blotting. As input, 1% of each lysate was loaded. Endogenous Cep152 protein was immunoprecipitated with Cep152 (fragment) antibody. As control, random IgGs (Santa Cruz Biotechnology, Inc.) were used.
Antibodies and indirect immunofluorescence
Cells grown on coverslips were fixed with ice-cold methanol for 10 min at −20°C. They were washed with PBS and blocked with 2% BSA/PBS for 30 min. Cells were incubated with primary antibodies for 1 h and with secondary antibodies for 30 min. DNA was stained with Hoechst. Between each step, cells were washed three times with 2% BSA/PBS. All incubations took place at room temperature. Images were taken with a spinning-disc confocal (Ultra-View; PerkinElmer) objective on an inverted microscope (Ti; Nikon) connected to an electron multiplying charge-coupled device camera (Hamamatsu Photonics). Deconvolution was applied for visualizing the flower-like procentriole formation (Huygens Essentials) with a 100× NA 1.0 oil objective. Z stacks were taken at 150-nm intervals, and maximum intensity projections were displayed. Images were later cropped in Photoshop (Adobe). Mouse anti–γ-tubulin (GTU-88) was purchased from Sigma-Aldrich, and rabbit anti–α-tubulin (ab18251) was obtained from Abcam. Rabbit anti–centrin-2 (N-17) antibodies were obtained from Santa Cruz Biotechnology, Inc., and mouse anti–centrin-2 was obtained from J. Salisbury (Mayo Clinic, Rochester, MN). Mouse anti-GT335 was provided by C. Janke (Institut Curie, Paris, France), rabbit anti-hSAS6 and rabbit anti-CPAP were obtained from P. Gönczy, rabbit anti-Cep135 was obtained from R. Kuriyama (University of Minnesota, Minneapolis, MN), and rabbit anti-CP110 was obtained from B. Dynlacht (New York University, New York, NY). Alexa Fluor 488 and 594 anti–mouse, Alexa Fluor 488, 594, and 405 anti–rabbit, and Alexa Fluor 488 anti–rat were purchased from Invitrogen. Each independent counting of cells in this study includes ≥150 cells. Evaluation of the monopolar spindle phenotype was based on counting 50 cells per experiment.
Immunogold EM
U2OS cells were grown on coverslips, fixed with 2% formaldehyde, and permeabilized with 0.05% saponin in PBS. Cells were incubated with Cep152 (Ab26) antibody and rabbit anti-IgG Nanogold antibody for 3 h each. Cells were further fixed with 2.5% glutaraldehyde in 50 µM cacodylate buffer, and Nanogold was silver enhanced with HQ silver (Nanoprobes). Cells were dehydrated and embedded in epoxy resin.
FRAP analysis
For FRAP analysis, cells were cultured on glass-bottom dishes (Ibidi Integral Biodiagnostics) in a top environmental chamber (Tokai Hit Stage; Spectra Services) and maintained at 37°C. FRAP was performed on a spinning-disc confocal unit (PerkinElmer) fitted on an inverted microscope (TE2000; Nikon) using a 100× NA 1.0 oil objective. A square region of interest (ROI) of 80 × 80 pixels centered on unsplit centrosomes was bleached with 40 iterations and 100% laser power (488-nm argon laser). Two images were taken before bleaching with a 2-s interval. An image was taken every 1.5 s (488-nm argon laser at 4% power) after bleaching for the six initial recordings and then every 3 s over a 100–120-s period. For each time point, images of four z stacks of 0.5 µm optical section spacing were collected; the highest intensity recording of these four images was regarded as the in-focus intensity of the ROI. Centrosomes with excessive z-axis movements that get out of the focal range by the end of the recording time were excluded from the analysis. Fluorescence intensity of the photobleached ROI was determined using Volocity FRAP image acquisition software. For processing and correction of time-lapse data, a standard algorithm (Khodjakov and Rieder, 1999) was used as follows: Ic = scaling factor × ([Ir − Ib]/[If − Ib]), where Ic is the corrected image, Ir is the noncorrected raw object fluorescent intensity, Ib is the background fluorescent intensity outside of the cells, If is the bright reference of the fluorescent intensity of a cluster of cells, and the scaling factor is the brightest pixel value in the bright reference image (after subtraction of the Ib). After this normalization, mean fluorescence intensities of the prebleaching images were set as 100%, and the subsequent relative recovery percentages were calculated for the remaining time points. Images were processed in Photoshop, and t1/2 values were determined using Volocity. The p-value for GFP-Plk4 recovery on the centrosome upon GL2 and O2 siRNA-treated samples was calculated using a two-tailed Student’s t test.
siRNAs
siRNAs used in this study were directed against the following sequences: firefly luciferase (GL2), 5′-AACGTACGCGGAATACTTCGA-3′; Plk4 O1, 5′-AACTATCTTGGAGCTTTATAA-3′; Plk4 O2, 5′-CTGGTAGTACTAGTTCACCTA-3′; Cdk2, 5′-AAGATGGACGGAGCTTGTTAT-3′; Cep152 O1, 5′-CAGCTCTTTGAGGCTTATGAG-3′; and Cep152 O2, 5′-GCGGATCCAACTGGAAATCTA-3′. All siRNAs were purchased from Applied Biosystems. Cells transfected with Plk4 siRNA were analyzed 2 d after transfection. Cells transfected with Cdk2 or Cep152 siRNAs were analyzed 3 d after transfection.
