


Abstract
The emotif database is a collection of more than 170 000 highly specific and sensitive protein sequence motifs representing conserved biochemical properties and biological functions. These protein motifs are derived from 7697 sequence alignments in the BLOCKS+ database (released on June 23, 2000) and all 8244 protein sequence alignments in the PRINTS database (version 27.0) using the emotif-maker algorithm developed by Nevill-Manning et al. (Nevill-Manning,C.G., Wu,T.D. and Brutlag,D.L. (1998) Proc. Natl Acad. Sci. USA, 95, 5865–5871; Nevill-Manning,C.G., Sethi,K.S., Wu,T.D. and Brutlag,D.L. (1997) ISMB-97, 5, 202–209). Since the amino acids and the groups of amino acids in these sequence motifs represent critical positions conserved in evolution, search algorithms employing the emotif patterns can identify and classify more widely divergent sequences than methods based on global sequence similarity. The emotif protein pattern database is available at http://motif.stanford.edu/emotif/.
INTRODUCTION
Many newly discovered proteins do not share significant global sequence similarity to any known protein. These proteins, however, may contain modular domains that confer distinct structures and functions conserved throughout evolution. The conserved modules usually possess characteristic sequence elements, or sequence motifs, that only proteins containing the domains share. Many sequence analysis tools rely on discrete sequence motifs to classify newly discovered proteins into the proper families. The methods used to generate the discrete sequence motifs, therefore, play a vital role in determining the specificity and the sensitivity of the classification performed by these tools.
Most motif generation algorithms focus on finding a single ‘best’ motif for a given alignment of the homologous domain sequences from various species. These methods usually attempt to maximize sensitivity at the expense of specificity. Sequence analysis tools that use these specificity-compromised motifs are prone to classifying newly discovered proteins into the wrong families. For genomic-scale automated sequence classification to be effective, the expected number of misclassifications made by the discrete motifs must be minimized.
DATABASE GENERATION AND FORMAT
The emotif-maker program, previously known as emotif, is a systematic method for determining regular expression patterns, or discrete sequence motifs, from aligned sets of protein sequences. Unlike other methods that allow only one motif to be reported for a given protein sequence alignment, emotif-maker permits the methodical enumeration of multiple motifs for the same alignment such that motifs are generated from the complete alignment as well as subsets of that alignment. These multiple sequence alignment subsets usually represent subfamilies within the superfamily represented by the full alignment. Discrete sequence motifs constructed from these subsets can, therefore, classify novel sequences with higher specificity than the position-specific scoring matrices derived from entire multiple sequence alignments (1–3).
The emotif-maker program was applied to 7697 non-PRINTS protein sequence alignments from the BLOCKS+ database released on June 10, 2000, and all 8244 protein sequence alignments from the PRINTS database, version 27.0 (1,2). The BLOCKS+ database and the PRINTS database both have an extensive collection of ungapped multiple sequence alignments that cover various protein domains known to be biologically relevant. A total of 170 294 regular expression patterns were generated from 15 863 ungapped multiple sequence alignments. Since these discrete motifs come from subsets of the alignments as well as full alignments, they can complement the BLOCKS+ and the PRINTS position-specific scoring matrices by assigning novel sequences to potential subfamilies within the known superfamilies (1–3). The details of the emotif-maker algorithm have already been described by Nevill-Manning et al. (4,5). These highly specific and sensitive regular expression patterns, which will be referred to hereafter as emotifs, can support both genome-scale sequence classification as well as single protein analysis.
Since the emotif database is derived from ungapped multiple sequence alignments from the BLOCKS+ database and the PRINTS database, the emotif database will be updated following each release of the BLOCKS+ database and the PRINTS database. The database of emotifs exists as a single ASCII text file. Each line in the text file corresponds to a unique emotif record. The tab-delimited fields in an emotif record are, from left to right, the expected false positive frequency of the emotif, the accession number of the emotif’s parent alignment, the descriptive name of that alignment, the regular expression pattern of the emotif, and the sensitivity of the emotif. The emotifs have been sorted by their expected false positive frequencies, such that the emotif with the smallest expected false positive frequency resides at the beginning of the file.
The expected false positive frequency of an emotif, as summarized by equation 1, is the probability that the emotif will match a sequence of the same length by chance.
expected false positive frequency (motif) =
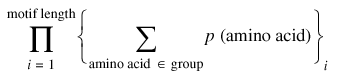 1
1
where group represents the set of amino acids allowed at position i of the emotif. This calculation assumes that the probability of finding a particular amino acid at a given position in a sequence, or p(amino acid), is equivalent to the distribution of that amino acid in the SWISS-PROT database and is independent of all other positions in the same sequence. By minimizing the expected false positive frequency of an emotif, the emotif-maker algorithm maximizes the specificity of that emotif.
The sensitivity of an emotif, defined as the percentage of sequences the emotif can match from its parent multiple sequence alignment, measures the emotif’s ability to detect the presence of a particular domain whose conserved sequence elements have been captured by the emotif. By maximizing an emotif’s coverage of its parent multiple sequence alignment, the emotif-maker algorithm maximizes the sensitivity of that emotif.
