

Abstract
In the title compound, C14H11N3O2, the dihedral angle between the mean planes of the benzotriazole ring system and the benzene ring of the salicylaldehyde group is 2.4 (2)°. There is an intramolecular O—H⋯N hydrogen bond which may influence the molecular conformation.
Related literature
For the application of N,N,O-tridentate Schiff-base metal complexes in the catalytic ring-opening polymerization of l-lactide, see: Wu et al. (2005 ▶); Chen et al. (2006 ▶). For a related structure, see: Li et al. (2009 ▶).
Experimental
Crystal data
C14H11N3O2
M r = 253.26
Monoclinic,
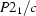
a = 12.2724 (5) Å
b = 14.5018 (5) Å
c = 6.8897 (3) Å
β = 91.571 (2)°
V = 1225.71 (8) Å3
Z = 4
Mo Kα radiation
μ = 0.10 mm−1
T = 296 K
0.34 × 0.31 × 0.23 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2008 ▶) T min = 0.972, T max = 0.977
13912 measured reflections
2946 independent reflections
1657 reflections with I > 2σ(I)
R int = 0.070
Refinement
R[F 2 > 2σ(F 2)] = 0.045
wR(F 2) = 0.146
S = 1.01
2946 reflections
172 parameters
H-atom parameters constrained
Δρmax = 0.21 e Å−3
Δρmin = −0.21 e Å−3
Data collection: APEX2 (Bruker, 2008 ▶); cell refinement: SAINT-Plus (Bruker, 2008 ▶); data reduction: SAINT-Plus; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810007233/lh5003sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810007233/lh5003Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1A⋯N1 | 0.85 | 1.94 | 2.588 (2) | 132 |
Acknowledgments
We gratefully acknowledge the financial support in part from the National Science Council, Taiwan (NSC97-2113-M-033-005-MY2) and in part from the project of the specific research fields in Chung Yuan Christian University, Taiwan (No. CYCU-98-CR—CH).
supplementary crystallographic information
Comment
Recently, NNO-tridentate Schiff-base zinc (Zn) and magnesium (Mg) complexes have been attracting considerable attention, mainly due to their applications in the catalytic ring-opening polymerization of L-lactide (Wu et al., 2005; Chen et al., 2006). These NNO-tridentate Schiff-base ligands were easily prepared by condensation from primary amine with the pendant arm of the amino group and various substituted salicylaldehyde derivatives in the presence of MgSO4. The additional amino group can be able to provide strong coordination to stabilize Zn or Mg atom and thereby stabilizes the zinc or magnesium alkoxide complex, without further disproportionation. Most recently, our group has successfully synthesized and structural characterized the amino-phenolate ligand derived from 4-methyl-2-(2H-benzotriazol-2-yl)-phenol (BTP-H) (Li et al., 2009). In order to develop more useful ligands originated from BTP derivates, our group is interested in the preparation of the multidentate Schiff-base ligand containing the benzotriazol group. Herein, we report the synthesis and crystal structure of the title compound, (I), a potential precursor for the preparation of the multidentate Schiff-base BTP ligands.
The molecular structure of (I) reveals the 5-methylsalicylaldehyde configuration with one benzotriazole functionalized group on the C2-position (Fig. 1). The dihedral angle between the planes of the benzotriazole unit and the benzene ring of the salicylaldehyde group is 2.4 (2)°. There is an intramolecular O—H···N hydrogen bond between the phenol and benzotriazole groups (Table 1). The distance of N1···H1A is substantially shorter than the van der Waals distance of 2.75 Å for the N and H distance. The distances in the benzotriazole-phenolate group are similar to those found in the crystal structure of 2-(2H-benzotriazol-2-yl)-6-((diethylamino)methyl)-4-methylphenol (Li et al., 2009).
