

Abstract
In the title compound, C11H14ClNO, the piperidine ring adopts a chair conformation: the hydroxyl substituent and the N-bound H atom occupy the axial positions, while the benzene ring occupies the equatorial position. In the crystal, the molecules are linked into a centrosymmetric tetramer through strong O—H⋯N and weak N—H⋯O hydrogen bonds; the N and O atoms act as both donor and acceptor for these interactions. The tetramers are further joined by hydrogen bonds into a layer parallel to (100).
Related literature
For related structures, see: De Camp & Ahmed (1972a
▶,b
▶); Friederich et al. (1993 ▶); Kimura & Okabayashi (1986 ▶). For details of the asymmetry parameters for chair conformations, see: Duax & Norton (1975 ▶). For a description of the Cambridge Structural Database, see: Allen (2002 ▶).
Experimental
Crystal data
C11H14ClNO
M r = 211.68
Monoclinic,
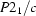
a = 11.3706 (10) Å
b = 9.5204 (8) Å
c = 10.6164 (9) Å
β = 108.458 (8)°
V = 1090.13 (16) Å3
Z = 4
Cu Kα radiation
μ = 2.83 mm−1
T = 295 K
0.3 × 0.2 × 0.15 mm
Data collection
Oxford Diffraction SuperNova, single source at offset, Atlas diffractometer
Absorption correction: multi-scan (CrysAlis PRO; Oxford Diffraction, 2009 ▶) T min = 0.401, T max = 0.654
4068 measured reflections
2190 independent reflections
2014 reflections with I > 2σ(I)
R int = 0.011
Refinement
R[F 2 > 2σ(F 2)] = 0.038
wR(F 2) = 0.111
S = 1.07
2190 reflections
183 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.32 e Å−3
Δρmin = −0.38 e Å−3
Data collection: CrysAlis PRO (Oxford Diffraction, 2009 ▶); cell refinement: CrysAlis PRO; data reduction: CrysAlis PRO; program(s) used to solve structure: SIR92 (Altomare et al., 1993 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: Stereochemical Workstation Operation Manual (Siemens, 1989 ▶) and Mercury (Macrae et al., 2008 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810004216/is2520sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810004216/is2520Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O4i | 0.89 (2) | 2.41 (2) | 3.2036 (16) | 147.2 (17) |
| O4—H4A⋯N1ii | 0.84 (2) | 1.97 (2) | 2.8089 (17) | 174 (2) |
Symmetry codes: (i) 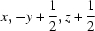 ; (ii)
; (ii) 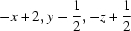 .
.
Acknowledgments
BPS thanks R. L. FineChem, Bangalore, India, for the gift of a sample of the title compound.
supplementary crystallographic information
Comment
The title compound, (1, Scheme 1), 4-(4-chlorophenyl)piperidin-4-ol is used as an intermediate for the synthesis of pharmaceuticals such as haloperidol (neuroleptic drug used to treat psychotic illnesses, extreme agitation, or Tourette's syndrome) and loperamide which is effective against diarrhea resulting from gastroenteritis or inflammatory bowel disease.
The piperidine ring adopts an almost ideal chair conformation (Fig. 1); the asymmetry parameters (Duax & Norton, 1975) are all smaller than 2.5°. The hydroxy group and N—H hydrogen atom occupy the axial positions [torsion angles: C2—C3—C4—O4 -64.46 (15)°, C6—C5—C4—O4 60.81 (15)°, C5—C6—N1—H1 65.0 (13)°, and C3—C2—N1—H1 -64.8 (14)°]. Such a mutual conformation of hydroxyl and phenyl groups is very typical, in the Cambridge Database (Allen, 2002; ver. 5.30 of Nov. 2008, last update Sep. 2009) there are 65 crystal structures of six-membered saturated rings with both OH and aromatic substituent in one position, only in three of them the hydroxyl group adopts the equatorial position [two polymorphs of (±)-β-1,2,5-trimethyl-4-phenylpiperidin-4-ol (De Camp & Ahmed, 1972a,b), cis-1,4-bis(4-bromophenyl)-1,4-dimethoxycyclohexane (Friederich et al., 1993), and cis-1-phenyl-3-piperidinocyclohexan-1-ol hydrochloride (Kimura & Okabayashi, 1986)].
