

Abstract
The reaction of propargyl bromide and 6-bromo-1,3-dihydroimidazo[4,5-b]pyridin-2-one in refluxing dimethylformamide yields the title compound, C12H8BrN3O, which features nitrogen-bound propadienyl and propynyl substituents. The imidazolopyridine fused ring is planar (r.m.s. deviation = 0.012 Å); the propadienyl chain is coplanar with the fused ring as it is conjugated with it, whereas the propynyl chain is not as the nitrogen-bound C atom is a methylene linkage. The acetylenic H atom is hydrogen bonded to the carbonyl O atom of an adjacent molecule, forming a helical chain runnning along the b axis.
Related literature
For the crystal structures of other imidazo[4,5-b]pyridin-2-ones, see: Kourafalos et al. (2002 ▶); Meanwell et al. (1995 ▶).
Experimental
Crystal data
C12H8BrN3O
M r = 290.12
Monoclinic,
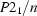
a = 9.6369 (4) Å
b = 9.3086 (4) Å
c = 13.5481 (5) Å
β = 99.123 (2)°
V = 1199.97 (8) Å3
Z = 4
Mo Kα radiation
μ = 3.41 mm−1
T = 293 K
0.55 × 0.35 × 0.30 mm
Data collection
Bruker X8 APEXII diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996 ▶) T min = 0.256, T max = 0.428
51411 measured reflections
3393 independent reflections
2577 reflections with I > 2σ(I)
R int = 0.040
Refinement
R[F 2 > 2σ(F 2)] = 0.031
wR(F 2) = 0.087
S = 1.02
3393 reflections
166 parameters
3 restraints
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.49 e Å−3
Δρmin = −0.63 e Å−3
Data collection: APEX2 (Bruker, 2008 ▶); cell refinement: SAINT (Bruker, 2008 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: X-SEED (Barbour, 2001 ▶); software used to prepare material for publication: publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810007695/hg2649sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810007695/hg2649Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C12—H12⋯O1i | 0.94 (1) | 2.22 (1) | 3.161 (3) | 173 (2) |
Symmetry code: (i)  .
.
Acknowledgments
We thank Université Sidi Mohamed Ben Abdallah, Université Mohammed V-Agdal and the University of Malaya for supporting this study.
supplementary crystallographic information
Experimental
To a solution of 6-bromo-1,3-dihydro-imidazo[4,5-b]pyridin-2-one (1 mmol), potassium carbonate (4 mmol) and tetra-n-butylammonium bromide (0.1 mmol) in DMF (20 ml) was added propargyl bromide (2.5 mmol). The solution was refluxed for 48 hours. After completion of the reaction (as monitored byTLC), the salt was filtered and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel by using an ethyl acetate/hexane (1/1) mixture as eluent. Slow evaporation of the solvent furnished colorless crystals.
Refinement
Carbon-bound H-atoms were placed in calculated positions (C—H 0.93 to 0.97 Å) and were included in the refinement in the riding model approximation, with U(H) set to 1.2U(C). The terminal acetylenic and allenic H-atoms were located in a difference Fourier map, and were refined with a distance restraint of C–H 0.95±0.01 Å; their temperature factors were refined.
Figures
Fig. 1.

Thermal ellipsoid plot (Barbour, 2001) of C12H8BrN3O at the 50% probability level; hydrogen atoms are drawn as spheres of arbitrary radius.
