

Abstract
In the crystal structure of the title compound, C5H8N2O3, the molecules exist in the zwitterionic form. The pyrrolidine ring adopts an envelope conformation with the unsubstituted endocyclic C atom situated at the flap. The other four endocyclic atoms are coplanar with the exocyclic carbonyl O atom, with an r.m.s. deviation from the mean plane of 0.06 Å. The carboxylate substituent is located axially, while the ammonium group occupies an equatorial position. In the crystal structure, the molecules are linked through N—H⋯O hydrogen bonds, forming a three-dimensional network.
Related literature
For molecular recognition in N-methyl amino acids and proline residues, see: Dugave & Demange (2003 ▶). For the construction of modified amino acids, see: Dumy et al. (1997 ▶); Keller et al. (1998 ▶); Mutter et al. (1999 ▶); Tuchscherer & Mutter (2001 ▶); Paul et al. (1992 ▶). For pyroglutamic acid derivatives, see: Zabrocki et al. (1988 ▶); Kaczmarek et al. (2005 ▶). For the preparation of the title compound, see: Kaczmarek et al. (2001 ▶); Kaczmarek (2009 ▶). For asymmetry parameters, see: Griffin et al. (1984 ▶).
Experimental
Crystal data
C5H8N2O3
M r = 144.13
Orthorhombic,
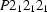
a = 5.9790 (3) Å
b = 9.3665 (4) Å
c = 11.3809 (5) Å
V = 637.36 (5) Å3
Z = 4
Cu Kα radiation
μ = 1.08 mm−1
T = 293 K
0.40 × 0.40 × 0.10 mm
Data collection
Bruker SMART APEX diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2003 ▶) T min = 0.707, T max = 0.900
7227 measured reflections
1169 independent reflections
1168 reflections with I > 2σ(I)
R int = 0.030
Refinement
R[F 2 > 2σ(F 2)] = 0.027
wR(F 2) = 0.069
S = 1.08
1169 reflections
125 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.13 e Å−3
Δρmin = −0.17 e Å−3
Absolute structure: Flack (1983 ▶), 461 Friedel pairs
Flack parameter: 0.1 (2)
Data collection: SMART (Bruker, 2003 ▶); cell refinement: SAINT-Plus (Bruker, 2003 ▶); data reduction: SAINT-Plus; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL and Mercury (Macrae et al., 2008 ▶); software used to prepare material for publication: SHELXTL and publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810004277/bt5187sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810004277/bt5187Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O2i | 0.82 (2) | 2.07 (2) | 2.8535 (15) | 161.2 (18) |
| N2—H2⋯O1ii | 0.88 (2) | 1.87 (2) | 2.7346 (16) | 168.5 (17) |
| N2—H3⋯O1iii | 0.897 (17) | 1.886 (17) | 2.7788 (14) | 173.4 (17) |
| N2—H4⋯O2iv | 0.868 (17) | 1.935 (17) | 2.7967 (15) | 172.3 (17) |
Symmetry codes: (i) 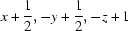 ; (ii)
; (ii) 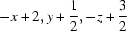 ; (iii)
; (iii) 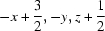 ; (iv)
; (iv) 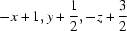 .
.
Acknowledgments
Financial support from the Ministry of Science and Higher Education, Poland (project No. 2P05F00129) is gratefully acknowledged.
supplementary crystallographic information
Comment
N-methyl amino acids and proline residues in the peptide chain may cause the cis-trans isomerisation of the amide bond and lead to conformational changes, which influence the molecular recognition (Dugave & Demange, 2003). Importance of the cis-amide bonds for the peptide bioactivity led to the construction of modified amino acids, which could lock a peptide bond in the cis-geometry (Dumy et al., 1997; Keller et al., 1998; Mutter et al., 1999; Tuchscherer & Mutter, 2001). In particular, Paul et al. (1992) designed mimetics of the cis-peptide bond based on the substitituted pyroglutamic acid residue. In contrast with a tetrazole replacement for the peptide bond, the pyroglutamic acid derivatives are more rigid (Zabrocki et al., 1988). Their carboxylic group could be either donor or acceptor of hydrogen bond without invloving the polypeptide main chain amide moieties (Kaczmarek et al., 2005).
The 4-aminopyroglutamic acid is a particularly useful residue for building the conformationally restricted peptide chains. Depending on the absolute configuration at both chiral centers it may be applied to construct the VIa or VIb β-turn mimetics.