The relatively strong and directional O—H···N hydrogen bonds join the molecules of 1, related by two-fold screw axis, into the chains along [010] directions. These chains are interconnected by far weaker N—H···O hydrogen bonds. These two kinds of contacts form centrosymmetric tetramers of the molecules (Fig. 2). In the crystal structures there are the hydrogen-bonded layers of molecules, created by interconnecting chains, in the bc plane (Fig. 3a). There are no directional interactions between neighbouring layers (Fig. 3b).
Experimental
The title compound was obtained as a gift sample from R. L. Fine Chem, Bangalore, India. X-ray quality crystals were obtained by a slow evaporation from an ethyl acetate solution (m.p. 410–413 K).
Refinement
Hydrogen atoms were found in the subsequent difference Fourier maps, and freely refined.
Figures
Fig. 1.

Anisotropic ellipsoid representation of the compound 1 together with atom labelling scheme. The ellipsoids are drawn at 50% probability level, hydrogen atoms are depicted as spheres with arbitrary radii.
Fig. 2.

Hydrogen-bonded tetramer [symmetry codes: (i) x, 1/2 - y, 1/2 + z; (ii) 2 - x, 1 - y, 1 - z; (iii) 2 - x, 1/2 + y, 1/2 - z].
Fig. 3.

The packing of the molecules of 1. (a) Hydrogen-boded layer; (b) the packing as seen along the y-direction.
Crystal data
| C11H14ClNO | F(000) = 448 |
| Mr = 211.68 | Dx = 1.290 Mg m−3 |
| Monoclinic, P21/c | Cu Kα radiation, λ = 1.54178 Å |
| Hall symbol: -P 2ybc | Cell parameters from 3304 reflections |
| a = 11.3706 (10) Å | θ = 4.1–75.2° |
| b = 9.5204 (8) Å | µ = 2.83 mm−1 |
| c = 10.6164 (9) Å | T = 295 K |
| β = 108.458 (8)° | Prism, yellow |
| V = 1090.13 (16) Å3 | 0.3 × 0.2 × 0.15 mm |
| Z = 4 |
Data collection
| Oxford Diffraction SuperNova, single source at offset, Atlas diffractometer | 2190 independent reflections |
| Radiation source: SuperNova (Cu) X-ray Source | 2014 reflections with I > 2σ(I) |
| mirror | Rint = 0.011 |
| Detector resolution: 10.5357 pixels mm-1 | θmax = 75.3°, θmin = 4.1° |
| ω scans | h = −13→14 |
| Absorption correction: multi-scan (CrysAlis PRO; Oxford Diffraction, 2009) | k = −11→7 |
| Tmin = 0.401, Tmax = 0.654 | l = −12→13 |
| 4068 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.038 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.111 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.07 | w = 1/[σ2(Fo2) + (0.0549P)2 + 0.250P] where P = (Fo2 + 2Fc2)/3 |
| 2190 reflections | (Δ/σ)max < 0.001 |
| 183 parameters | Δρmax = 0.32 e Å−3 |
| 0 restraints | Δρmin = −0.38 e Å−3 |
