

Abstract
The title compound, C13H18BNO4, was readily obtained from the reaction of methyl 4-boronobenzene acetate with ethanolamine. A combination of intermolecular N—H⋯O hydrogen bonds and C—H⋯π interactions leads to the pairwise association of molecules.
Related literature
For background to the biological importance of boron, see: Warrington (1923 ▶); Jabbour et al. (2004 ▶). For the use of boron-containing reagents in synthetic chemistry, see: Miyaura & Suzuki (1995 ▶); Corey et al. (1987 ▶); Liu et al. (2007 ▶); Jung & Lazarova (1999 ▶); Chan et al. (1998 ▶); Evans et al. (1998 ▶); Lam et al. (1998 ▶). For related structures, see: Rettig & Trotter (1975 ▶); Wang & Georghiou (2002 ▶).
Experimental
Crystal data
C13H18BNO4
M r = 263.10
Orthorhombic,
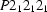
a = 8.3776 (11) Å
b = 8.9269 (11) Å
c = 17.369 (2) Å
V = 1299.0 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.10 mm−1
T = 153 K
0.30 × 0.09 × 0.06 mm
Data collection
Rigaku Saturn diffractometer
Absorption correction: numerical (NUMABS; Higashi, 1999 ▶) T min = 0.985, T max = 0.997
16363 measured reflections
1725 independent reflections
1707 reflections with I > 2σ(I)
R int = 0.037
Refinement
R[F 2 > 2σ(F 2)] = 0.043
wR(F 2) = 0.112
S = 1.17
1725 reflections
173 parameters
H-atom parameters constrained
Δρmax = 0.20 e Å−3
Δρmin = −0.22 e Å−3
Data collection: CrystalClear (Rigaku, 2002 ▶); cell refinement: CrystalClear; data reduction: CrystalClear; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 (Farrugia, 1997 ▶); software used to prepare material for publication: publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810037864/fj2337sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810037864/fj2337Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
Cg3 is the centroid of the C1–C6 ring.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O2i | 0.93 | 2.06 | 2.921 (2) | 154 |
| C10—H10B⋯Cg3ii | 0.99 | 2.65 | 3.618 (2) | 166 |
Symmetry codes: (i) 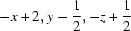 ; (ii)
; (ii) 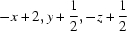 .
.
Acknowledgments
We thank the NSERC and the Department of Chemistry for financial support. We are also grateful to the Ministry for Higher Education, Egypt, for a scholarship to ALZ.
supplementary crystallographic information
Comment
Boron is known to be an important trace element in higher plants (Warrington, 1923) and boron-containing compounds have been shown to have a range of diverse biological acvtivities (Jabbour et al., 2004). The use of boron-containing reagents is also widespread in synthetic chemistry and this is due mainly to the pioneering work of H. C. Brown and coworkers and Suzuki and his coworkers (Miyaura & Suzuki, 1995). Corey, Bakshi and Shibata (Corey et al., 1987) discovered that a chrial oxazaborolidine ("CBS" reagent) which contains both boron-nitrogen and boron-oxygen bonds was capable of effecting enantioselective reduction of prochiral ketones, imines, and oximes to produce chiral alcohols, amines, and amino alcohols in excellent yields and ee's. Corey's group has also shown that chiral oxazaborolidine-aluminium bromide complexes (Liu et al. 2007) are also effective catalysts for enantioselective Diels-Alder reactions. In principle, oxazaborolidines are derived from reactions of a boronic acid and aminoalcohols and a less well known application of oxazaborolidines is to facilitate the conversion of a pinacolatoborane, by mild acid-catalysis (Jung & Lazarova, 1999), to the corresponding boronic acid, a key step for cupric acetate promoted coupling of an arylboronic acids with phenols (Chan et al., 1998; Evans et al., 1998; Lam et al. 1998).
