

Abstract
In the title compound, C7H11N2 +·C7H5O2 −, the 2-amino-4,6-dimethylpyridinium cation and the benzoate anion are linked by two N—H⋯O hydrogen bonds, forming an R 2 2(8) ring motif. The H atoms in both the methyl groups are rotationally disordered, with fixed site occupancies of 0.50. In the crystal structure, the molecules are stabilized by intermolecular N—H⋯O hydrogen bonds. A π–π interaction, with a centroid–centroid distance of 3.661 (2) Å, is also observed.
Related literature
For the biological activity of Schiff bases with azomethine linkages, see Dhar & Taploo (1982 ▶). For hydrogen bonding, see: Jeffrey (1997 ▶); Jeffrey & Saenger (1991 ▶). For graph-set descriptions of hydrogen-bond ring motifs, see: Bernstein et al. (1995 ▶).
Experimental
Crystal data
C7H11N2 +·C7H5O2 −
M r = 244.29
Monoclinic,
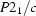
a = 7.5362 (16) Å
b = 22.937 (4) Å
c = 8.2124 (14) Å
β = 109.820 (2)°
V = 1335.5 (4) Å3
Z = 4
Mo Kα radiation
μ = 0.08 mm−1
T = 296 K
0.57 × 0.23 × 0.05 mm
Data collection
Bruker SMART APEXII CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS;p Bruker, 2009 ▶) T min = 0.954, T max = 0.996
7639 measured reflections
2336 independent reflections
1527 reflections with I > 2σ(I)
R int = 0.029
Refinement
R[F 2 > 2σ(F 2)] = 0.044
wR(F 2) = 0.127
S = 1.02
2336 reflections
199 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.10 e Å−3
Δρmin = −0.14 e Å−3
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810034811/fj2327sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810034811/fj2327Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1N1⋯O2 | 1.04 | 1.65 | 2.683 (2) | 172 |
| N2—H1N2⋯O1 | 1.01 | 1.78 | 2.779 (2) | 171 |
| N2—H2N2⋯O1i | 0.90 | 1.97 | 2.853 (2) | 168 |
Symmetry code: (i) 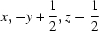 .
.
Acknowledgments
This research was supported by Universiti Sains Malaysia (USM) under the University Research grant (No. 1001/PFARMASI/815004) and the Ministry of Science, Technology and Innovation through an R&D Initiative Grant (09-05-IFN-MEB 004). HKF and MMR also thank USM for the Research University Grant (No. 1001/PFIZIK/811160). MRA gratefully acknowledges a PhD scholarship from Universiti Malaysia Sarawak.
supplementary crystallographic information
Comment
This compound is derived from 2-amino-4,6-dimethylpyridine and benzaldehyde. Schiff bases provide more potential sites for both chemical and biological activities of compounds. Schiff bases with azomethine linkage were used as anti-infectious agents (Dhar et al., 1982). Pyridine and its derivatives play an important role in heterocyclic chemistry (Jeffrey, 1997). They are often involved in hydrogen-bond interactions (Jeffrey & Saenger, 1991).
The title 1:1 adduct compound contains an 2-amino-4,6-dimethylpyridinium cation and benzoate anion in the asymmetric unit. The parameters in (I), (Fig. 1), are within normal ranges. The 2-amino-4,6-dimethylpyridinium cation is planar with the maximum deviation of 0.005 (2)Å for atom C9. The H atoms of the methyl groups are disordered over two positions and with fixed site-occupancy factors of 0.50:0.50 for both of the methyl groups. The carboxylate group in benzoate anion is slightly twisted and make a dihedral angle of 7.2 (1)° with the attached benzene ring.
The 2-amino-4,6-dimethylpyridinium cation and benzoate anion groups are linked together by intermolecular N1—H1N1···O2 and N2—H1N2···O1 interactions (Table 1) forming an R22(8) ring motif. In the crystal structure, the molecules stabilized by intermolecular N—H···O hydrogen bonds (Table 1) and π-π interactions with Cg1—Cg2 = 3.661 (2)Å (Cg1 = N1/C8-C12, Cg2 = C1-C6).