The title compound may be obtained by two different methods elaborated by us, i.e. by electrophilic amination reaction of N-protected (S)-pyroglutamate ester, which gives separable 9:1 mixture of (2S,4R) and (2S,4S) diastereoisomers (Kaczmarek et al., 2001) or through Michael addition of dehydroalanine derivatives to sodium salt of N-benzyloxycarbonylaminomalonate ester, which gives after hydrolysis and decarboxylation mixture of all four possible stereomers. The details of the last reaction and resolution of stereoisomers will be described elsewhere (Kaczmarek, 2009).
A view of the title compound is given in Fig. 1. The molecule has two chiral centres viz. C3 and C5. Their absolute configurations follow from the synthetic procedure and are R and S, respectively.
The pyrrolidine ring adopts an envelope conformation with N1, C2, C3 and C5 almost coplanar and the C4 situated at the flap.
Additionally, the former four endocyclic atoms are coplanar with the exocyclic carbonyl oxygen, the average r.m.s. deviation from the mean plane is 0.06 Å.
The three lowest ring asymmetry parameters (Griffin et al., 1984) are: CS(C4) = 1.26 (14), C2(C2) = 11.92 (14), C2(N1) = 15.46 (14)°. The carboxylate substituent is located axially in conformation stabilized by the short N1···O2 contact [2.787 (2) Å], while the ammonium group occupies equatorial position.
In the crystal each molecule is linked through N—H ···O hydrogen bonds with eight adjacent molecules, their deatils are shown in Table 2 and Fig. 2.
Experimental
An optically pure (ee>99%) N'-benzyloxycarbonyl protected precursor of the title compound was hydrogenated in methanol solution over 10% palladium on charcoal, which resulted in precipitation of the final product. After filtration of solids final product was washed out of the catalyst with the aim of water. The (2S,4R)-4-aminopyroglutamic acid crystals were grown from this water solution by slow evaporation.
Refinement
All H atoms were located in difference Fourier maps and refined freely.
Figures
Fig. 1.

Molecule of the title compound. Displacement ellipsoids are drawn at the 50% probability level.
Fig. 2.

View of hydrogen bonding in the crystal of the title compound. Symmetry codes: I (x, y, z); II (x + 1/2,-y+1/2,-z+1); III (-x + 2, y + 1/2,-z+3/2); IV (-x + 1, y + 1/2,-z+3/2); V (-x + 3/2,-y, z + 1/2).
Crystal data
| C5H8N2O3 | Dx = 1.502 Mg m−3 |
| Mr = 144.13 | Melting point: 423(2) K |
| Orthorhombic, P212121 | Cu Kα radiation, λ = 1.54178 Å |
| Hall symbol: P 2ac 2ab | Cell parameters from 7056 reflections |
| a = 5.9790 (3) Å | θ = 6.1–70.8° |
| b = 9.3665 (4) Å | µ = 1.08 mm−1 |
| c = 11.3809 (5) Å | T = 293 K |
| V = 637.36 (5) Å3 | Prism, colourless |
| Z = 4 | 0.40 × 0.40 × 0.10 mm |
| F(000) = 304 |
Data collection
| Bruker SMART APEX diffractometer | 1169 independent reflections |
| Radiation source: fine-focus sealed tube | 1168 reflections with I > 2σ(I) |
| graphite | Rint = 0.030 |
| ω scans | θmax = 70.8°, θmin = 6.1° |
| Absorption correction: multi-scan (SADABS; Bruker, 2003) | h = −6→5 |
| Tmin = 0.707, Tmax = 0.900 | k = −11→11 |
| 7227 measured reflections | l = −13→13 |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H atoms treated by a mixture of independent and constrained refinement |