There has been only one reported X-ray crystallographic study of the structure of a diethanolamine ester of a phenylboronic acid (1) (Rettig & Trotter, 1975). This compound which was named as B-phenyl-diptychboroxazolidine (alternative names: Tetrahydro-[1,3,2]oxaza-borol[2,3-b][1,3,2]oxazaborole; [[2,2'-(Imino-kN)bis[ethanolato-kO]](2-)]phenylboron) was measured on a diffractometer with Cu Ka radiation and it was revealed to be non-centrosymmetric and in the P21 space group. The absolute configuration of the enatiomorphic crystal was determined in this study. In connection with our own work (Wang & Georghiou, 2002), crystals of (2), the corresponding diethanolamine ester of the 4-boronic acid derivative of methyl phenylacetate, were obtained and the structure of the molecule is reported here.
Methyl p-[[2,2'-iminobis[ethanolato]](2-)-N,O,O']phenylacetateboron (2; Figure 1) crystallized in the non-centrosymmetric space group P212121, however, data collection was performed using molybdenum radiation, and the absolute configuration could not be determined due to the lack of an atom with significant anomalous dispersion. Intermolecular hydrogen bonding between N1—H1···O2i (N1···O2i = 2.921 (2) Å) and C—H···π interactions between C10—H10B···Cg3ii (C10···Cg3ii= 3.618 (2); where Cg3 is the centroid of C1—C6) leads to the pair-wise association of molecules (Figure 2). These molecular associates are related via the twofold screw axes in the crystal structure (viewed perpendicular to the b axis in Figure 3).
Experimental
To a solution of PdCl2(dppf) (160 mg, 0.18 mmol) in dioxane (24 ml) was added methyl 4-(trifluoroacetoxyacetate (1.58 g, 5.93 mmol), Et3N (2.49 ml, 17.8 mmol) and 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.30 ml, 8.9 mmol). After stirring for 20 h at 100 oC, the reaction mixture was extracted with benzene. The extract was purified by flash column chromatography (silica gel, 10%EtOAc in hexanes) to afford the arylboronate (1.47 g, 90%) 1HNMR (500 MHz, CDCl3) 1.34 (s, 12H), 3.64 (s, 2H), 3.67(s, 3H), 7.27(d, J=10 Hz, 2H), 7.77(d, J=10 Hz, 2H). To a solution of the arylboronate (1.47 g, 5.3 mmol) in diethylether (53 ml) was added diethanolamine (0.6 ml, 5.8 mmol) in 2-propanol (10 ml). The resulting mixture was stirred at ambient temperature for 72 h, the reaction mixture was then filtered and the solid was washed with diethyl ether to give cyclic aminoarylboronate (1.08 g, 77%) as a colorless powder. Crystals suitable for X-ray diffraction analysis were obtained by crystallization from ethyl acetate solution. 1H NMR (500 MHz, CDCl3) 2.49–2.51(m, 2H), 2.96–3.03(m, 2H), 3.58(s, 2H), 3.63(s, 3H), 3.70–3.72(m, 2H), 3.79–3.74(m, 2H), 5.83(s, 1H), 7.14(d, J=7.5 Hz, 2H), 7.43(d, J=7.5 Hz, 2H); 13C NMR 41.0, 51.1, 51.8, 63.2, 128.3, 132.7, 132.8, 172.6.
Refinement
H atoms were found in difference Fourier maps and subsequently placed in idealized positions with constrained distances and with Uiso(H) values set to either 1.2Ueq or 1.5Ueq of the attached atom. They were refined on a riding model. All non-hydrogen atoms were refined anisotropically. This crystal was a weak anomalous scatterer collected with MoKa radiation, therefore, Friedel mates were merged (MERG 4) and absolute configuration was not determined.
Figures
Fig. 1.

The molecular structure of (2), with atom labels and 50% probability displacement ellipsoids for non-H atoms.
Fig. 2.

Intermolecular hydrogen bonds (long dashes) and C—H···π interactions (short dashes) between two associated molecules.
Fig. 3.