Experimental
The title compound (I) was synthesized by the following procedures (Fig. 2): In a 100 ml round bottom flask was placed with 4-methyl-2-(2H-benzotriazol-2-yl)phenol (4.50 g, 20.0 mmol) and hexamethylenetetramine (5.60 g, 40 mmol). To this was added trifluoroacetic acid (24 ml, 0.30 mol) and the yellow solution became hot. The resulting mixture was heated to 418 K under reflux for 18 h, during which time the solution colour turned the yellow to dark brown/black. The hot solution was poured into 4 N HCl(aq) (40 ml) and stirred for another 2 h, during which time the solids were formed. The mixture was placed at 253 K overnight and the solids were filtered. The mixture was then extracted with dichloromethane (3 x 150 ml) and the organic layers were dried over MgSO4. The final solution was removed the solvent under vacuum to give yellow solids. Yield: 4.40 g (87 %). Single crystals suitable for X-ray diffraction were obtained from a saturated solution of the title compound in Et2O.
Refinement
The H atoms were placed in idealized positions and constrained to ride on their parent atoms, with C–H = 0.93 Å with Uiso(H) = 1.2 Ueq(C) for phenyl hydrogen; 0.96 Å with Uiso(H) = 1.5 Ueq(C) for CH3 group; 0.93 Å with Uiso(H) = 1.2 Ueq(C) for CHO group; O–H = 0.85 Å with Uiso(H) = 1.2 Ueq(C).
Figures
Fig. 1.

The molecular structure of (I) with the atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. The dashed lines indicates a hydrogen bond.
Fig. 2.

The synthetic procedure of the title compound I.
Crystal data
| C14H11N3O2 | F(000) = 528 |
| Mr = 253.26 | Dx = 1.372 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 1657 reflections |
| a = 12.2724 (5) Å | θ = 1.7–28.3° |
| b = 14.5018 (5) Å | µ = 0.10 mm−1 |
| c = 6.8897 (3) Å | T = 296 K |
| β = 91.571 (2)° | Columnar, yellow |
| V = 1225.71 (8) Å3 | 0.34 × 0.31 × 0.23 mm |
| Z = 4 |
Data collection
| Bruker APEXII CCD diffractometer | 2946 independent reflections |
| Radiation source: fine-focus sealed tube | 1657 reflections with I > 2σ(I) |
| graphite | Rint = 0.070 |
| Detector resolution: 8.3333 pixels mm-1 | θmax = 28.3°, θmin = 1.7° |
| φ and ω scans | h = −16→16 |
| Absorption correction: multi-scan (SADABS; Bruker, 2008) | k = −19→19 |
| Tmin = 0.972, Tmax = 0.977 | l = −7→9 |
| 13912 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.045 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.146 | H-atom parameters constrained |
| S = 1.01 | w = 1/[σ2(Fo2) + (0.075P)2] where P = (Fo2 + 2Fc2)/3 |
| 2946 reflections | (Δ/σ)max < 0.001 |