| R[F2 > 2σ(F2)] = 0.027 | w = 1/[σ2(Fo2) + (0.0468P)2 + 0.0681P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.069 | (Δ/σ)max < 0.001 |
| S = 1.08 | Δρmax = 0.13 e Å−3 |
| 1169 reflections | Δρmin = −0.17 e Å−3 |
| 125 parameters | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 0 restraints | Extinction coefficient: 0.047 (3) |
| Primary atom site location: structure-invariant direct methods | Absolute structure: Flack (1983), 461 Friedel pairs |
| Secondary atom site location: difference Fourier map | Flack parameter: 0.1 (2) |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.85679 (17) | −0.10762 (9) | 0.57781 (8) | 0.0373 (3) | |
| H2 | 0.838 (4) | 0.3509 (18) | 0.9130 (15) | 0.042 (4)* | |
| O2 | 0.57914 (17) | 0.04638 (10) | 0.55325 (8) | 0.0383 (3) | |
| O3 | 0.6519 (2) | 0.46234 (9) | 0.68254 (10) | 0.0469 (3) | |
| N1 | 0.8566 (2) | 0.27436 (11) | 0.61237 (10) | 0.0353 (3) | |
| H1 | 0.917 (4) | 0.3094 (19) | 0.5549 (17) | 0.054 (5)* | |
| C1 | 0.7725 (2) | 0.01571 (13) | 0.58351 (10) | 0.0283 (3) | |
| C5 | 0.9163 (2) | 0.12935 (13) | 0.64357 (11) | 0.0304 (3) | |
| H51 | 1.078 (3) | 0.1140 (16) | 0.6258 (13) | 0.032 (4)* | |
| C4 | 0.8646 (3) | 0.12504 (13) | 0.77672 (11) | 0.0340 (3) | |
| H41 | 0.807 (3) | 0.0354 (17) | 0.8009 (15) | 0.045 (5)* | |
| H42 | 0.995 (4) | 0.156 (2) | 0.8147 (18) | 0.056 (5)* | |
| C3 | 0.6848 (2) | 0.23810 (12) | 0.79144 (11) | 0.0297 (3) | |
| H31 | 0.541 (3) | 0.1976 (15) | 0.7877 (14) | 0.030 (4)* | |
| N2 | 0.7038 (2) | 0.31400 (11) | 0.90532 (10) | 0.0310 (3) | |
| H4 | 0.607 (3) | 0.3822 (18) | 0.9140 (14) | 0.036 (4)* | |
| H3 | 0.695 (3) | 0.249 (2) | 0.9630 (14) | 0.042 (4)* | |
| C2 | 0.7255 (2) | 0.34135 (13) | 0.68928 (11) | 0.0319 (3) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0425 (6) | 0.0262 (4) | 0.0432 (5) | 0.0027 (4) | −0.0079 (4) | −0.0062 (4) |
| O2 | 0.0334 (6) | 0.0347 (5) | 0.0466 (5) | −0.0016 (4) | −0.0093 (4) | 0.0089 (4) |
| O3 | 0.0561 (7) | 0.0309 (5) | 0.0536 (6) | 0.0133 (5) | 0.0046 (5) | 0.0104 (4) |
| N1 | 0.0451 (7) | 0.0234 (5) | 0.0374 (6) | −0.0044 (5) | 0.0072 (5) | 0.0050 (4) |
| C1 | 0.0331 (7) | 0.0267 (6) | 0.0251 (5) | −0.0021 (4) | 0.0001 (5) | 0.0034 (4) |
| C5 | 0.0311 (7) | 0.0241 (6) | 0.0361 (6) | −0.0008 (5) | 0.0004 (5) | 0.0004 (5) |
| C4 | 0.0435 (8) | 0.0250 (6) | 0.0335 (6) | 0.0040 (5) | −0.0074 (6) | 0.0002 (5) |
| C3 | 0.0294 (7) | 0.0257 (5) | 0.0340 (6) | −0.0027 (5) | −0.0020 (4) | 0.0037 (5) |
| N2 | 0.0327 (7) | 0.0257 (5) | 0.0345 (5) | 0.0022 (5) | 0.0027 (4) | 0.0029 (4) |
| C2 | 0.0321 (7) | 0.0270 (6) | 0.0367 (6) | −0.0015 (5) | −0.0022 (5) | 0.0044 (5) |
Geometric parameters (Å, °)
| O1—C1 | 1.2621 (16) | C4—C3 | 1.5187 (19) |
| O2—C1 | 1.2399 (17) | C4—H41 | 0.949 (17) |
| O3—C2 | 1.2179 (16) | C4—H42 | 0.94 (2) |
| N1—C2 | 1.3321 (17) | C3—N2 | 1.4826 (16) |
| N1—C5 | 1.4485 (15) | C3—C2 | 1.5318 (16) |
| N1—H1 | 0.82 (2) | C3—H31 | 0.939 (17) |
| C1—C5 | 1.5295 (17) | N2—H2 | 0.88 (2) |
| C5—C4 | 1.5470 (17) | N2—H4 | 0.867 (18) |
| C5—H51 | 0.997 (17) | N2—H3 | 0.898 (18) |
| C2—N1—C5 | 115.19 (10) | C3—C4—H41 | 109.0 (11) |
| O3—C2—N1 | 127.55 (12) | C5—C4—H41 | 112.3 (10) |
| O3—C2—C3 | 125.30 (12) | C3—C4—H42 | 108.7 (13) |
