

Abstract
In the title molecule {systematic name: (5S)-5-[(β-d-glucopyranosyloxy)methyl]furan-2(5H)-one}, C11H16O8, the five-membered ring is essentially planar, the maximum deviation being 0.0151 (13) Å for the O atom. The six-membered ring adopts a chair conformation with puckering parameters Q = 0.581 (2) Å, θ = 9.0 (2)° and ϕ = 39.7 (13)°, and with all of the substituents of the glucoside unit having normal equatorial orientations. The crystal structure is stabilized by extensive O—H⋯O and C—H⋯O hydrogen bonding, resulting in a three-dimensional network.
Related literature
For background to ranunculin, see: Hill & van Heyningen (1951 ▶); Bai et al. (1996 ▶); Benn & Yelland (1968 ▶); Boll (1968 ▶); Camps et al. (1982 ▶); Fang et al. (1989 ▶). For chemical and spectrometric data for closely related, simple butenolides, see: Perry et al. (1996 ▶). For comparison bond distances, see: Allen et al. (1987 ▶). For puckering parameters, see: Cremer & Pople (1975 ▶).
Experimental
Crystal data
C11H16O8
M r = 276.24
Monoclinic,
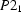
a = 5.7944 (4) Å
b = 6.9359 (3) Å
c = 15.0491 (10) Å
β = 97.895 (2)°
V = 599.08 (6) Å3
Z = 2
Mo Kα radiation
μ = 0.13 mm−1
T = 173 K
0.30 × 0.24 × 0.02 mm
Data collection
Nonius KappaCCD diffractometer with APEXII CCD
Absorption correction: multi-scan (SORTAV; Blessing, 1997 ▶) T min = 0.961, T max = 0.997
1926 measured reflections
1133 independent reflections
1112 reflections with I > 2σ(I)
R int = 0.018
Refinement
R[F 2 > 2σ(F 2)] = 0.026
wR(F 2) = 0.069
S = 1.04
1133 reflections
184 parameters
1 restraint
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.18 e Å−3
Δρmin = −0.16 e Å−3
Data collection: COLLECT (Hooft, 1998 ▶); cell refinement: DENZO (Otwinowski & Minor, 1997 ▶); data reduction: SCALEPACK (Otwinowski & Minor, 1997 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810034847/fl2315sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810034847/fl2315Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O5—H5O⋯O8i | 0.86 (3) | 2.17 (3) | 2.910 (2) | 144 (3) |
| O6—H6O⋯O5ii | 0.92 (3) | 1.94 (3) | 2.824 (2) | 162 (3) |
| O7—H7O⋯O6ii | 0.84 (3) | 1.84 (3) | 2.668 (2) | 173 (3) |
| O8—H8O⋯O2iii | 0.88 (3) | 1.96 (3) | 2.830 (2) | 167 (3) |
| O6—H6O⋯O7 | 0.92 (3) | 2.56 (3) | 2.894 (2) | 102 (2) |
Symmetry codes: (i) 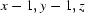 ; (ii)
; (ii) 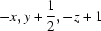 ; (iii)
; (iii) 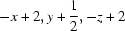 .
.
supplementary crystallographic information
Comment
Ranunculin (I) is the glycosidic precursor of the vesicant protoanemonin present in numerous species of Rannunculaceae and is especially associated with the burning sensation on chewing leaves of buttercup plants. It was first obtained in crystalline form by Hill and van Heyningen (1951) who established its gross structure and showed that it undergoes enzymatic cleavage by β-glucosidase to yield the aglucone, which underwent easy dehydration to protoanemonin. These processes were shown to occur readily under autolytic conditions (Bai et al., 1996). The S-stereochemistry of the dihydrofuranone ring was deduced by Benn and Yelland (1968), and Boll (1968), as shown in the schematic diagram, and later confirmed by synthesis (Camps et al., 1982; Fang et al., 1989). Our sample of (I) was a natural product, and as such had been biosynthesised in the plant (and not made in a laboratory). The only stereoisomer which occurs naturally is the D-isomer, and both that and the anomeric configuration of the glycoosidic bond in (I) were established by cleavage of the glycoside by β-D-glucosidases. The X-ray structure reported here provides the simplest unequivocal proof of that stereochemistry since the chirality follows from that of the D-glucopyranosyl moiety.