| 172 parameters | Δρmax = 0.21 e Å−3 |
| 0 restraints | Δρmin = −0.20 e Å−3 |
Special details
| Experimental. 1H NMR (CDCl3, ppm): δ 11.88 (s, 1H, PhOH),10.51 (s, 1H, PhCHO), 8.36 (s, 1H, PhH), 7.94 (d, 2H, PhH), 7.68 (s, 1H, PhH), 7.50 (d, 2H, PhH), 2.41 (s, 3H, PhCH3). |
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.50858 (9) | 0.36518 (6) | 0.26655 (17) | 0.0578 (3) | |
| H1A | 0.5768 | 0.3681 | 0.2490 | 0.069* | |
| O2 | 0.18809 (10) | 0.37151 (8) | 0.3322 (2) | 0.0816 (4) | |
| N1 | 0.68476 (10) | 0.27325 (8) | 0.20289 (19) | 0.0488 (3) | |
| N2 | 0.62690 (10) | 0.19539 (8) | 0.22480 (18) | 0.0458 (3) | |
| N3 | 0.68234 (10) | 0.11695 (8) | 0.21302 (19) | 0.0517 (4) | |
| C1 | 0.45880 (12) | 0.28244 (9) | 0.2757 (2) | 0.0435 (4) | |
| C2 | 0.51319 (12) | 0.19766 (9) | 0.2580 (2) | 0.0428 (4) | |
| C3 | 0.45656 (12) | 0.11543 (10) | 0.2707 (2) | 0.0471 (4) | |
| H3B | 0.4938 | 0.0600 | 0.2579 | 0.057* | |
| C4 | 0.34523 (12) | 0.11386 (10) | 0.3022 (2) | 0.0490 (4) | |
| C5 | 0.29195 (12) | 0.19740 (10) | 0.3180 (2) | 0.0493 (4) | |
| H5A | 0.2173 | 0.1977 | 0.3381 | 0.059* | |
| C6 | 0.34674 (12) | 0.28099 (10) | 0.3048 (2) | 0.0456 (4) | |
| C7 | 0.78649 (11) | 0.24258 (10) | 0.1756 (2) | 0.0476 (4) | |
| C8 | 0.88414 (13) | 0.29132 (12) | 0.1448 (2) | 0.0578 (5) | |
| H8A | 0.8858 | 0.3554 | 0.1408 | 0.069* | |
| C9 | 0.97489 (13) | 0.24055 (13) | 0.1215 (2) | 0.0637 (5) | |
| H9A | 1.0405 | 0.2706 | 0.1011 | 0.076* | |
| C10 | 0.97342 (14) | 0.14307 (13) | 0.1272 (3) | 0.0672 (5) | |
| H10A | 1.0382 | 0.1111 | 0.1099 | 0.081* | |
| C11 | 0.88075 (13) | 0.09447 (12) | 0.1571 (2) | 0.0622 (5) | |
| H11A | 0.8806 | 0.0304 | 0.1608 | 0.075* | |
| C12 | 0.78537 (12) | 0.14584 (11) | 0.1820 (2) | 0.0491 (4) | |
| C13 | 0.28498 (14) | 0.02373 (11) | 0.3176 (3) | 0.0687 (5) | |
| H13A | 0.2093 | 0.0356 | 0.3392 | 0.103* | |
| H13B | 0.3153 | −0.0114 | 0.4241 | 0.103* | |
| H13C | 0.2919 | −0.0105 | 0.1993 | 0.103* | |
| C14 | 0.28544 (14) | 0.36772 (11) | 0.3179 (2) | 0.0578 (5) | |
| H14A | 0.3241 | 0.4228 | 0.3150 | 0.069* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0533 (6) | 0.0414 (6) | 0.0792 (9) | −0.0065 (5) | 0.0092 (5) | −0.0014 (5) |
| O2 | 0.0565 (8) | 0.0613 (8) | 0.1276 (13) | 0.0086 (6) | 0.0154 (7) | −0.0057 (7) |
| N1 | 0.0475 (7) | 0.0449 (7) | 0.0540 (9) | −0.0062 (6) | 0.0031 (6) | 0.0029 (6) |
| N2 | 0.0458 (7) | 0.0421 (7) | 0.0495 (9) | −0.0022 (5) | 0.0021 (6) | 0.0019 (5) |
| N3 | 0.0477 (7) | 0.0449 (7) | 0.0629 (10) | 0.0010 (6) | 0.0044 (6) | −0.0005 (6) |