SEARCH SOFTWARE
The emotif-search program, previously known as identify, provides a rapid and efficient search interface to the database of emotifs. Given a protein sequence, emotif-search will identify significant matches between subsequences within the query and the regular expression patterns from the emotif database. Significance is determined by the number of false positive emotif matches the user expects for a given protein sequence. As defined by equation 2, the expected number of false positive matches, or the expectation, for an emotif-search query is the number of matches by chance the user is willing to accept, given the length of the query sequence and the content of the emotif database.
expectation = [(query length) + 1] 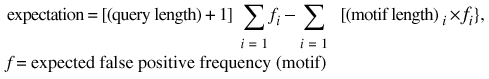 [(motif
length) i × fi},
[(motif
length) i × fi},
f = expected false positive frequency (motif) 2
where n is the number of emotifs the query sequence has encountered so far. The expectation for a query rises as the number of comparisons the emotif-search program performs increases. The emotifs have been sorted by their expected false positive frequencies, such that the more specific emotifs, or the emotifs with the smaller expected false positive frequencies, are compared against a query sequence before the less specific emotifs, or the emotifs with the larger expected false positive frequencies. As a result of this sorting, the emotif-search program guarantees that matches with the more specific emotifs are reported first.
The online version of emotif-search provides an easy-to-use interface for single-protein analysis. After emotif-search receives a cutoff expectation value and a query protein sequence over the network, it will return emotif matches as long as the expectation for the query remains below the user-specified cutoff. A sample output of emotif-search online is shown in Figure 1.
Figure 1.

The protein sequence of type II human hexokinase (P52789) was submitted to emotif-search online. The cutoff expectation for the query was set at 1e–7. The matches were ranked in order, so that matches with the more specific emotifs were reported first. Along with each hit, emotif-search calculates the expectation for the query based on the fraction of the emotif database it has already run against the input sequence. In addition emotif-search determines the probability that the query sequence has not matched a single emotif from the ones processed so far by chance. The reporting of these two values allows the user to rerun the search with a lower value of the cutoff expectation for a given query. The black line between the sixth and the seventh emotif match is a sample readjustment performed on this particular sample query. The cutoff expectation was lowered from 1e–7 to 1e–15. Only the first six hits were considered significant by the new, more stringent cutoff.
APPLICATION
To facilitate efficient automated genomic-scale protein classification, emotif-search and all the necessary preprocessing programs have been repackaged into the emotif Batch Analysis Suite, or ebas. The core of this software bundle is a Makefile script that has streamlined and automated the steps involved in running the database of emotifs against multiple ORFs, genomes, and proteomes. ebas accepts FASTA-formatted ORF sequences, FASTA-formatted genomic DNA sequences, and FASTA-formatted protein sequences as input. ebas assumes the ORFs are reported in the right frame; therefore, ebas will only translate one frame of the ORF sequences. Genomic DNA sequences, on the other hand, may or may not code for proteins; ebas, therefore, will translate all six frames of the genomic DNA sequences. The input protein and translated sequences are then piped to emotif-search for automated classification. ebas has been applied to coding sequences derived from the Arabidopsis thaliana genomic sequences; the coding sequence derivation was performed by C. J. Palm at the Stanford DNA Sequencing and Technology Center using the Genscan program (6). The results from ebas may be accessed at http://baggage.stanford.edu/group/arabprotein/.
CONCLUSION
The extensive collection of ungapped multiple sequence alignments in the BLOCKS+ database and the PRINTS database covers various protein domains known to be biologically relevant. From these multiple sequence alignments discrete patterns like emotifs or position-specific scoring matrices like those available through the BLOCKS+ database and the PRINTS database may both be generated (1–3). emotifs have the advantage over the position-specific scoring matrices derived from the entire multiple sequence alignments in that the emotifs are able to classify novel proteins into protein subfamilies. In addition emotifs focus on just those highly conserved residues; this focus greatly improves the signal-to-noise ratios of the emotifs and enhances the specificity of the automated sequence classification. The emotif database, therefore, can complement the BLOCKS+ and the PRINTS position-specific scoring matrices in identifying novel protein sequences.
AVAILABILITY
The emotif database is available at http://motif.stanford.edu/emotif/ for interactive use. Local installation of the emotif database and the associated software is also available upon request. Not-for-profit institutions may contact Douglas L. Brutlag (brutlag@stanford.edu) for a royalty-free license; for-profit institutions should contact Richard Scholes (rich.scholes@stanford.edu).
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by a grant from the National Genome Research Institute R01 HG02235-07. J.Y.H. was supported by the Cell and Molecular Biology training grant 2T32GM07276-26 from the National Institutes of Health.
References
- 1.Henikoff J.G., Greene,E.A., Pietrokovski,S. and Henikoff,S. (2000) Increased coverage of protein families with the Blocks Database servers. Nucleic Acids Res., 28, 228–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Attwood T.K., Croning,M.D.R., Flower,D.R., Lewis,A.P., Mabey,J.E., Scordis,P., Selley,J.N. and Wright,W. (2000) PRINTS-S: the database formerly known as PRINTS. Nucleic Acids Res., 28, 225–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu T.D., Nevill-Manning,C.G. and Brutlag,D.L. (2000) Fast probabilistic analysis of sequence function using scoring matrices. Bioinformatics, 16, 233–244. [DOI] [PubMed] [Google Scholar]
- 4.Nevill-Manning C.G., Wu,T.D. and Brutlag,D.L. (1998) Highly specific protein sequence motifs for genome analysis. Proc. Natl Acad. Sci. USA, 95, 5865–5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nevill-Manning C.G., Sethi,K.S., Wu,T.D. and Brutlag,D.L. (1997) Enumerating and Ranking Discrete Motifs. ISMB-97, 5, 202–209. [PubMed]
- 6.Palm C.J., Federspiel,N.A. and Davis,R.W. (2000) DAtA: Database of Arabidopsis thaliana Annotation. Nucleic Acids Res., 28, 102–103. [DOI] [PMC free article] [PubMed] [Google Scholar]