Unit cell viewed perpendicular to the b axis, showing the pair-wise ordering of molecules in the crystal lattice. (Cell axes: a = red, b = green, c = blue)
Crystal data
| C13H18BNO4 | Dx = 1.345 Mg m−3 |
| Mr = 263.10 | Melting point = 452–453 K |
| Orthorhombic, P212121 | Mo Kα radiation, λ = 0.71075 Å |
| Hall symbol: P 2ac 2ab | Cell parameters from 5108 reflections |
| a = 8.3776 (11) Å | θ = 2.6–30.6° |
| b = 8.9269 (11) Å | µ = 0.10 mm−1 |
| c = 17.369 (2) Å | T = 153 K |
| V = 1299.0 (3) Å3 | Platelet, colorless |
| Z = 4 | 0.30 × 0.09 × 0.06 mm |
| F(000) = 560 |
Data collection
| Rigaku Saturn diffractometer | 1725 independent reflections |
| Radiation source: fine-focus sealed tube | 1707 reflections with I > 2σ(I) |
| graphite - Rigaku SHINE | Rint = 0.037 |
| Detector resolution: 14.63 pixels mm-1 | θmax = 27.5°, θmin = 2.6° |
| ω scans | h = −10→10 |
| Absorption correction: numerical (NUMABS; Higashi, 1999) | k = −11→11 |
| Tmin = 0.985, Tmax = 0.997 | l = −22→22 |
| 16363 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.043 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.112 | H-atom parameters constrained |
| S = 1.17 | w = 1/[σ2(Fo2) + (0.0529P)2 + 0.4126P] where P = (Fo2 + 2Fc2)/3 |
| 1725 reflections | (Δ/σ)max < 0.001 |
| 173 parameters | Δρmax = 0.20 e Å−3 |
| 0 restraints | Δρmin = −0.22 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.The crystal was a weak anomalous scatterer collected with Mo Kα radiation. Friedel mates were merged (MERG 4) and the absolute configuration was not determined. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 1.00908 (19) | 0.47529 (18) | 0.35582 (9) | 0.0281 (4) | |
| O2 | 0.85897 (19) | 0.47478 (17) | 0.23631 (9) | 0.0268 (4) | |
| O3 | 0.5232 (2) | −0.2735 (2) | 0.39057 (11) | 0.0422 (5) | |
| O4 | 0.6510 (3) | −0.2494 (3) | 0.50205 (13) | 0.0617 (7) | |
| N1 | 1.0730 (2) | 0.2952 (2) | 0.25504 (10) | 0.0249 (4) | |
| H1 | 1.0599 | 0.1919 | 0.2577 | 0.030* | |
| C1 | 0.7998 (3) | 0.2734 (2) | 0.34181 (12) | 0.0243 (4) | |
| C2 | 0.6576 (3) | 0.2247 (3) | 0.30806 (13) | 0.0253 (4) | |
| H2 | 0.6285 | 0.2624 | 0.2589 | 0.030* | |
| C3 | 0.5568 (3) | 0.1220 (3) | 0.34475 (13) | 0.0261 (5) | |
| H3 | 0.4626 | 0.0888 | 0.3195 | 0.031* | |
| C4 | 0.5932 (3) | 0.0679 (3) | 0.41775 (13) | 0.0263 (5) | |
| C5 | 0.7326 (3) | 0.1174 (3) | 0.45272 (13) | 0.0296 (5) | |
| H5 | 0.7589 | 0.0827 | 0.5028 | 0.035* | |