Crystal data
| C12H8BrN3O | F(000) = 576 |
| Mr = 290.12 | Dx = 1.606 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 9975 reflections |
| a = 9.6369 (4) Å | θ = 2.4–25.6° |
| b = 9.3086 (4) Å | µ = 3.41 mm−1 |
| c = 13.5481 (5) Å | T = 293 K |
| β = 99.123 (2)° | Irregular, colorless |
| V = 1199.97 (8) Å3 | 0.55 × 0.35 × 0.30 mm |
| Z = 4 |
Data collection
| Bruker X8 APEXII diffractometer | 3393 independent reflections |
| Radiation source: fine-focus sealed tube | 2577 reflections with I > 2σ(I) |
| graphite | Rint = 0.040 |
| φ and ω scans | θmax = 29.7°, θmin = 2.7° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1996) | h = −13→13 |
| Tmin = 0.256, Tmax = 0.428 | k = −12→12 |
| 51411 measured reflections | l = −18→18 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.031 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.087 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.02 | w = 1/[σ2(Fo2) + (0.0413P)2 + 0.4635P] where P = (Fo2 + 2Fc2)/3 |
| 3393 reflections | (Δ/σ)max = 0.001 |
| 166 parameters | Δρmax = 0.49 e Å−3 |
| 3 restraints | Δρmin = −0.63 e Å−3 |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.40648 (3) | 0.39876 (3) | 0.231050 (17) | 0.06785 (11) | |
| O1 | 0.49655 (17) | 0.95432 (17) | 0.64205 (11) | 0.0577 (4) | |
| N1 | 0.21863 (16) | 0.64109 (19) | 0.42594 (13) | 0.0453 (4) | |
| N2 | 0.54742 (15) | 0.78896 (16) | 0.52363 (11) | 0.0395 (3) | |
| N3 | 0.32605 (16) | 0.81244 (17) | 0.54674 (11) | 0.0417 (3) | |
| C1 | 0.2477 (2) | 0.5495 (2) | 0.35479 (15) | 0.0490 (4) | |
| H1 | 0.1744 | 0.4972 | 0.3188 | 0.059* | |
| C2 | 0.3815 (2) | 0.53039 (19) | 0.33326 (13) | 0.0435 (4) | |
| C3 | 0.4981 (2) | 0.60309 (19) | 0.38454 (13) | 0.0399 (4) | |
| H3 | 0.5887 | 0.5895 | 0.3708 | 0.048* | |
| C4 | 0.46792 (17) | 0.69616 (18) | 0.45684 (12) | 0.0346 (3) | |
| C5 | 0.32815 (18) | 0.71031 (18) | 0.47294 (12) | 0.0365 (3) | |
| C6 | 0.6938 (2) | 0.8150 (2) | 0.53701 (16) | 0.0503 (5) | |
| H6 | 0.7302 | 0.8828 | 0.5845 | 0.060* | |
| C7 | 0.7806 (2) | 0.7510 (2) | 0.48776 (17) | 0.0529 (5) | |
| C8 | 0.8709 (3) | 0.6872 (4) | 0.4413 (2) | 0.0750 (8) | |
| H81 | 0.916 (3) | 0.602 (2) | 0.467 (3) | 0.106 (12)* | |
| H82 | 0.890 (3) | 0.722 (3) | 0.3788 (13) | 0.089 (10)* | |
| C9 | 0.4605 (2) | 0.8628 (2) | 0.57909 (14) | 0.0416 (4) | |
| C10 | 0.2020 (2) | 0.8584 (2) | 0.58740 (16) | 0.0509 (5) | |
| H10A | 0.1252 | 0.8736 | 0.5328 | 0.061* | |
| H10B | 0.2218 | 0.9494 | 0.6217 | 0.061* | |
| C11 | 0.1586 (2) | 0.7538 (2) | 0.65707 (15) | 0.0521 (5) | |