| C1 | 0.0505 (9) | 0.0395 (7) | 0.0405 (9) | −0.0044 (6) | 0.0012 (7) | −0.0002 (6) |
| C2 | 0.0433 (8) | 0.0437 (8) | 0.0414 (9) | −0.0004 (6) | 0.0013 (6) | 0.0014 (6) |
| C3 | 0.0482 (9) | 0.0399 (8) | 0.0532 (10) | 0.0018 (6) | 0.0012 (7) | 0.0019 (6) |
| C4 | 0.0491 (9) | 0.0438 (8) | 0.0542 (11) | −0.0018 (6) | 0.0017 (7) | 0.0028 (7) |
| C5 | 0.0444 (8) | 0.0508 (9) | 0.0530 (10) | −0.0006 (6) | 0.0046 (7) | 0.0009 (7) |
| C6 | 0.0484 (9) | 0.0435 (8) | 0.0448 (10) | 0.0002 (6) | 0.0018 (7) | 0.0000 (6) |
| C7 | 0.0466 (9) | 0.0543 (9) | 0.0420 (9) | −0.0041 (7) | 0.0025 (7) | −0.0001 (7) |
| C8 | 0.0539 (10) | 0.0615 (10) | 0.0583 (12) | −0.0110 (8) | 0.0053 (8) | 0.0019 (8) |
| C9 | 0.0510 (10) | 0.0747 (12) | 0.0657 (13) | −0.0113 (9) | 0.0071 (8) | 0.0008 (9) |
| C10 | 0.0478 (9) | 0.0776 (12) | 0.0766 (14) | 0.0042 (9) | 0.0070 (8) | −0.0052 (9) |
| C11 | 0.0516 (10) | 0.0583 (10) | 0.0771 (13) | 0.0046 (8) | 0.0076 (8) | −0.0044 (8) |
| C12 | 0.0463 (9) | 0.0514 (9) | 0.0497 (10) | −0.0021 (7) | 0.0021 (7) | −0.0006 (7) |
| C13 | 0.0560 (10) | 0.0495 (10) | 0.1008 (15) | −0.0060 (8) | 0.0074 (9) | 0.0064 (9) |
| C14 | 0.0567 (10) | 0.0478 (9) | 0.0693 (12) | −0.0005 (7) | 0.0078 (8) | −0.0033 (7) |
Geometric parameters (Å, °)
| O1—C1 | 1.3488 (15) | C5—H5A | 0.9300 |
| O1—H1A | 0.8500 | C6—C14 | 1.470 (2) |
| O2—C14 | 1.2026 (18) | C7—C12 | 1.404 (2) |
| N1—C7 | 1.3434 (18) | C7—C8 | 1.412 (2) |
| N1—N2 | 1.3445 (16) | C8—C9 | 1.348 (2) |
| N2—N3 | 1.3292 (16) | C8—H8A | 0.9300 |
| N2—C2 | 1.4206 (18) | C9—C10 | 1.414 (3) |
| N3—C12 | 1.3545 (18) | C9—H9A | 0.9300 |
| C1—C6 | 1.395 (2) | C10—C11 | 1.358 (2) |
| C1—C2 | 1.4058 (19) | C10—H10A | 0.9300 |
| C2—C3 | 1.3842 (18) | C11—C12 | 1.402 (2) |
| C3—C4 | 1.389 (2) | C11—H11A | 0.9300 |
| C3—H3B | 0.9300 | C13—H13A | 0.9600 |
| C4—C5 | 1.3823 (19) | C13—H13B | 0.9600 |
| C4—C13 | 1.507 (2) | C13—H13C | 0.9600 |
| C5—C6 | 1.3905 (19) | C14—H14A | 0.9300 |
| C1—O1—H1A | 120.0 | C12—C7—C8 | 120.93 (14) |
| C7—N1—N2 | 103.50 (12) | C9—C8—C7 | 116.85 (16) |
| N3—N2—N1 | 116.04 (12) | C9—C8—H8A | 121.6 |
| N3—N2—C2 | 122.44 (11) | C7—C8—H8A | 121.6 |
| N1—N2—C2 | 121.52 (11) | C8—C9—C10 | 122.10 (15) |
| N2—N3—C12 | 103.09 (11) | C8—C9—H9A | 119.0 |
| O1—C1—C6 | 118.00 (13) | C10—C9—H9A | 119.0 |
| O1—C1—C2 | 123.87 (13) | C11—C10—C9 | 122.29 (16) |
| C6—C1—C2 | 118.13 (13) | C11—C10—H10A | 118.9 |
| C3—C2—C1 | 120.50 (14) | C9—C10—H10A | 118.9 |
| C3—C2—N2 | 119.17 (12) | C10—C11—C12 | 116.62 (16) |
| C1—C2—N2 | 120.33 (12) | C10—C11—H11A | 121.7 |
| C2—C3—C4 | 121.44 (13) | C12—C11—H11A | 121.7 |