Experimental
An ethanol solution (20 ml) of 2-amino-4,6-dimethylpyridine (1.22 g, Aldrich) and benzaldehyde (1.06 g, Merck) were mixed, heated on a hot plate and stirred with a magnetic stirrer. The reaction mixture was refluxed for 4h. The resulting condensation solution was allowed to cool slowly at room temperature to form brownish materials. Purification was done using thin layer chromatography (TLC) and silica gel column chromatography (CC) eluted by chloroform:methanol and n-hexane:ethyl acetate solvent system. Finally the pure compound was recrystallized in ethanol which afforded the C7H11N2+.C7H5O2- salt.
Refinement
N bound H atoms was located from a difference Fourier map and were refined using a riding model, with Uiso(H) = 1.2Ueq(N). The methyl hydrogen atoms were located from the difference Fourier map and refined freely with the parent atom [Uiso(H) = 1.5Ueq(C)]. The rest of the hydrogen atoms were positioned geometrically and refined as riding model [Uiso(H) = 1.2Ueq(C)].
Figures
Fig. 1.

The molecular structure, showing 30% probability displacement ellipsoids and the atom-numbering scheme. Hydrogen atoms are shown as spheres of arbitrary radius. Dashed lines indicate hydrogen bonds.
Fig. 2.

The crystal packing of (I) viewed along the c axis. Dashed lines indicate hydrogen bonds. H atoms not involved in the hydrogen bond interactions have been omitted for clarity.
Crystal data
| C7H11N2+·C7H5O2− | F(000) = 520 |
| Mr = 244.29 | Dx = 1.215 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 1881 reflections |
| a = 7.5362 (16) Å | θ = 2.8–30.1° |
| b = 22.937 (4) Å | µ = 0.08 mm−1 |
| c = 8.2124 (14) Å | T = 296 K |
| β = 109.820 (2)° | Plate, colourless |
| V = 1335.5 (4) Å3 | 0.57 × 0.23 × 0.05 mm |
| Z = 4 |
Data collection
| Bruker SMART APEXII CCD area-detector diffractometer | 2336 independent reflections |
| Radiation source: fine-focus sealed tube | 1527 reflections with I > 2σ(I) |
| graphite | Rint = 0.029 |
| φ and ω scans | θmax = 25.0°, θmin = 2.8° |
| Absorption correction: multi-scan (SADABS;p Bruker, 2009) | h = −8→8 |
| Tmin = 0.954, Tmax = 0.996 | k = −27→26 |
| 7639 measured reflections | l = −9→9 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.044 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.127 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.02 | w = 1/[σ2(Fo2) + (0.0636P)2 + 0.0715P] where P = (Fo2 + 2Fc2)/3 |
| 2336 reflections | (Δ/σ)max < 0.001 |
| 199 parameters | Δρmax = 0.10 e Å−3 |
| 0 restraints | Δρmin = −0.14 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| O1 | 0.5868 (2) | 0.30973 (6) | 0.23891 (18) | 0.0874 (5) | |
| O2 | 0.5079 (2) | 0.40213 (6) | 0.18422 (17) | 0.0833 (5) | |
| C1 | 0.6431 (3) | 0.42956 (9) | 0.5357 (3) | 0.0703 (5) | |