| C12 | 0.1208 (3) | 0.6715 (3) | 0.71186 (19) | 0.0694 (7) | |
| H12 | 0.093 (3) | 0.601 (2) | 0.7550 (18) | 0.088 (11)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.0918 (2) | 0.05653 (15) | 0.05116 (14) | 0.01376 (12) | −0.00107 (12) | −0.01710 (9) |
| O1 | 0.0645 (9) | 0.0499 (8) | 0.0563 (9) | 0.0031 (7) | 0.0024 (7) | −0.0158 (7) |
| N1 | 0.0357 (8) | 0.0481 (9) | 0.0513 (9) | −0.0027 (7) | 0.0046 (7) | 0.0022 (7) |
| N2 | 0.0367 (7) | 0.0396 (8) | 0.0417 (7) | −0.0023 (6) | 0.0050 (6) | −0.0021 (6) |
| N3 | 0.0417 (8) | 0.0425 (8) | 0.0431 (8) | 0.0011 (6) | 0.0133 (6) | −0.0014 (6) |
| C1 | 0.0469 (10) | 0.0461 (10) | 0.0504 (10) | −0.0050 (8) | −0.0035 (8) | −0.0017 (8) |
| C2 | 0.0555 (11) | 0.0369 (9) | 0.0362 (8) | 0.0034 (8) | 0.0016 (7) | −0.0014 (7) |
| C3 | 0.0408 (9) | 0.0411 (9) | 0.0385 (8) | 0.0031 (7) | 0.0085 (7) | 0.0024 (7) |
| C4 | 0.0351 (8) | 0.0350 (8) | 0.0334 (7) | −0.0008 (6) | 0.0051 (6) | 0.0060 (6) |
| C5 | 0.0386 (8) | 0.0350 (8) | 0.0364 (8) | 0.0006 (7) | 0.0079 (6) | 0.0057 (6) |
| C6 | 0.0381 (9) | 0.0515 (11) | 0.0592 (12) | −0.0078 (8) | 0.0015 (8) | −0.0032 (9) |
| C7 | 0.0343 (9) | 0.0597 (12) | 0.0628 (12) | −0.0064 (9) | 0.0017 (9) | 0.0098 (10) |
| C8 | 0.0396 (11) | 0.101 (2) | 0.0867 (19) | 0.0039 (13) | 0.0176 (12) | 0.0060 (17) |
| C9 | 0.0474 (10) | 0.0370 (9) | 0.0402 (9) | 0.0029 (7) | 0.0061 (7) | 0.0014 (7) |
| C10 | 0.0512 (11) | 0.0491 (11) | 0.0570 (12) | 0.0097 (9) | 0.0224 (9) | 0.0029 (9) |
| C11 | 0.0510 (11) | 0.0614 (12) | 0.0461 (10) | 0.0007 (9) | 0.0149 (8) | −0.0033 (9) |
| C12 | 0.0748 (16) | 0.0808 (18) | 0.0555 (13) | −0.0120 (14) | 0.0197 (12) | 0.0087 (12) |
Geometric parameters (Å, °)
| Br1—C2 | 1.8928 (18) | C3—C4 | 1.373 (2) |
| O1—C9 | 1.216 (2) | C3—H3 | 0.9300 |
| N1—C5 | 1.312 (2) | C4—C5 | 1.404 (2) |
| N1—C1 | 1.349 (3) | C6—C7 | 1.295 (3) |
| N2—C4 | 1.390 (2) | C6—H6 | 0.9300 |
| N2—C9 | 1.393 (2) | C7—C8 | 1.296 (4) |
| N2—C6 | 1.414 (2) | C8—H81 | 0.95 (1) |
| N3—C5 | 1.382 (2) | C8—H82 | 0.95 (1) |
| N3—C9 | 1.382 (2) | C10—C11 | 1.463 (3) |
| N3—C10 | 1.458 (2) | C10—H10A | 0.9700 |
| C1—C2 | 1.378 (3) | C10—H10B | 0.9700 |
| C1—H1 | 0.9300 | C11—C12 | 1.164 (3) |
| C2—C3 | 1.398 (3) | C12—H12 | 0.94 (1) |
| C5—N1—C1 | 114.50 (16) | N1—C5—C4 | 126.59 (17) |
| C4—N2—C9 | 109.96 (14) | N3—C5—C4 | 107.53 (15) |
| C4—N2—C6 | 128.72 (16) | C7—C6—N2 | 124.62 (19) |
| C9—N2—C6 | 121.29 (16) | C7—C6—H6 | 117.7 |
| C5—N3—C9 | 110.03 (15) | N2—C6—H6 | 117.7 |
| C5—N3—C10 | 125.63 (16) | C8—C7—C6 | 178.0 (3) |