Special details
| Geometry. All s.u.'s (except the s.u. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell s.u.'s are taken into account individually in the estimation of s.u.'s in distances, angles and torsion angles; correlations between s.u.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell s.u.'s is used for estimating s.u.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.92832 (13) | 0.42199 (14) | 0.33836 (13) | 0.0530 (3) | |
| H1 | 0.8930 (19) | 0.430 (2) | 0.402 (2) | 0.069 (5)* | |
| C2 | 0.83116 (16) | 0.40784 (17) | 0.20917 (16) | 0.0537 (4) | |
| H21 | 0.8738 (17) | 0.4069 (19) | 0.1413 (18) | 0.058 (5)* | |
| H22 | 0.7767 (18) | 0.491 (2) | 0.1973 (18) | 0.065 (5)* | |
| C3 | 0.75467 (14) | 0.27404 (16) | 0.19461 (16) | 0.0507 (3) | |
| H31 | 0.7086 (19) | 0.277 (2) | 0.255 (2) | 0.072 (6)* | |
| H32 | 0.6977 (17) | 0.2675 (19) | 0.0997 (19) | 0.061 (5)* | |
| C4 | 0.83678 (12) | 0.14272 (14) | 0.22350 (12) | 0.0414 (3) | |
| O4 | 0.89540 (10) | 0.13797 (11) | 0.12224 (9) | 0.0476 (3) | |
| H4A | 0.949 (2) | 0.073 (2) | 0.140 (2) | 0.074 (6)* | |
| C5 | 0.93538 (14) | 0.16101 (17) | 0.36010 (13) | 0.0462 (3) | |
| H51 | 0.9930 (16) | 0.0777 (19) | 0.3783 (16) | 0.052 (4)* | |
| H52 | 0.8963 (17) | 0.1649 (19) | 0.4293 (18) | 0.058 (5)* | |
| C6 | 1.00732 (15) | 0.29679 (18) | 0.36784 (15) | 0.0534 (4) | |
| H61 | 1.0557 (17) | 0.2932 (19) | 0.3036 (18) | 0.059 (5)* | |
| H62 | 1.0676 (18) | 0.307 (2) | 0.457 (2) | 0.066 (5)* | |
| C41 | 0.75873 (12) | 0.01087 (15) | 0.21490 (13) | 0.0433 (3) | |
| C42 | 0.76509 (17) | −0.0736 (2) | 0.32296 (16) | 0.0604 (4) | |
| H42 | 0.818 (2) | −0.051 (2) | 0.409 (2) | 0.080 (6)* | |
| C43 | 0.69364 (19) | −0.1934 (2) | 0.3105 (2) | 0.0715 (5) | |
| H43 | 0.699 (2) | −0.251 (2) | 0.384 (2) | 0.084 (6)* | |
| C44 | 0.61247 (15) | −0.22890 (18) | 0.18927 (19) | 0.0600 (4) | |
| Cl44 | 0.52022 (5) | −0.37870 (6) | 0.17414 (7) | 0.0901 (2) | |
| C45 | 0.60313 (16) | −0.14761 (19) | 0.07924 (18) | 0.0601 (4) | |
| H45 | 0.550 (2) | −0.174 (2) | −0.007 (2) | 0.081 (6)* | |
| C46 | 0.67646 (14) | −0.02996 (18) | 0.09244 (15) | 0.0536 (4) | |
| H46 | 0.6735 (18) | 0.028 (2) | 0.014 (2) | 0.072 (6)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0619 (8) | 0.0540 (7) | 0.0485 (7) | −0.0073 (6) | 0.0250 (6) | −0.0050 (5) |
| C2 | 0.0587 (9) | 0.0478 (8) | 0.0548 (8) | 0.0021 (7) | 0.0181 (7) | 0.0037 (6) |
| C3 | 0.0468 (7) | 0.0509 (8) | 0.0539 (8) | 0.0038 (6) | 0.0152 (6) | 0.0030 (6) |