| H1A | 0.5807 | 0.4584 | 0.4578 | 0.084* | |
| C2 | 0.7115 (3) | 0.44196 (11) | 0.7090 (3) | 0.0868 (6) | |
| H2A | 0.6943 | 0.4789 | 0.7480 | 0.104* | |
| C3 | 0.8046 (3) | 0.40029 (12) | 0.8240 (3) | 0.0924 (7) | |
| H3A | 0.8500 | 0.4087 | 0.9418 | 0.111* | |
| C4 | 0.8316 (3) | 0.34597 (11) | 0.7670 (3) | 0.0872 (7) | |
| H4A | 0.8968 | 0.3177 | 0.8460 | 0.105* | |
| C5 | 0.7621 (3) | 0.33309 (9) | 0.5921 (3) | 0.0722 (5) | |
| H5A | 0.7807 | 0.2962 | 0.5535 | 0.087* | |
| C6 | 0.6651 (2) | 0.37496 (8) | 0.4747 (2) | 0.0583 (5) | |
| C7 | 0.5808 (3) | 0.36135 (8) | 0.2857 (2) | 0.0635 (5) | |
| N1 | 0.33910 (19) | 0.38091 (6) | −0.15464 (18) | 0.0574 (4) | |
| H1N1 | 0.3994 | 0.3858 | −0.0211 | 0.069* | |
| N2 | 0.4633 (2) | 0.28852 (6) | −0.1151 (2) | 0.0734 (5) | |
| H1N2 | 0.5218 | 0.2955 | 0.0136 | 0.088* | |
| H2N2 | 0.4965 | 0.2603 | −0.1752 | 0.088* | |
| C8 | 0.2395 (2) | 0.42572 (7) | −0.2537 (2) | 0.0616 (5) | |
| C9 | 0.1624 (3) | 0.41807 (9) | −0.4262 (3) | 0.0703 (5) | |
| H9A | 0.0956 | 0.4485 | −0.4947 | 0.084* | |
| C10 | 0.1811 (3) | 0.36533 (9) | −0.5041 (2) | 0.0692 (5) | |
| C11 | 0.2816 (3) | 0.32162 (8) | −0.4016 (2) | 0.0660 (5) | |
| H11A | 0.2959 | 0.2863 | −0.4511 | 0.079* | |
| C12 | 0.3632 (3) | 0.32926 (7) | −0.2236 (2) | 0.0582 (5) | |
| C13 | 0.2269 (4) | 0.47999 (10) | −0.1583 (4) | 0.0817 (7) | |
| H13A | 0.086 (9) | 0.490 (3) | −0.181 (9) | 0.123* | 0.50 |
| H13B | 0.285 (10) | 0.475 (3) | −0.023 (9) | 0.123* | 0.50 |
| H13C | 0.298 (10) | 0.513 (2) | −0.195 (8) | 0.123* | 0.50 |
| H13D | 0.155 (12) | 0.506 (3) | −0.232 (7) | 0.123* | 0.50 |
| H13E | 0.344 (8) | 0.494 (3) | −0.087 (10) | 0.123* | 0.50 |
| H13F | 0.184 (10) | 0.470 (3) | −0.045 (10) | 0.123* | 0.50 |
| C14 | 0.0940 (5) | 0.3577 (2) | −0.6963 (3) | 0.0984 (8) | |
| H14A | 0.143 (11) | 0.323 (5) | −0.738 (10) | 0.148* | 0.50 |
| H14B | 0.117 (14) | 0.392 (3) | −0.758 (11) | 0.148* | 0.50 |
| H14C | −0.041 (12) | 0.347 (3) | −0.724 (9) | 0.148* | 0.50 |
| H14D | 0.071 (13) | 0.319 (5) | −0.722 (11) | 0.148* | 0.50 |
| H14E | 0.179 (10) | 0.376 (5) | −0.759 (10) | 0.148* | 0.50 |
| H14F | −0.037 (11) | 0.382 (3) | −0.749 (9) | 0.148* | 0.50 |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.1331 (13) | 0.0527 (8) | 0.0720 (9) | −0.0037 (8) | 0.0288 (8) | 0.0009 (7) |
| O2 | 0.1135 (12) | 0.0558 (8) | 0.0663 (9) | −0.0009 (7) | 0.0119 (8) | 0.0059 (7) |
| C1 | 0.0710 (12) | 0.0710 (13) | 0.0675 (13) | 0.0012 (10) | 0.0216 (10) | −0.0006 (10) |
| C2 | 0.0918 (16) | 0.0927 (16) | 0.0724 (15) | 0.0031 (12) | 0.0231 (12) | −0.0119 (13) |
| C3 | 0.0939 (16) | 0.114 (2) | 0.0648 (14) | −0.0117 (14) | 0.0210 (12) | −0.0052 (14) |
| C4 | 0.0785 (14) | 0.0990 (18) | 0.0745 (15) | −0.0042 (12) | 0.0135 (11) | 0.0235 (13) |