Online supplemental material
Fig. S1 shows characterization of Cep152 and Plk4 antibodies. Fig. S2 shows analysis of Cep152 localization to centrosomes. Fig. S3 shows that Cep152 has distinct sites for Plk4 binding and centriolar localization. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201007107/DC1.
Acknowledgments
We thank B. Dynlacht, D. Görlich, J. Salisbury, P. Gönczy, C. Janke, and R. Kuriyama for reagents and U. Engel and C. Ackermann (Nikon Imaging Center, Heidelberg, Germany) for equipment and assistance.
This work was supported by the Deutsche José Carreras-Leukämiestiftung (grant DJCLS R07/21v to I. Hoffmann).
Footnotes
Abbreviations used in this paper:
- MBP
- maltose-binding protein
- PB
- Polo-box
- PCM
- pericentriolar material
References
- Andersen J.S., Wilkinson C.J., Mayor T., Mortensen P., Nigg E.A., Mann M. 2003. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 426:570–574 10.1038/nature02166 [DOI] [PubMed] [Google Scholar]
- Basto R., Brunk K., Vinadogrova T., Peel N., Franz A., Khodjakov A., Raff J.W. 2008. Centrosome amplification can initiate tumorigenesis in flies. Cell. 133:1032–1042 10.1016/j.cell.2008.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt-Dias M., Glover D.M. 2007. Centrosome biogenesis and function: centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 8:451–463 10.1038/nrm2180 [DOI] [PubMed] [Google Scholar]
- Bettencourt-Dias M., Rodrigues-Martins A., Carpenter L., Riparbelli M., Lehmann L., Gatt M.K., Carmo N., Balloux F., Callaini G., Glover D.M. 2005. SAK/PLK4 is required for centriole duplication and flagella development. Curr. Biol. 15:2199–2207 10.1016/j.cub.2005.11.042 [DOI] [PubMed] [Google Scholar]
- Blachon S., Gopalakrishnan J., Omori Y., Polyanovsky A., Church A., Nicastro D., Malicki J., Avidor-Reiss T. 2008. Drosophila asterless and vertebrate Cep152 Are orthologs essential for centriole duplication. Genetics. 180:2081–2094 10.1534/genetics.108.095141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobinnec Y., Moudjou M., Fouquet J.P., Desbruyères E., Eddé B., Bornens M. 1998. Glutamylation of centriole and cytoplasmic tubulin in proliferating non-neuronal cells. Cell Motil. Cytoskeleton. 39:223–232 [DOI] [PubMed] [Google Scholar]
- Chang J., Cizmecioglu O., Hoffmann I., Rhee K. 2010. PLK2 phosphorylation is critical for CPAP function in procentriole formation during the centrosome cycle. EMBO J. 29:2395–2406 10.1038/emboj.2010.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha-Ferreira I., Rodrigues-Martins A., Bento I., Riparbelli M., Zhang W., Laue E., Callaini G., Glover D.M., Bettencourt-Dias M. 2009. The SCF/Slimb ubiquitin ligase limits centrosome amplification through degradation of SAK/PLK4. Curr. Biol. 19:43–49 10.1016/j.cub.2008.11.037 [DOI] [PubMed] [Google Scholar]
- Elia A.E., Cantley L.C., Yaffe M.B. 2003a. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 299:1228–1231 10.1126/science.1079079 [DOI] [PubMed] [Google Scholar]
- Elia A.E., Rellos P., Haire L.F., Chao J.W., Ivins F.J., Hoepker K., Mohammad D., Cantley L.C., Smerdon S.J., Yaffe M.B. 2003b. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell. 115:83–95 10.1016/S0092-8674(03)00725-6 [DOI] [PubMed] [Google Scholar]
- Ganem N.J., Godinho S.A., Pellman D. 2009. A mechanism linking extra centrosomes to chromosomal instability. Nature. 460:278–282 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habedanck R., Stierhof Y.D., Wilkinson C.J., Nigg E.A. 2005. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 7:1140–1146 10.1038/ncb1320 [DOI] [PubMed] [Google Scholar]
- Hinchcliffe E.H., Sluder G. 2001. “It takes two to tango”: understanding how centrosome duplication is regulated throughout the cell cycle. Genes Dev. 15:1167–1181 10.1101/gad.894001 [DOI] [PubMed] [Google Scholar]
- Hinchcliffe E.H., Li C., Thompson E.A., Maller J.L., Sluder G. 1999. Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science. 283:851–854 10.1126/science.283.5403.851 [DOI] [PubMed] [Google Scholar]