The molecular structure of (I) is presented in Fig. 1. The five membered ring, O1/C2—C5, is essentially planar with the maximum deviation of any atom being 0.0151 (13) Å for O1. The six-membered ring adopts a chair conformation with puckering parameters (Cremer & Pople, 1975): Q = 0.581 (2) Å, θ = 9.0 (2)° and φ = 39.7 (13)°, with all of the substituents of the glucoside unit having normal equatorial orientations. The 5-membered ring folds back away from the six-membered ring as reflected by the torsion angle C6—O3—C1—C5 -162.86 (17)°. The bond distances and angles are as expected (Allen, 2002). The structure is stabilized by extensive O—H···O and C—H···O hydrogen bonding resulting in a three diemsional network (Table 1 & Fig. 2).
There are two other, closely related, simple butenolides known, though their structures rest on chemical and spectrometric data, i.e., without X-ray crystallographic support. They are the (5R,6R) and (5S,6R) steeroisomers of 5-([1-β-D-glucopyranosyloxy]ethyl)-2(5H)-furanone (Perry et al., 1996); the glycosidic precursor of (Z)-5-ethylidene-2(5H)-furanone, a homologue of protoanemonin in Halocarpus biformis juvenile foliage.
Experimental
The details of the isolation, and some of the physical properties, of our sample of (I) have been reported previously (Benn & Yelland 1968). Suitable crystals of the title compound for X-ray study were grown from a solution in aqueous ethanol (ca 1:20) in the form of plates.
Refinement
An absolute structure could not be established reliably because of insufficient anomalous scattering effects. Therefore, 792 Friedel pairs were merged. The H-atoms were located from difference maps and were included in the refinements at geometrically idealized positions with C—H distances = 0.95, 0.99 and 1.00 Å for aryl, methylene and methine type H-atoms, respectively; the positions of hydroxyl H-atoms were allowed to refine freely. The H-atoms were assigned Uiso = 1.2 and 1.5 × Ueq of the parent C and O-atoms, respectively. The final difference map was free of chemically significant features.
Figures
Fig. 1.

ORTEP-3 (Farrugia, 1997) drawing of the title compound with displacement ellipsoids plotted at 50% probability level.
Fig. 2.

Unit cell packing of the title compound showing intermolecular hydrogen bonds of O—H···O; H-atoms not involved in hydrogen bonds have been excluded.
Crystal data
| C11H16O8 | F(000) = 292 |
| Mr = 276.24 | Dx = 1.531 Mg m−3 |
| Monoclinic, P21 | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: P 2yb | Cell parameters from 1560 reflections |
| a = 5.7944 (4) Å | θ = 1.0–30.0° |
| b = 6.9359 (3) Å | µ = 0.13 mm−1 |
| c = 15.0491 (10) Å | T = 173 K |