| C6 | 0.8342 (3) | 0.2173 (3) | 0.41551 (13) | 0.0276 (5) | |
| H6 | 0.9294 | 0.2482 | 0.4406 | 0.033* | |
| C7 | 1.1685 (3) | 0.4963 (3) | 0.33084 (14) | 0.0302 (5) | |
| H7A | 1.2382 | 0.5254 | 0.3744 | 0.036* | |
| H7B | 1.1743 | 0.5747 | 0.2906 | 0.036* | |
| C8 | 1.2172 (3) | 0.3441 (3) | 0.29856 (15) | 0.0313 (5) | |
| H8A | 1.3108 | 0.3534 | 0.2641 | 0.038* | |
| H8B | 1.2427 | 0.2729 | 0.3405 | 0.038* | |
| C9 | 0.8982 (3) | 0.4085 (3) | 0.16438 (13) | 0.0283 (5) | |
| H9A | 0.8939 | 0.4839 | 0.1226 | 0.034* | |
| H9B | 0.8231 | 0.3263 | 0.1520 | 0.034* | |
| C10 | 1.0671 (3) | 0.3484 (3) | 0.17388 (14) | 0.0297 (5) | |
| H10A | 1.0875 | 0.2650 | 0.1376 | 0.036* | |
| H10B | 1.1469 | 0.4283 | 0.1649 | 0.036* | |
| C11 | 0.4878 (3) | −0.0470 (3) | 0.45767 (13) | 0.0287 (5) | |
| H11A | 0.4717 | −0.0173 | 0.5120 | 0.034* | |
| H11B | 0.3821 | −0.0498 | 0.4323 | 0.034* | |
| C12 | 0.5624 (3) | −0.1999 (3) | 0.45463 (15) | 0.0330 (5) | |
| C13 | 0.5926 (4) | −0.4222 (3) | 0.3835 (2) | 0.0499 (8) | |
| H13A | 0.7093 | −0.4143 | 0.3831 | 0.060* | |
| H13B | 0.5563 | −0.4686 | 0.3354 | 0.060* | |
| H13C | 0.5589 | −0.4840 | 0.4272 | 0.060* | |
| B1 | 0.9234 (3) | 0.3847 (3) | 0.30025 (14) | 0.0236 (5) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0291 (8) | 0.0257 (8) | 0.0296 (8) | −0.0045 (7) | 0.0027 (6) | −0.0029 (7) |
| O2 | 0.0318 (8) | 0.0203 (7) | 0.0283 (8) | 0.0029 (7) | 0.0023 (7) | 0.0014 (6) |
| O3 | 0.0412 (10) | 0.0352 (10) | 0.0503 (10) | 0.0057 (9) | −0.0049 (9) | −0.0132 (9) |
| O4 | 0.0875 (17) | 0.0522 (12) | 0.0453 (11) | 0.0243 (14) | −0.0164 (12) | 0.0011 (9) |
| N1 | 0.0271 (9) | 0.0181 (8) | 0.0295 (9) | 0.0004 (8) | 0.0027 (8) | 0.0024 (8) |
| C1 | 0.0254 (10) | 0.0195 (10) | 0.0279 (10) | 0.0014 (9) | 0.0055 (8) | −0.0020 (9) |
| C2 | 0.0263 (10) | 0.0228 (10) | 0.0268 (10) | 0.0030 (9) | 0.0023 (9) | 0.0005 (9) |
| C3 | 0.0232 (10) | 0.0234 (10) | 0.0318 (11) | 0.0010 (9) | 0.0011 (9) | −0.0018 (9) |
| C4 | 0.0289 (10) | 0.0213 (10) | 0.0286 (10) | 0.0009 (9) | 0.0075 (9) | −0.0026 (9) |
| C5 | 0.0360 (12) | 0.0291 (11) | 0.0236 (10) | −0.0050 (10) | 0.0014 (10) | 0.0013 (9) |
| C6 | 0.0282 (10) | 0.0270 (11) | 0.0276 (10) | −0.0031 (10) | 0.0000 (9) | −0.0030 (9) |
| C7 | 0.0285 (11) | 0.0286 (12) | 0.0334 (12) | −0.0054 (10) | −0.0020 (9) | 0.0032 (10) |
| C8 | 0.0262 (11) | 0.0284 (12) | 0.0393 (13) | −0.0005 (10) | −0.0021 (10) | 0.0054 (10) |