| C9—N3—C10 | 124.33 (16) | C7—C8—H81 | 121 (2) |
| N1—C1—C2 | 122.72 (18) | C7—C8—H82 | 120.7 (19) |
| N1—C1—H1 | 118.6 | H81—C8—H82 | 118 (3) |
| C2—C1—H1 | 118.6 | O1—C9—N3 | 127.55 (18) |
| C1—C2—C3 | 122.49 (17) | O1—C9—N2 | 126.48 (18) |
| C1—C2—Br1 | 118.07 (14) | N3—C9—N2 | 105.97 (15) |
| C3—C2—Br1 | 119.43 (14) | N3—C10—C11 | 112.56 (17) |
| C4—C3—C2 | 114.56 (16) | N3—C10—H10A | 109.1 |
| C4—C3—H3 | 122.7 | C11—C10—H10A | 109.1 |
| C2—C3—H3 | 122.7 | N3—C10—H10B | 109.1 |
| C3—C4—N2 | 134.39 (16) | C11—C10—H10B | 109.1 |
| C3—C4—C5 | 119.12 (16) | H10A—C10—H10B | 107.8 |
| N2—C4—C5 | 106.49 (15) | C12—C11—C10 | 178.4 (3) |
| N1—C5—N3 | 125.87 (16) | C11—C12—H12 | 177.2 (19) |
| C5—N1—C1—C2 | 0.0 (3) | C3—C4—C5—N1 | 0.9 (3) |
| N1—C1—C2—C3 | 0.9 (3) | N2—C4—C5—N1 | −179.58 (17) |
| N1—C1—C2—Br1 | −179.76 (15) | C3—C4—C5—N3 | −178.29 (15) |
| C1—C2—C3—C4 | −0.8 (3) | N2—C4—C5—N3 | 1.26 (18) |
| Br1—C2—C3—C4 | 179.81 (12) | C4—N2—C6—C7 | −1.4 (3) |
| C2—C3—C4—N2 | −179.36 (18) | C9—N2—C6—C7 | −179.6 (2) |
| C2—C3—C4—C5 | 0.0 (2) | C5—N3—C9—O1 | 178.90 (19) |
| C9—N2—C4—C3 | 178.04 (18) | C10—N3—C9—O1 | −2.7 (3) |
| C6—N2—C4—C3 | −0.3 (3) | C5—N3—C9—N2 | −0.2 (2) |
| C9—N2—C4—C5 | −1.41 (19) | C10—N3—C9—N2 | 178.17 (16) |
| C6—N2—C4—C5 | −179.75 (17) | C4—N2—C9—O1 | −178.10 (19) |
| C1—N1—C5—N3 | 178.16 (17) | C6—N2—C9—O1 | 0.4 (3) |
| C1—N1—C5—C4 | −0.9 (3) | C4—N2—C9—N3 | 1.00 (19) |
| C9—N3—C5—N1 | −179.84 (17) | C6—N2—C9—N3 | 179.49 (16) |
| C10—N3—C5—N1 | 1.8 (3) | C5—N3—C10—C11 | 76.4 (2) |
| C9—N3—C5—C4 | −0.67 (19) | C9—N3—C10—C11 | −101.7 (2) |
| C10—N3—C5—C4 | −179.00 (16) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C12—H12···O1i | 0.94 (1) | 2.22 (1) | 3.161 (3) | 173 (2) |
Symmetry codes: (i) −x+1/2, y−1/2, −z+3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HG2649).
References
- Barbour, L. J. (2001). J. Supramol. Chem.1, 189–191.
- Bruker (2008). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Kourafalos, V. N., Marakos, P., Pouli, N., Terzis, A. & Townsend, L. B. (2002). Heterocycles, 57, 2335–2343.
- Meanwell, N. A., Sit, S. Y., Gao, J. N., Wong, H. S., Gao, Q., St Laurent, D. R. & Balasubramanian, N. (1995). J. Org. Chem.50, 1565–1582.
- Sheldrick, G. M. (1996). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Westrip, S. P. (2010). publCIF In preparation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810007695/hg2649sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810007695/hg2649Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report