| N1—C2—C3 | 107.15 (11) | C5—C4—H42 | 106.1 (13) |
| N1—C5—C1 | 113.86 (10) | H41—C4—H42 | 116.4 (17) |
| N1—C5—C4 | 102.44 (10) | N2—C3—C4 | 112.11 (10) |
| C1—C5—C4 | 107.90 (10) | N2—C3—C2 | 110.40 (10) |
| C3—C4—C5 | 103.37 (10) | N2—C3—H31 | 107.7 (9) |
| C4—C3—C2 | 104.12 (10) | C4—C3—H31 | 111.1 (9) |
| C2—N1—H1 | 126.6 (13) | C2—C3—H31 | 111.5 (9) |
| C5—N1—H1 | 117.8 (13) | C3—N2—H2 | 110.3 (11) |
| O2—C1—O1 | 124.76 (12) | C3—N2—H4 | 113.6 (11) |
| O2—C1—C5 | 119.14 (11) | H2—N2—H4 | 107.9 (16) |
| O1—C1—C5 | 115.82 (11) | C3—N2—H3 | 108.0 (11) |
| N1—C5—H51 | 108.9 (9) | H2—N2—H3 | 104.4 (16) |
| C1—C5—H51 | 110.7 (9) | H4—N2—H3 | 112.3 (15) |
| C4—C5—H51 | 112.9 (8) | ||
| N1—C5—C4—C3 | 26.38 (13) | O2—C1—C5—N1 | −26.96 (16) |
| C5—C4—C3—C2 | −25.98 (13) | O1—C1—C5—N1 | 158.87 (11) |
| C5—N1—C2—O3 | −179.48 (14) | O2—C1—C5—C4 | 86.02 (13) |
| C5—N1—C2—C3 | 1.41 (16) | O1—C1—C5—C4 | −88.15 (13) |
| C4—C3—C2—N1 | 16.23 (14) | C1—C5—C4—C3 | −94.06 (11) |
| C4—C3—C2—O3 | −162.91 (14) | C5—C4—C3—N2 | −145.33 (10) |
| C2—N1—C5—C4 | −17.99 (15) | N2—C3—C2—O3 | −42.41 (18) |
| C2—N1—C5—C1 | 98.23 (14) | N2—C3—C2—N1 | 136.73 (12) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O2i | 0.82 (2) | 2.07 (2) | 2.8535 (15) | 161.2 (18) |
| N2—H2···O1ii | 0.88 (2) | 1.87 (2) | 2.7346 (16) | 168.5 (17) |
| N2—H3···O1iii | 0.897 (17) | 1.886 (17) | 2.7788 (14) | 173.4 (17) |
| N2—H4···O2iv | 0.868 (17) | 1.935 (17) | 2.7967 (15) | 172.3 (17) |
Symmetry codes: (i) x+1/2, −y+1/2, −z+1; (ii) −x+2, y+1/2, −z+3/2; (iii) −x+3/2, −y, z+1/2; (iv) −x+1, y+1/2, −z+3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: BT5187).
References
- Bruker (2003). SADABS, SAINT-Plus and SMART Bruker AXS Inc., Madison, Wisconsin, USA.
- Dugave, Ch. & Demange, L. (2003). Chem. Rev.103, 2475–2532. [DOI] [PubMed]
- Dumy, P., Keller, M., Ryan, D. E., Rohwedder, B., Wöhr, T. & Mutter, M. (1997). J. Am. Chem. Soc.119, 918–925.
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Griffin, J. F., Duax, W. L. & Weeks, C. M. (1984). Atlas of Steroid Structure, Vol. 2, p. 8. New York: IFI/Plenum.
- Kaczmarek, K. (2009). Private communication.
- Kaczmarek, K., Kaleta, M., Chung, N. N., Schiller, P. W. & Zabrocki, J. (2001). Acta Biochimica Pol.48, 1159–1163. [PubMed]
- Kaczmarek, K., Wolf, W. M. & Zabrocki, J. (2005). Acta Cryst. E61, o629–o631.
- Keller, M., Sager, C., Dumy, P., Schutkowski, M., Fischer, G. S. & Mutter, M. (1998). J. Am. Chem. Soc.120, 2714–2720.
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst.41, 466–470.
- Mutter, M., Wöhr, T., Gioria, S. & Keller, M. (1999). Biopolymers (Peptide Science)51, 121–128. [DOI] [PubMed]
- Paul, P. K. C., Burney, P. A., Campbell, M. M. & Osguthorpe, D. J. (1992). Bioorg. & Med. Chem. Lett.2, 141–144.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Tuchscherer, G. & Mutter, M. (2001). Chimia, 55, 306–313.
- Westrip, S. P. (2010). publCIF In preparation.
- Zabrocki, J., Smith, G. D., Dunbar, J. B., Ijima, H. & Marshall, G. R. (1988). J. Am. Chem. Soc.110, 5875–5880.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810004277/bt5187sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810004277/bt5187Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report