- Holland A.J., Lan W., Niessen S., Hoover H., Cleveland D.W. 2010. Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J. Cell Biol. 188:191–198 10.1083/jcb.200911102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäkel S., Görlich D. 1998. Importin beta, transportin, RanBP5 and RanBP7 mediate nuclear import of ribosomal proteins in mammalian cells. EMBO J. 17:4491–4502 10.1093/emboj/17.15.4491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodjakov A., Rieder C.L. 1999. The sudden recruitment of γ-tubulin to the centrosome at the onset of mitosis and its dynamic exchange throughout the cell cycle, do not require microtubules. J. Cell Biol. 146:585–596 10.1083/jcb.146.3.585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleylein-Sohn J., Westendorf J., Le Clech M., Habedanck R., Stierhof Y.D., Nigg E.A. 2007. Plk4-induced centriole biogenesis in human cells. Dev. Cell. 13:190–202 10.1016/j.devcel.2007.07.002 [DOI] [PubMed] [Google Scholar]
- Ko M.A., Rosario C.O., Hudson J.W., Kulkarni S., Pollett A., Dennis J.W., Swallow C.J. 2005. Plk4 haploinsufficiency causes mitotic infidelity and carcinogenesis. Nat. Genet. 37:883–888 10.1038/ng1605 [DOI] [PubMed] [Google Scholar]
- Kohlmaier G., Loncarek J., Meng X., McEwen B.F., Mogensen M.M., Spektor A., Dynlacht B.D., Khodjakov A., Gönczy P. 2009. Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr. Biol. 19:1012–1018 10.1016/j.cub.2009.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey K.R., Jackson P.K., Stearns T. 1999. Cyclin-dependent kinase control of centrosome duplication. Proc. Natl. Acad. Sci. USA. 96:2817–2822 10.1073/pnas.96.6.2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidel S., Delattre M., Cerutti L., Baumer K., Gönczy P. 2005. SAS-6 defines a protein family required for centrosome duplication in C. elegans and in human cells. Nat. Cell Biol. 7:115–125 10.1038/ncb1220 [DOI] [PubMed] [Google Scholar]
- Leung G.C., Hudson J.W., Kozarova A., Davidson A., Dennis J.W., Sicheri F. 2002. The Sak polo-box comprises a structural domain sufficient for mitotic subcellular localization. Nat. Struct. Biol. 9:719–724 10.1038/nsb848 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y., Hayashi K., Nishida E. 1999. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr. Biol. 9:429–432 10.1016/S0960-9822(99)80191-2 [DOI] [PubMed] [Google Scholar]
- Meraldi P., Lukas J., Fry A.M., Bartek J., Nigg E.A. 1999. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat. Cell Biol. 1:88–93 10.1038/10054 [DOI] [PubMed] [Google Scholar]
- Moudjou M., Bornens M. 1998. Method of centrosome isolation from cultured animal cells. Cell biology: A Laboratory Handbook. Second edition Celis J.E., editor Academic Press, San Diego: 111–119 [Google Scholar]
- Nigg E.A. 2006. Origins and consequences of centrosome aberrations in human cancers. Int. J. Cancer. 119:2717–2723 10.1002/ijc.22245 [DOI] [PubMed] [Google Scholar]
- Nigg E.A. 2007. Centrosome duplication: of rules and licenses. Trends Cell Biol. 17:215–221 10.1016/j.tcb.2007.03.003 [DOI] [PubMed] [Google Scholar]
- Ohta T., Essner R., Ryu J.H., Palazzo R.E., Uetake Y., Kuriyama R. 2002. Characterization of Cep135, a novel coiled-coil centrosomal protein involved in microtubule organization in mammalian cells. J. Cell Biol. 156:87–99 10.1083/jcb.200108088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel N., Stevens N.R., Basto R., Raff J.W. 2007. Overexpressing centriole-replication proteins in vivo induces centriole overduplication and de novo formation. Curr. Biol. 17:834–843 10.1016/j.cub.2007.04.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers G.C., Rusan N.M., Roberts D.M., Peifer M., Rogers S.L. 2009. The SCFSlimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. J. Cell Biol. 184:225–239 10.1083/jcb.200808049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T.I., Kleylein-Sohn J., Westendorf J., Le Clech M., Lavoie S.B., Stierhof Y.D., Nigg E.A. 2009. Control of centriole length by CPAP and CP110. Curr. Biol. 19:1005–1011 10.1016/j.cub.2009.05.016 [DOI] [PubMed] [Google Scholar]
- Strnad P., Gönczy P. 2008. Mechanisms of procentriole formation. Trends Cell Biol. 18:389–396 10.1016/j.tcb.2008.06.004 [DOI] [PubMed] [Google Scholar]
- Tang C.J., Fu R.H., Wu K.S., Hsu W.B., Tang T.K. 2009. CPAP is a cell-cycle regulated protein that controls centriole length. Nat. Cell Biol. 11:825–831 10.1038/ncb1889 [DOI] [PubMed] [Google Scholar]
- Varmark H., Llamazares S., Rebollo E., Lange B., Reina J., Schwarz H., Gonzalez C. 2007. Asterless is a centriolar protein required for centrosome function and embryo development in Drosophila. Curr. Biol. 17:1735–1745 10.1016/j.cub.2007.09.031 [DOI] [PubMed] [Google Scholar]