| β = 97.895 (2)° | Plate, colorless |
| V = 599.08 (6) Å3 | 0.30 × 0.24 × 0.02 mm |
| Z = 2 |
Data collection
| Nonius APEXII CCD [APEXII is a Bruker machine - is this a KappaCCD upgraded with an APEXII CCD?]diffractometer | 1133 independent reflections |
| Radiation source: fine-focus sealed tube | 1112 reflections with I > 2σ(I) |
| graphite | Rint = 0.018 |
| φ & ω scans | θmax = 25.0°, θmin = 3.2° |
| Absorption correction: multi-scan (SORTAV; Blessing, 1997) | h = −6→6 |
| Tmin = 0.961, Tmax = 0.997 | k = −6→8 |
| 1926 measured reflections | l = −17→17 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.026 | Hydrogen site location: difference Fourier map |
| wR(F2) = 0.069 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.04 | w = 1/[σ2(Fo2) + (0.040P)2 + 0.1617P] where P = (Fo2 + 2Fc2)/3 |
| 1133 reflections | (Δ/σ)max = 0.002 |
| 184 parameters | Δρmax = 0.18 e Å−3 |
| 1 restraint | Δρmin = −0.16 e Å−3 |
Special details
| Experimental. NMR data (400 MHz, 1H; 100 MHz 13C) for a solution in D2O containing sodium 3-trimethylsilylpropionate-2,3 - d4 as reference: δH (400 MHz) 7.77 (1H, dd, J = 1.5 and 5.8 Hz, H-4), 6.3 (1H, dd, J = 2.1 and 5.8 Hz, H-3), 5.47 (1H, m), 4.48 (1H, d, J = 7.9 Hz, H-1'), 4.30 (1H, dd, J = 3.2 and 12.2 Hz, H-6 A), 3.95 (1H, dd, J = 5.8 and 12.2 Hz, H-6B), 3.91 (1H, dd, J = 2.1 and 12.5 Hz, H-6A'), 3.72 (1H, dd, J = 5.8 and 12.5 Hz, H-6B'), 3.48 (1H, dd, dd, J = ca 9 Hz H-3'), 3.43 (1H, m, H-5'), 3.37 (1H, dd, J = ca 9 Hz, H=4'), and 3.25 (1H, dd, J = 7.9 and 9.2 Hz, H-2'); δC 179.2 s (C-2), 158.5 d (C-4), 124.7 d (C-3), 105.6 d (C-1'), 86.9 d (C-5), 78.7 d (C-5'), 78.3 d (C-3'), 75.6 d (C-2'), 72.2 d (C-4'), 71.7 t (C-6), 63.3 t (C-6'). |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.9146 (3) | −0.1112 (2) | 0.96815 (9) | 0.0225 (4) | |
| O2 | 1.0618 (3) | −0.0493 (3) | 1.11025 (10) | 0.0309 (4) | |
| O3 | 0.4499 (2) | 0.0377 (2) | 0.79524 (9) | 0.0200 (3) | |
| O4 | 0.5209 (2) | 0.3269 (2) | 0.73224 (9) | 0.0188 (3) | |
| O5 | 0.1227 (3) | −0.0568 (2) | 0.63887 (10) | 0.0241 (4) | |
| H5O | 0.085 (5) | −0.121 (5) | 0.6833 (19) | 0.036* | |
| O6 | −0.0241 (2) | 0.2692 (2) | 0.53178 (10) | 0.0208 (4) | |
| H6O | −0.025 (5) | 0.336 (5) | 0.479 (2) | 0.031* | |
| O7 | 0.3667 (3) | 0.5126 (3) | 0.50561 (9) | 0.0215 (3) | |
| H7O | 0.259 (5) | 0.591 (5) | 0.4895 (18) | 0.032* | |
| O8 | 0.8805 (3) | 0.6376 (3) | 0.72096 (10) | 0.0277 (4) | |
| H8O | 0.893 (5) | 0.563 (5) | 0.769 (2) | 0.042* | |
| C1 | 0.6798 (3) | 0.0332 (4) | 0.84401 (13) | 0.0216 (5) | |
| H1A | 0.7191 | 0.1601 | 0.8721 | 0.026* | |
| H1B | 0.7947 | 0.0036 | 0.8030 | 0.026* | |
| C2 | 0.8918 (4) | −0.0724 (3) | 1.05518 (13) | 0.0225 (5) | |