| C5 | 0.0718 (12) | 0.0666 (12) | 0.0748 (14) | −0.0054 (9) | 0.0204 (10) | 0.0104 (10) |
| C6 | 0.0551 (10) | 0.0569 (11) | 0.0638 (11) | −0.0076 (8) | 0.0212 (8) | 0.0039 (9) |
| C7 | 0.0713 (12) | 0.0520 (11) | 0.0673 (12) | −0.0098 (9) | 0.0238 (9) | 0.0053 (10) |
| N1 | 0.0639 (9) | 0.0481 (8) | 0.0611 (9) | −0.0003 (7) | 0.0223 (7) | 0.0034 (7) |
| N2 | 0.1069 (13) | 0.0532 (9) | 0.0645 (10) | 0.0139 (8) | 0.0348 (9) | 0.0070 (8) |
| C8 | 0.0552 (10) | 0.0549 (11) | 0.0730 (13) | 0.0005 (8) | 0.0192 (9) | 0.0079 (9) |
| C9 | 0.0616 (11) | 0.0704 (13) | 0.0755 (14) | 0.0094 (9) | 0.0187 (10) | 0.0136 (10) |
| C10 | 0.0580 (11) | 0.0871 (14) | 0.0641 (12) | 0.0035 (10) | 0.0228 (9) | 0.0068 (11) |
| C11 | 0.0721 (12) | 0.0667 (12) | 0.0664 (12) | 0.0017 (9) | 0.0328 (10) | −0.0042 (9) |
| C12 | 0.0643 (11) | 0.0513 (10) | 0.0650 (12) | 0.0000 (8) | 0.0297 (9) | 0.0049 (9) |
| C13 | 0.0840 (17) | 0.0544 (13) | 0.0937 (19) | 0.0083 (11) | 0.0132 (14) | −0.0013 (12) |
| C14 | 0.093 (2) | 0.130 (3) | 0.0671 (15) | 0.014 (2) | 0.0203 (14) | 0.0021 (16) |
Geometric parameters (Å, °)
| O1—C7 | 1.250 (2) | C8—C9 | 1.348 (3) |
| O2—C7 | 1.249 (2) | C8—C13 | 1.491 (3) |
| C1—C2 | 1.369 (3) | C9—C10 | 1.398 (3) |
| C1—C6 | 1.380 (3) | C9—H9A | 0.9300 |
| C1—H1A | 0.9300 | C10—C11 | 1.362 (3) |
| C2—C3 | 1.360 (3) | C10—C14 | 1.500 (3) |
| C2—H2A | 0.9300 | C11—C12 | 1.391 (3) |
| C3—C4 | 1.370 (3) | C11—H11A | 0.9300 |
| C3—H3A | 0.9300 | C13—H13A | 1.04 (6) |
| C4—C5 | 1.384 (3) | C13—H13B | 1.06 (7) |
| C4—H4A | 0.9300 | C13—H13C | 1.04 (5) |
| C5—C6 | 1.381 (3) | C13—H13D | 0.89 (6) |
| C5—H5A | 0.9300 | C13—H13E | 0.93 (6) |
| C6—C7 | 1.497 (3) | C13—H13F | 1.11 (7) |
| N1—C12 | 1.352 (2) | C14—H14A | 0.99 (9) |
| N1—C8 | 1.367 (2) | C14—H14B | 0.98 (10) |
| N1—H1N1 | 1.0414 | C14—H14C | 1.00 (8) |
| N2—C12 | 1.335 (2) | C14—H14D | 0.90 (10) |
| N2—H1N2 | 1.0101 | C14—H14E | 1.03 (10) |
| N2—H2N2 | 0.9002 | C14—H14F | 1.09 (7) |
| C2—C1—C6 | 121.1 (2) | N1—C12—C11 | 118.47 (16) |
| C2—C1—H1A | 119.4 | C8—C13—H13A | 109 (3) |
| C6—C1—H1A | 119.4 | C8—C13—H13B | 112 (3) |
| C3—C2—C1 | 120.0 (2) | H13A—C13—H13B | 104 (4) |
| C3—C2—H2A | 120.0 | C8—C13—H13C | 109 (3) |
| C1—C2—H2A | 120.0 | H13A—C13—H13C | 113 (4) |
| C2—C3—C4 | 120.2 (2) | H13B—C13—H13C | 109 (4) |
| C2—C3—H3A | 119.9 | C8—C13—H13D | 109 (4) |
| C4—C3—H3A | 119.9 | H13A—C13—H13D | 52 (4) |
| C3—C4—C5 | 120.1 (2) | H13B—C13—H13D | 137 (5) |
| C3—C4—H4A | 120.0 | H13C—C13—H13D | 65 (4) |
| C5—C4—H4A | 120.0 | C8—C13—H13E | 113 (4) |
| C6—C5—C4 | 120.0 (2) | H13A—C13—H13E | 137 (4) |
| C6—C5—H5A | 120.0 | H13B—C13—H13E | 54 (4) |
| C4—C5—H5A | 120.0 | H13C—C13—H13E | 57 (4) |
| C1—C6—C5 | 118.58 (18) | H13D—C13—H13E | 116 (5) |
| C1—C6—C7 | 120.30 (16) | C8—C13—H13F | 111 (3) |