| C9 | 0.0349 (12) | 0.0233 (11) | 0.0266 (11) | −0.0002 (10) | −0.0020 (9) | 0.0003 (9) |
| C10 | 0.0347 (12) | 0.0259 (11) | 0.0285 (11) | 0.0016 (10) | 0.0062 (10) | 0.0010 (9) |
| C11 | 0.0308 (11) | 0.0261 (11) | 0.0291 (11) | −0.0023 (9) | 0.0051 (9) | −0.0008 (9) |
| C12 | 0.0345 (12) | 0.0316 (12) | 0.0330 (11) | −0.0013 (11) | 0.0057 (10) | 0.0010 (10) |
| C13 | 0.0444 (15) | 0.0340 (14) | 0.071 (2) | 0.0026 (13) | 0.0060 (15) | −0.0162 (15) |
| B1 | 0.0261 (12) | 0.0188 (11) | 0.0258 (11) | 0.0004 (10) | 0.0020 (10) | −0.0009 (9) |
Geometric parameters (Å, °)
| O1—C7 | 1.417 (3) | C5—C6 | 1.392 (3) |
| O1—B1 | 1.449 (3) | C5—H5 | 0.9500 |
| O2—C9 | 1.421 (3) | C6—H6 | 0.9500 |
| O2—B1 | 1.474 (3) | C7—C8 | 1.525 (4) |
| O3—C12 | 1.333 (3) | C7—H7A | 0.9900 |
| O3—C13 | 1.455 (4) | C7—H7B | 0.9900 |
| O4—C12 | 1.193 (3) | C8—H8A | 0.9900 |
| N1—C10 | 1.488 (3) | C8—H8B | 0.9900 |
| N1—C8 | 1.491 (3) | C9—C10 | 1.523 (3) |
| N1—B1 | 1.681 (3) | C9—H9A | 0.9900 |
| N1—H1 | 0.9300 | C9—H9B | 0.9900 |
| C1—C2 | 1.397 (3) | C10—H10A | 0.9900 |
| C1—C6 | 1.405 (3) | C10—H10B | 0.9900 |
| C1—B1 | 1.607 (3) | C11—C12 | 1.502 (3) |
| C2—C3 | 1.400 (3) | C11—H11A | 0.9900 |
| C2—H2 | 0.9500 | C11—H11B | 0.9900 |
| C3—C4 | 1.391 (3) | C13—H13A | 0.9800 |
| C3—H3 | 0.9500 | C13—H13B | 0.9800 |
| C4—C5 | 1.388 (3) | C13—H13C | 0.9800 |
| C4—C11 | 1.520 (3) | ||
| C7—O1—B1 | 109.67 (18) | N1—C8—H8B | 111.1 |
| C9—O2—B1 | 110.55 (17) | C7—C8—H8B | 111.1 |
| C12—O3—C13 | 114.9 (2) | H8A—C8—H8B | 109.1 |
| C10—N1—C8 | 114.44 (19) | O2—C9—C10 | 105.45 (19) |
| C10—N1—B1 | 105.44 (17) | O2—C9—H9A | 110.7 |
| C8—N1—B1 | 103.17 (17) | C10—C9—H9A | 110.7 |
| C10—N1—H1 | 111.1 | O2—C9—H9B | 110.7 |
| C8—N1—H1 | 111.1 | C10—C9—H9B | 110.7 |
| B1—N1—H1 | 111.1 | H9A—C9—H9B | 108.8 |
| C2—C1—C6 | 116.5 (2) | N1—C10—C9 | 104.23 (19) |
| C2—C1—B1 | 123.6 (2) | N1—C10—H10A | 110.9 |
| C6—C1—B1 | 119.9 (2) | C9—C10—H10A | 110.9 |
| C1—C2—C3 | 121.8 (2) | N1—C10—H10B | 110.9 |
| C1—C2—H2 | 119.1 | C9—C10—H10B | 110.9 |
| C3—C2—H2 | 119.1 | H10A—C10—H10B | 108.9 |
| C4—C3—C2 | 120.7 (2) | C12—C11—C4 | 110.81 (19) |
| C4—C3—H3 | 119.7 | C12—C11—H11A | 109.5 |
| C2—C3—H3 | 119.7 | C4—C11—H11A | 109.5 |
| C5—C4—C3 | 118.2 (2) | C12—C11—H11B | 109.5 |
| C5—C4—C11 | 120.3 (2) | C4—C11—H11B | 109.5 |
| C3—C4—C11 | 121.5 (2) | H11A—C11—H11B | 108.1 |
| C4—C5—C6 | 121.0 (2) | O4—C12—O3 | 123.2 (3) |