| C3 | 0.6434 (4) | −0.0666 (4) | 1.06406 (14) | 0.0252 (5) | |
| H3 | 0.5794 | −0.0486 | 1.1183 | 0.030* | |
| C4 | 0.5229 (4) | −0.0912 (3) | 0.98301 (14) | 0.0243 (5) | |
| H4 | 0.3578 | −0.0895 | 0.9699 | 0.029* | |
| C5 | 0.6862 (4) | −0.1216 (3) | 0.91570 (13) | 0.0204 (5) | |
| H5 | 0.6609 | −0.2517 | 0.8874 | 0.025* | |
| C6 | 0.4469 (4) | 0.1343 (3) | 0.71336 (13) | 0.0178 (4) | |
| H6 | 0.5552 | 0.0694 | 0.6765 | 0.021* | |
| C7 | 0.2004 (3) | 0.1338 (3) | 0.66293 (13) | 0.0176 (4) | |
| H7 | 0.0906 | 0.1980 | 0.6994 | 0.021* | |
| C8 | 0.2093 (3) | 0.2433 (3) | 0.57590 (13) | 0.0167 (4) | |
| H8 | 0.2985 | 0.1652 | 0.5363 | 0.020* | |
| C9 | 0.3238 (3) | 0.4413 (3) | 0.59065 (13) | 0.0172 (4) | |
| H9 | 0.2164 | 0.5310 | 0.6170 | 0.021* | |
| C10 | 0.5559 (3) | 0.4305 (3) | 0.65204 (13) | 0.0180 (4) | |
| H10 | 0.6738 | 0.3622 | 0.6208 | 0.022* | |
| C11 | 0.6423 (4) | 0.6302 (3) | 0.67948 (14) | 0.0218 (5) | |
| H11A | 0.6248 | 0.7137 | 0.6256 | 0.026* | |
| H11B | 0.5424 | 0.6840 | 0.7217 | 0.026* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0203 (7) | 0.0294 (9) | 0.0169 (7) | 0.0038 (6) | −0.0002 (5) | 0.0024 (6) |
| O2 | 0.0327 (8) | 0.0329 (10) | 0.0233 (8) | 0.0036 (8) | −0.0095 (7) | −0.0014 (8) |
| O3 | 0.0212 (7) | 0.0242 (8) | 0.0137 (7) | −0.0011 (7) | −0.0005 (5) | 0.0038 (6) |
| O4 | 0.0226 (7) | 0.0190 (8) | 0.0143 (7) | −0.0026 (6) | 0.0005 (5) | −0.0001 (6) |
| O5 | 0.0317 (8) | 0.0223 (8) | 0.0172 (7) | −0.0077 (7) | −0.0007 (6) | 0.0020 (7) |
| O6 | 0.0177 (7) | 0.0259 (8) | 0.0174 (7) | −0.0007 (6) | −0.0027 (6) | 0.0030 (7) |
| O7 | 0.0218 (7) | 0.0257 (8) | 0.0172 (7) | 0.0016 (7) | 0.0036 (6) | 0.0063 (7) |
| O8 | 0.0262 (8) | 0.0330 (9) | 0.0227 (8) | −0.0102 (8) | −0.0010 (6) | 0.0034 (8) |
| C1 | 0.0200 (10) | 0.0262 (12) | 0.0175 (10) | −0.0020 (10) | −0.0010 (8) | 0.0041 (9) |
| C2 | 0.0302 (11) | 0.0188 (10) | 0.0175 (9) | 0.0063 (10) | 0.0000 (9) | 0.0026 (9) |
| C3 | 0.0306 (11) | 0.0257 (11) | 0.0201 (10) | 0.0037 (10) | 0.0064 (8) | 0.0025 (10) |
| C4 | 0.0235 (10) | 0.0234 (12) | 0.0264 (12) | 0.0004 (10) | 0.0044 (9) | 0.0071 (10) |
| C5 | 0.0217 (10) | 0.0214 (11) | 0.0168 (10) | −0.0001 (9) | −0.0023 (8) | 0.0016 (9) |
| C6 | 0.0216 (10) | 0.0179 (10) | 0.0138 (9) | −0.0003 (9) | 0.0020 (7) | 0.0016 (9) |
| C7 | 0.0190 (10) | 0.0188 (10) | 0.0151 (9) | −0.0014 (9) | 0.0025 (7) | 0.0003 (9) |
| C8 | 0.0152 (10) | 0.0207 (10) | 0.0135 (9) | 0.0003 (9) | −0.0001 (8) | −0.0008 (8) |
| C9 | 0.0185 (10) | 0.0191 (10) | 0.0143 (9) | 0.0010 (9) | 0.0038 (7) | 0.0010 (9) |
| C10 | 0.0180 (10) | 0.0216 (10) | 0.0147 (9) | −0.0004 (10) | 0.0038 (8) | 0.0017 (9) |
| C11 | 0.0253 (11) | 0.0206 (11) | 0.0189 (10) | −0.0016 (9) | 0.0004 (8) | 0.0006 (10) |