| C2—C3—H3B | 119.3 | N3—C12—C11 | 129.87 (15) |
| C4—C3—H3B | 119.3 | N3—C12—C7 | 108.91 (12) |
| C5—C4—C3 | 117.86 (13) | C11—C12—C7 | 121.22 (13) |
| C5—C4—C13 | 121.36 (14) | C4—C13—H13A | 109.5 |
| C3—C4—C13 | 120.78 (13) | C4—C13—H13B | 109.5 |
| C4—C5—C6 | 121.88 (14) | H13A—C13—H13B | 109.5 |
| C4—C5—H5A | 119.1 | C4—C13—H13C | 109.5 |
| C6—C5—H5A | 119.1 | H13A—C13—H13C | 109.5 |
| C5—C6—C1 | 120.19 (13) | H13B—C13—H13C | 109.5 |
| C5—C6—C14 | 119.52 (14) | O2—C14—C6 | 123.75 (15) |
| C1—C6—C14 | 120.28 (13) | O2—C14—H14A | 118.1 |
| N1—C7—C12 | 108.46 (12) | C6—C14—H14A | 118.1 |
| N1—C7—C8 | 130.62 (15) | ||
| C7—N1—N2—N3 | −0.35 (17) | C2—C1—C6—C5 | 0.8 (2) |
| C7—N1—N2—C2 | 179.82 (12) | O1—C1—C6—C14 | 2.1 (2) |
| N1—N2—N3—C12 | 0.42 (17) | C2—C1—C6—C14 | −178.04 (13) |
| C2—N2—N3—C12 | −179.75 (12) | N2—N1—C7—C12 | 0.12 (15) |
| O1—C1—C2—C3 | 179.41 (13) | N2—N1—C7—C8 | −179.78 (15) |
| C6—C1—C2—C3 | −0.5 (2) | N1—C7—C8—C9 | −179.89 (14) |
| O1—C1—C2—N2 | −1.3 (2) | C12—C7—C8—C9 | 0.2 (2) |
| C6—C1—C2—N2 | 178.88 (12) | C7—C8—C9—C10 | 0.1 (2) |
| N3—N2—C2—C3 | −2.7 (2) | C8—C9—C10—C11 | −0.2 (3) |
| N1—N2—C2—C3 | 177.08 (13) | C9—C10—C11—C12 | 0.1 (2) |
| N3—N2—C2—C1 | 177.91 (13) | N2—N3—C12—C11 | −179.89 (15) |
| N1—N2—C2—C1 | −2.3 (2) | N2—N3—C12—C7 | −0.30 (16) |
| C1—C2—C3—C4 | −0.4 (3) | C10—C11—C12—N3 | 179.71 (16) |
| N2—C2—C3—C4 | −179.71 (13) | C10—C11—C12—C7 | 0.2 (2) |
| C2—C3—C4—C5 | 0.8 (2) | N1—C7—C12—N3 | 0.12 (16) |
| C2—C3—C4—C13 | −179.41 (15) | C8—C7—C12—N3 | −179.97 (14) |
| C3—C4—C5—C6 | −0.5 (2) | N1—C7—C12—C11 | 179.75 (14) |
| C13—C4—C5—C6 | 179.77 (14) | C8—C7—C12—C11 | −0.3 (2) |
| C4—C5—C6—C1 | −0.4 (3) | C5—C6—C14—O2 | −3.1 (3) |
| C4—C5—C6—C14 | 178.51 (14) | C1—C6—C14—O2 | 175.78 (16) |
| O1—C1—C6—C5 | −179.06 (13) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1A···N1 | 0.85 | 1.94 | 2.588 (2) | 132 |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: LH5003).
References
- Bruker (2008). APEX2, SADABS and SAINT-Plus Bruker AXS Inc., Madison, Wisconsin, USA.
- Chen, H.-Y., Tang, H.-Y. & Lin, C.-C. (2006). Macromolecules, 39, 3745–3752.
- Li, J.-Y., Liu, Y.-C., Lin, C.-H. & Ko, B.-T. (2009). Acta Cryst. E65, o2475. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Wu, J.-C., Huang, B.-H., Hsueh, M.-L., Lai, S.-L. & Lin, C.-C. (2005). Polymer, 46, 9784–9792.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810007233/lh5003sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810007233/lh5003Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report