| C4 | 0.0446 (7) | 0.0485 (7) | 0.0339 (6) | 0.0023 (5) | 0.0163 (5) | 0.0033 (5) |
| O4 | 0.0558 (6) | 0.0550 (6) | 0.0377 (5) | 0.0044 (5) | 0.0227 (4) | 0.0064 (4) |
| C5 | 0.0492 (7) | 0.0539 (8) | 0.0356 (6) | −0.0010 (6) | 0.0136 (5) | 0.0032 (5) |
| C6 | 0.0508 (8) | 0.0627 (9) | 0.0451 (7) | −0.0073 (7) | 0.0129 (6) | −0.0003 (7) |
| C41 | 0.0435 (6) | 0.0479 (7) | 0.0407 (6) | 0.0032 (6) | 0.0167 (5) | 0.0004 (5) |
| C42 | 0.0649 (10) | 0.0668 (10) | 0.0459 (8) | −0.0133 (8) | 0.0123 (7) | 0.0086 (7) |
| C43 | 0.0746 (11) | 0.0722 (12) | 0.0672 (10) | −0.0159 (9) | 0.0216 (9) | 0.0162 (9) |
| C44 | 0.0495 (8) | 0.0533 (9) | 0.0805 (11) | −0.0037 (7) | 0.0253 (8) | −0.0041 (8) |
| Cl44 | 0.0732 (3) | 0.0695 (3) | 0.1295 (5) | −0.0240 (2) | 0.0349 (3) | −0.0071 (3) |
| C45 | 0.0515 (8) | 0.0656 (10) | 0.0609 (9) | −0.0039 (7) | 0.0145 (7) | −0.0124 (8) |
| C46 | 0.0540 (8) | 0.0600 (9) | 0.0453 (7) | −0.0006 (7) | 0.0138 (6) | −0.0004 (7) |
Geometric parameters (Å, °)
| N1—C6 | 1.465 (2) | C5—H52 | 0.972 (19) |
| N1—C2 | 1.470 (2) | C6—H61 | 1.003 (19) |
| N1—H1 | 0.89 (2) | C6—H62 | 0.99 (2) |
| C2—C3 | 1.522 (2) | C41—C42 | 1.384 (2) |
| C2—H21 | 0.987 (19) | C41—C46 | 1.395 (2) |
| C2—H22 | 0.99 (2) | C42—C43 | 1.382 (3) |
| C3—C4 | 1.5322 (19) | C42—H42 | 0.95 (2) |
| C3—H31 | 0.95 (2) | C43—C44 | 1.368 (3) |
| C3—H32 | 1.013 (18) | C43—H43 | 0.94 (2) |
| C4—O4 | 1.4337 (15) | C44—C45 | 1.377 (3) |
| C4—C41 | 1.5233 (19) | C44—Cl44 | 1.7473 (17) |
| C4—C5 | 1.5365 (18) | C45—C46 | 1.377 (2) |
| O4—H4A | 0.84 (2) | C45—H45 | 0.96 (2) |
| C5—C6 | 1.518 (2) | C46—H46 | 0.99 (2) |
| C5—H51 | 1.008 (17) | ||
| C6—N1—C2 | 110.71 (12) | C4—C5—H52 | 110.2 (11) |
| C6—N1—H1 | 107.4 (13) | H51—C5—H52 | 108.1 (14) |
| C2—N1—H1 | 109.3 (13) | N1—C6—C5 | 113.44 (13) |
| N1—C2—C3 | 114.07 (13) | N1—C6—H61 | 108.3 (10) |
| N1—C2—H21 | 106.5 (10) | C5—C6—H61 | 109.6 (10) |
| C3—C2—H21 | 108.4 (11) | N1—C6—H62 | 108.6 (11) |
| N1—C2—H22 | 107.6 (11) | C5—C6—H62 | 109.5 (11) |
| C3—C2—H22 | 110.0 (11) | H61—C6—H62 | 107.2 (15) |
| H21—C2—H22 | 110.3 (15) | C42—C41—C46 | 117.02 (14) |
| C2—C3—C4 | 111.72 (12) | C42—C41—C4 | 123.54 (13) |
| C2—C3—H31 | 109.2 (13) | C46—C41—C4 | 119.44 (12) |
| C4—C3—H31 | 108.6 (12) | C43—C42—C41 | 121.70 (15) |
| C2—C3—H32 | 108.5 (10) | C43—C42—H42 | 117.1 (13) |
| C4—C3—H32 | 107.8 (10) | C41—C42—H42 | 121.2 (13) |
| H31—C3—H32 | 111.1 (15) | C44—C43—C42 | 119.67 (16) |
| O4—C4—C41 | 109.23 (10) | C44—C43—H43 | 119.2 (14) |
| O4—C4—C3 | 105.91 (11) | C42—C43—H43 | 121.1 (14) |