| C5—C6—C7 | 121.10 (17) | H13A—C13—H13F | 68 (4) |
| O2—C7—O1 | 123.88 (17) | H13C—C13—H13F | 136 (4) |
| O2—C7—C6 | 118.14 (16) | H13D—C13—H13F | 115 (5) |
| O1—C7—C6 | 117.98 (16) | H13E—C13—H13F | 92 (5) |
| C12—N1—C8 | 122.30 (16) | C10—C14—H14A | 112 (5) |
| C12—N1—H1N1 | 117.7 | C10—C14—H14B | 111 (5) |
| C8—N1—H1N1 | 120.0 | H14A—C14—H14B | 108 (6) |
| C12—N2—H1N2 | 122.4 | C10—C14—H14C | 108 (4) |
| C12—N2—H2N2 | 109.7 | H14A—C14—H14C | 102 (7) |
| H1N2—N2—H2N2 | 125.7 | H14B—C14—H14C | 115 (6) |
| C9—C8—N1 | 118.79 (17) | C10—C14—H14D | 110 (6) |
| C9—C8—C13 | 125.41 (18) | H14B—C14—H14D | 135 (7) |
| N1—C8—C13 | 115.79 (18) | H14C—C14—H14D | 67 (5) |
| C8—C9—C10 | 121.30 (17) | C10—C14—H14E | 111 (4) |
| C8—C9—H9A | 119.4 | H14A—C14—H14E | 77 (5) |
| C10—C9—H9A | 119.4 | H14C—C14—H14E | 138 (5) |
| C11—C10—C9 | 118.37 (18) | H14D—C14—H14E | 112 (7) |
| C11—C10—C14 | 121.2 (2) | C10—C14—H14F | 112 (3) |
| C9—C10—C14 | 120.5 (2) | H14A—C14—H14F | 132 (6) |
| C10—C11—C12 | 120.77 (18) | H14B—C14—H14F | 72 (5) |
| C10—C11—H11A | 119.6 | H14C—C14—H14F | 46 (4) |
| C12—C11—H11A | 119.6 | H14D—C14—H14F | 109 (7) |
| N2—C12—N1 | 117.34 (16) | H14E—C14—H14F | 104 (6) |
| N2—C12—C11 | 124.19 (17) | ||
| C6—C1—C2—C3 | 0.7 (3) | C12—N1—C8—C9 | 0.1 (2) |
| C1—C2—C3—C4 | 0.5 (4) | C12—N1—C8—C13 | −179.6 (2) |
| C2—C3—C4—C5 | −0.8 (4) | N1—C8—C9—C10 | 0.7 (3) |
| C3—C4—C5—C6 | −0.1 (3) | C13—C8—C9—C10 | −179.7 (2) |
| C2—C1—C6—C5 | −1.6 (3) | C8—C9—C10—C11 | −0.8 (3) |
| C2—C1—C6—C7 | 176.91 (18) | C8—C9—C10—C14 | 179.9 (2) |
| C4—C5—C6—C1 | 1.3 (3) | C9—C10—C11—C12 | 0.2 (3) |
| C4—C5—C6—C7 | −177.18 (18) | C14—C10—C11—C12 | 179.5 (2) |
| C1—C6—C7—O2 | 7.2 (3) | C8—N1—C12—N2 | 179.88 (16) |
| C5—C6—C7—O2 | −174.36 (18) | C8—N1—C12—C11 | −0.6 (2) |
| C1—C6—C7—O1 | −172.47 (18) | C10—C11—C12—N2 | 179.94 (18) |
| C5—C6—C7—O1 | 6.0 (3) | C10—C11—C12—N1 | 0.5 (3) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N1···O2 | 1.04 | 1.65 | 2.683 (2) | 172 |
| N2—H1N2···O1 | 1.01 | 1.78 | 2.779 (2) | 171 |
| N2—H2N2···O1i | 0.90 | 1.97 | 2.853 (2) | 168 |
Symmetry codes: (i) x, −y+1/2, z−1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: FJ2327).
References
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl.34, 1555–1573.
- Bruker (2009). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Dhar, D. N. & Taploo, C. L. (1982). J. Sci. Ind. Res 41, 501–506.
- Jeffrey, G. A. (1997). An Introduction to Hydrogen Bonding Oxford University Press.
- Jeffrey, G. A. & Saenger, W. (1991). Hydrogen Bonding in Biological Structures Berlin: Springer.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810034811/fj2327sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810034811/fj2327Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report