Geometric parameters (Å, °)
| O1—C2 | 1.361 (2) | C2—C3 | 1.464 (3) |
| O1—C5 | 1.447 (2) | C3—C4 | 1.331 (3) |
| O2—C2 | 1.208 (3) | C3—H3 | 0.9500 |
| O3—C6 | 1.401 (2) | C4—C5 | 1.493 (3) |
| O3—C1 | 1.430 (2) | C4—H4 | 0.9500 |
| O4—C6 | 1.419 (3) | C5—H5 | 1.0000 |
| O4—C10 | 1.443 (2) | C6—C7 | 1.523 (3) |
| O5—C7 | 1.427 (3) | C6—H6 | 1.0000 |
| O5—H5O | 0.86 (3) | C7—C8 | 1.521 (3) |
| O6—C8 | 1.434 (2) | C7—H7 | 1.0000 |
| O6—H6O | 0.92 (3) | C8—C9 | 1.528 (3) |
| O7—C9 | 1.425 (2) | C8—H8 | 1.0000 |
| O7—H7O | 0.84 (3) | C9—C10 | 1.525 (2) |
| O8—C11 | 1.435 (3) | C9—H9 | 1.0000 |
| O8—H8O | 0.88 (3) | C10—C11 | 1.511 (3) |
| C1—C5 | 1.519 (3) | C10—H10 | 1.0000 |
| C1—H1A | 0.9900 | C11—H11A | 0.9900 |
| C1—H1B | 0.9900 | C11—H11B | 0.9900 |
| C2—O1—C5 | 109.40 (15) | O4—C6—H6 | 109.9 |
| C6—O3—C1 | 111.09 (15) | C7—C6—H6 | 109.9 |
| C6—O4—C10 | 112.00 (15) | O5—C7—C8 | 106.92 (16) |
| C7—O5—H5O | 113 (2) | O5—C7—C6 | 111.72 (17) |
| C8—O6—H6O | 110.7 (18) | C8—C7—C6 | 106.69 (16) |
| C9—O7—H7O | 105.7 (19) | O5—C7—H7 | 110.5 |
| C11—O8—H8O | 107 (2) | C8—C7—H7 | 110.5 |
| O3—C1—C5 | 108.08 (17) | C6—C7—H7 | 110.5 |
| O3—C1—H1A | 110.1 | O6—C8—C7 | 108.63 (16) |
| C5—C1—H1A | 110.1 | O6—C8—C9 | 108.56 (17) |
| O3—C1—H1B | 110.1 | C7—C8—C9 | 112.90 (16) |
| C5—C1—H1B | 110.1 | O6—C8—H8 | 108.9 |
| H1A—C1—H1B | 108.4 | C7—C8—H8 | 108.9 |
| O2—C2—O1 | 120.56 (19) | C9—C8—H8 | 108.9 |
| O2—C2—C3 | 130.8 (2) | O7—C9—C10 | 108.24 (15) |
| O1—C2—C3 | 108.66 (17) | O7—C9—C8 | 107.89 (16) |
| C4—C3—C2 | 108.1 (2) | C10—C9—C8 | 111.93 (17) |
| C4—C3—H3 | 125.9 | O7—C9—H9 | 109.6 |
| C2—C3—H3 | 125.9 | C10—C9—H9 | 109.6 |
| C3—C4—C5 | 109.8 (2) | C8—C9—H9 | 109.6 |
| C3—C4—H4 | 125.1 | O4—C10—C11 | 107.91 (16) |
| C5—C4—H4 | 125.1 | O4—C10—C9 | 108.49 (15) |
| O1—C5—C4 | 103.91 (16) | C11—C10—C9 | 110.63 (17) |
| O1—C5—C1 | 106.48 (16) | O4—C10—H10 | 109.9 |
| C4—C5—C1 | 115.19 (19) | C11—C10—H10 | 109.9 |
| O1—C5—H5 | 110.3 | C9—C10—H10 | 109.9 |
| C4—C5—H5 | 110.3 | O8—C11—C10 | 114.47 (18) |
| C1—C5—H5 | 110.3 | O8—C11—H11A | 108.6 |
| O3—C6—O4 | 107.90 (15) | C10—C11—H11A | 108.6 |
| O3—C6—C7 | 109.54 (16) | O8—C11—H11B | 108.6 |
| O4—C6—C7 | 109.80 (16) | C10—C11—H11B | 108.6 |
| O3—C6—H6 | 109.9 | H11A—C11—H11B | 107.6 |
| C6—O3—C1—C5 | −162.86 (17) | O3—C6—C7—C8 | 179.11 (17) |
| C5—O1—C2—O2 | −176.8 (2) | O4—C6—C7—C8 | 60.8 (2) |
| C5—O1—C2—C3 | 3.2 (2) | O5—C7—C8—O6 | 68.0 (2) |
| O2—C2—C3—C4 | 176.8 (3) | C6—C7—C8—O6 | −172.30 (16) |
| O1—C2—C3—C4 | −3.3 (3) | O5—C7—C8—C9 | −171.50 (16) |
| C2—C3—C4—C5 | 2.0 (3) | C6—C7—C8—C9 | −51.8 (2) |
| C2—O1—C5—C4 | −2.0 (2) | O6—C8—C9—O7 | −71.42 (19) |