| C41—C4—C3 | 110.72 (11) | C43—C44—C45 | 120.47 (16) |
| O4—C4—C5 | 109.74 (11) | C43—C44—Cl44 | 119.66 (14) |
| C41—C4—C5 | 112.78 (11) | C45—C44—Cl44 | 119.87 (14) |
| C3—C4—C5 | 108.23 (12) | C46—C45—C44 | 119.30 (15) |
| C4—O4—H4A | 109.3 (14) | C46—C45—H45 | 119.4 (13) |
| C6—C5—C4 | 111.55 (12) | C44—C45—H45 | 121.3 (13) |
| C6—C5—H51 | 110.7 (10) | C45—C46—C41 | 121.82 (15) |
| C4—C5—H51 | 109.1 (9) | C45—C46—H46 | 120.8 (12) |
| C6—C5—H52 | 107.1 (11) | C41—C46—H46 | 117.4 (12) |
| C6—N1—C2—C3 | 53.23 (17) | O4—C4—C41—C46 | −49.35 (16) |
| N1—C2—C3—C4 | −54.44 (18) | C3—C4—C41—C46 | 66.91 (16) |
| C2—C3—C4—O4 | −64.46 (15) | C5—C4—C41—C46 | −171.65 (13) |
| C2—C3—C4—C41 | 177.25 (12) | C46—C41—C42—C43 | 0.2 (3) |
| C2—C3—C4—C5 | 53.16 (15) | C4—C41—C42—C43 | −179.21 (17) |
| O4—C4—C5—C6 | 60.81 (15) | C41—C42—C43—C44 | −1.3 (3) |
| C41—C4—C5—C6 | −177.17 (11) | C42—C43—C44—C45 | 1.2 (3) |
| C3—C4—C5—C6 | −54.33 (15) | C42—C43—C44—Cl44 | −178.91 (16) |
| C2—N1—C6—C5 | −54.19 (16) | C43—C44—C45—C46 | 0.0 (3) |
| C4—C5—C6—N1 | 56.56 (16) | Cl44—C44—C45—C46 | −179.90 (13) |
| O4—C4—C41—C42 | 130.07 (15) | C44—C45—C46—C41 | −1.1 (3) |
| C3—C4—C41—C42 | −113.68 (16) | C42—C41—C46—C45 | 1.0 (2) |
| C5—C4—C41—C42 | 7.76 (19) | C4—C41—C46—C45 | −179.54 (14) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O4i | 0.89 (2) | 2.41 (2) | 3.2036 (16) | 147.2 (17) |
| O4—H4A···N1ii | 0.84 (2) | 1.97 (2) | 2.8089 (17) | 174 (2) |
Symmetry codes: (i) x, −y+1/2, z+1/2; (ii) −x+2, y−1/2, −z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: IS2520).
References
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Altomare, A., Cascarano, G., Giacovazzo, C. & Guagliardi, A. (1993). J. Appl. Cryst.26, 343–350.
- De Camp, W. H. & Ahmed, F. R. (1972a). Acta Cryst. B28, 1796–1800.
- De Camp, W. H. & Ahmed, F. R. (1972b). Acta Cryst. B28, 3484–3489.
- Duax, W. L. & Norton, D. A. (1975). Atlas of Steroid Structures New York: Plenum.
- Friederich, R., Nieger, M. & Vögtle, F. (1993). Chem. Ber.126, 1723–1732.
- Kimura, M. & Okabayashi, I. (1986). J. Heterocycl. Chem.23, 1287–1289.
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst.41, 466–470.
- Oxford Diffraction (2009). CrysAlis PRO Oxford Diffraction Ltd. Yarnton, England.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Siemens (1989). Stereochemical Workstation Operation Manual Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810004216/is2520sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810004216/is2520Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report