| C4—C5—H5 | 119.5 | O4—C12—C11 | 124.8 (3) |
| C6—C5—H5 | 119.5 | O3—C12—C11 | 112.0 (2) |
| C5—C6—C1 | 121.7 (2) | O3—C13—H13A | 109.5 |
| C5—C6—H6 | 119.1 | O3—C13—H13B | 109.5 |
| C1—C6—H6 | 119.1 | H13A—C13—H13B | 109.5 |
| O1—C7—C8 | 104.28 (19) | O3—C13—H13C | 109.5 |
| O1—C7—H7A | 110.9 | H13A—C13—H13C | 109.5 |
| C8—C7—H7A | 110.9 | H13B—C13—H13C | 109.5 |
| O1—C7—H7B | 110.9 | O1—B1—O2 | 112.28 (19) |
| C8—C7—H7B | 110.9 | O1—B1—C1 | 111.41 (18) |
| H7A—C7—H7B | 108.9 | O2—B1—C1 | 116.1 (2) |
| N1—C8—C7 | 103.34 (19) | O1—B1—N1 | 101.97 (18) |
| N1—C8—H8A | 111.1 | O2—B1—N1 | 100.39 (17) |
| C7—C8—H8A | 111.1 | C1—B1—N1 | 113.35 (18) |
Hydrogen-bond geometry (Å, °)
| Cg3 is the centroid of the C1–C6 ring. |
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O2i | 0.93 | 2.06 | 2.921 (2) | 154 |
| C10—H10B···Cg3ii | 0.99 | 2.65 | 3.618 (2) | 166 |
Symmetry codes: (i) −x+2, y−1/2, −z+1/2; (ii) −x+2, y+1/2, −z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: FJ2337).
References
- Chan, D. M. T., Monaco, K. L., Wang, R. P. & Winters, M. P. (1998). Tetrahedron Lett.39, 2933–2936.
- Corey, E. J., Bakshi, R. K. & Shibata, S. (1987). J. Am. Chem. Soc.109, 5553–5554.
- Evans, D. A., Katz, J. L. & West, T. R. (1998). Tetrahedron Lett.39, 2937–2940.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Higashi, T. (1999). NUMABS Rigaku Corporation, Tokyo.
- Jabbour, A., Steinberg, D., Valery, M. D., Moussaieff, A., Zaks, B. & Srebnik, M. (2004). J. Med. Chem.47, 2409–2410. [DOI] [PubMed]
- Jung, M. E. & Lazarova, T. I. (1999). J. Org. Chem.64, 2976–2977. [DOI] [PubMed]
- Lam, P. Y. S., Clark, C. G., Saubern, S., Adams, J., Winters, M. P., Chan, D. M. T. & Combs, A. (1998). Tetrahedron Lett.39, 2941–2944.
- Liu, D., Canales, E. & Corey, E. J. (2007). J. Am. Chem. Soc.129, 1498–1499. [DOI] [PubMed]
- Miyaura, N. & Suzuki, A. (1995). Chem. Rev.95, 2457–2483.
- Rettig, S. J. & Trotter, J. (1975). Can. J. Chem.53, 1393–1401.
- Rigaku (2002). CrystalClear Rigaku Corporation, Tokyo, Japan.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Wang, Y.-C. & Georghiou, P. E. (2002). Org. Lett.4, 2675–2678. [DOI] [PubMed]
- Warrington, K. (1923). Ann. Bot.37, 629–672.
- Westrip, S. P. (2010). J. Appl. Cryst.43, 920–925.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810037864/fj2337sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810037864/fj2337Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report