| C2—O1—C5—C1 | 120.06 (19) | C7—C8—C9—O7 | 168.07 (16) |
| C3—C4—C5—O1 | −0.1 (3) | O6—C8—C9—C10 | 169.60 (14) |
| C3—C4—C5—C1 | −116.2 (2) | C7—C8—C9—C10 | 49.1 (2) |
| O3—C1—C5—O1 | −175.41 (16) | C6—O4—C10—C11 | −177.97 (16) |
| O3—C1—C5—C4 | −60.8 (2) | C6—O4—C10—C9 | 62.1 (2) |
| C1—O3—C6—O4 | −61.2 (2) | O7—C9—C10—O4 | −169.84 (18) |
| C1—O3—C6—C7 | 179.26 (17) | C8—C9—C10—O4 | −51.1 (2) |
| C10—O4—C6—O3 | 171.64 (15) | O7—C9—C10—C11 | 72.0 (2) |
| C10—O4—C6—C7 | −69.02 (19) | C8—C9—C10—C11 | −169.25 (17) |
| O3—C6—C7—O5 | −64.4 (2) | O4—C10—C11—O8 | 74.2 (2) |
| O4—C6—C7—O5 | 177.31 (17) | C9—C10—C11—O8 | −167.27 (16) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O5—H5O···O8i | 0.86 (3) | 2.17 (3) | 2.910 (2) | 144 (3) |
| O6—H6O···O5ii | 0.92 (3) | 1.94 (3) | 2.824 (2) | 162 (3) |
| O7—H7O···O6ii | 0.84 (3) | 1.84 (3) | 2.668 (2) | 173 (3) |
| O8—H8O···O2iii | 0.88 (3) | 1.96 (3) | 2.830 (2) | 167 (3) |
| C3—H3···O4iv | 0.95 | 2.55 | 3.413 (3) | 151 |
| C4—H4···O1v | 0.95 | 2.57 | 3.504 (3) | 168 |
| C8—H8···O7vi | 1.00 | 2.37 | 3.306 (3) | 155 |
| C1—H1A···O2iii | 0.99 | 2.38 | 3.289 (3) | 153 |
| C10—H10···O6vii | 1.00 | 2.43 | 3.415 (3) | 167 |
| O6—H6O···O7 | 0.92 (3) | 2.56 (3) | 2.894 (2) | 102 (2) |
| C11—H11A···O7 | 0.99 | 2.59 | 2.986 (3) | 104 |
Symmetry codes: (i) x−1, y−1, z; (ii) −x, y+1/2, −z+1; (iii) −x+2, y+1/2, −z+2; (iv) −x+1, y−1/2, −z+2; (v) x−1, y, z; (vi) −x+1, y−1/2, −z+1; (vii) x+1, y, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: FL2315).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Bai, Y., Benn, M. H., Majak, W. & McDiarmid, R. (1996). J. Agric. Food Chem.44, 2235–2238.
- Benn, M. H. & Yelland, L. J. (1968). Can. J. Chem.46, 729–732.
- Blessing, R. H. (1997). J. Appl. Cryst.30, 421–426.
- Boll, P. M. (1968). Acta Chem. Scand.22, 3245–3250.
- Camps, P., Cardellach, J., Font, J., Ortuno, R. M. & Ponsati, O. (1982). Tetrahedron, 38, 2395–2402.
- Cremer, D. & Pople, J. A. (1975). J. Am. Chem. Soc.97, 1354–1358.
- Fang, Z., Zhou, J. & Huang, L. (1989). Yaoxue Xuebao, 24, 182–188.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Hill, R. & van Heyningen, R. (1951). Biochem. J.49, 332–335. [DOI] [PMC free article] [PubMed]
- Hooft, R. (1998). COLLECT Nonius BV, Delft, The Netherlands.
- Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr & R. M. Sweet, pp. 307–326. New York: Academic Press.
- Perry, N. B., Benn, M. H., Forster, L. M., Routledge, A. & Weavers, R. T. (1996). Phytochemistry, 42, 453–459. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810034847/fl2315sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810034847/fl2315Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report