

Abstract
As influenza viruses have developed resistance towards current drugs, new inhibitors that prevent viral replication through different inhibitory mechanisms are useful. In this study, we developed a screening procedure to search for new antiinfluenza inhibitors from 1,200,000 compounds and identified previously reported as well as new antiinfluenza compounds. Several antiinfluenza compounds were inhibitory to the influenza RNA-dependent RNA polymerase (RdRP), including nucleozin and its analogs. The most potent nucleozin analog, 3061 (FA-2), inhibited the replication of the influenza A/WSN/33 (H1N1) virus in MDCK cells at submicromolar concentrations and protected the lethal H1N1 infection of mice. Influenza variants resistant to 3061 (FA-2) were isolated and shown to have the mutation on nucleoprotein (NP) that is distinct from the recently reported resistant mutation of Y289H [Kao R, et al. (2010) Nat Biotechnol 28:600]. Recombinant influenza carrying the Y52H NP is also resistant to 3061 (FA-2), and NP aggregation induced by 3061 (FA-2) was identified as the most likely cause for inhibition. In addition, we identified another antiinfluenza RdRP inhibitor 367 which targets PB1 protein but not NP. A mutant resistant to 367 has H456P mutation at the PB1 protein and both the recombinant influenza and the RdRP expressing the PB1 H456P mutation have elevated resistance to 367. Our high-throughput screening (HTS) campaign thus resulted in the identification of antiinfluenza compounds targeting RdRP activity.
Keywords: high-throughput screening, antiinfluenza, influenza NP, influenza PB1, chemical genetics
Influenza virus infections, commonly called “flu,” can cause acute respiratory distress, resulting in morbidity and even excess mortality (1). Vaccinations remain the principle prophylactics for controlling influenza infections. Other prophylactic and therapeutic antiviral drugs are needed as well especially during an outbreak and for people with weaker immune systems, such as children, elderly, or individuals undergoing other medical treatments (2, 3).
Currently available drugs for influenza viruses are M2 channel blockers such as amantidine and rimantidine (4), and neuraminidase inhibitors including Oseltamivir and Zanamivir (5, 6). M2 channel blockers are inexpensive and readily available but are active only on influenza A strains; further, high percentages of circulating influenza strains have developed resistance to amantidine and rimantidine (7, 8). Viruses that are resistant to the neuraminidase inhibitor Oseltamivir have also been reported since 2005 (9, 10), and currently greater than 75% of influenza H1N1 viruses in Norway and many other countries are resistant to Oseltamivir (11). More surprisingly, the new 2009 H1N1 virus, also called S-OIV (Swine-Originated Influenza Virus), was susceptible to Oseltamivir initially (12), but developed Oseltamivir-resistant variants in 4 mo (13). In addition to the neuraminidase and the M2, influenza polymerase has been considered as a promising antiinfluenza drug target because its mode of action is very different from the human RNA polymerases (14, 15).
Our approach to identify new antiinfluenza compounds is to screen a large library of 1.2 million compounds for hits using a cell-based infection assay using a high-throughput system manufactured by GNF systems. The hits were further screened for compounds that inhibit viral yields by counting the number of infectious particles in a high-throughput mode. Among the hits that are inhibitory to viral replication, there are both known antiinfluenza agents and new inhibitors. A few of the identified hits are inhibitory in an influenza RNA-dependent RNA polymerase (RdRP) assay. One antiinfluenza compound, 3061 [recently named as FA-2 (16)], and structurally similar compounds with substituted isoxazolyl carbonyl piperazine skeleton, were active in the inhibition of influenza virus H1N1 (A/WSN/1933) (WSN) and the influenza RdRP activity both at submicromolar levels. Compound 3061 (FA-2) is structurally similar to nucleozin that was recently reported as an antiinfluenza inhibitor targeting the nucleoprotein (NP) because viruses with NP mutation Y289H are nucleozin resistant (16). To understand the antiviral mechanism of 3061 (FA-2), we also studied resistant mutants and found that those with Y52H NP mutation are 3061 (FA-2) resistant. In addition, we have identified another antiinfluenza compound, 367 that targets the PB1 protein and its mutation at H456P has elevated resistance to 367 in both antiinfluenza and the RdRP assays.
Results and Discussion
Identification of Antiinfluenza Hits by High-Throughput Screening of a Large Compound Library.
The screening method development and optimization are described in Figs. S1 and S2 of SI Text.
Among the identified antiinfluenza hits, some are established antiinfluenza drugs or compounds (SI Text and Fig. S3). Compounds identified as new antiinfluenza hits with IC50 values smaller than 5 μM are shown in Fig. 1A.
Fig. 1.

Structures and characterizations of new antiinfluenza compounds identified by HTS. (A) The structures and name of the identified antiinfluenza compounds are shown with their anti-WSN IC50 values next to their names. (B) The WSN viral yield reductions by inhibitors at indicated concentrations were measured at 24 h after infection with multiplication of infection at 0.01. Shown are Average ± Standard deviation (N = 3). (C) Inhibition of the RdRP activities by the antiinfluenza inhibitors were determined using transfected 293 cells expressing negative sensed luciferase RNA and the influenza RdRP subunit proteins. The relative activities refer to the RdRP reporter luciferase activities measured at different inhibitor concentrations relative to against the control measurements. Shown are Average ± Standard deviation (N = 3).
Characterization of the New Antiinfluenza Compounds Identified by HTS.
Among the six new antiinfluenza compounds identified, there are four apparent chemical skeletons. We used the four compounds 581, 788 (Nucleozin), 367, and 1075 that have greater antiinfluenza activities for more detailed studies. One of the compounds, 788 (Nucleozin), was recently reported as a potent antiinfluenza inhibitor and was named Nucleozin (16). These compounds markedly reduced the WSN H1N1 viral yields after a one-day treatment at 10 μM, and showed significant yield reduction at 1 μM (Fig. 1B). We explored the inhibition activities of the antiinfluenza compounds using three additional assays: influenza neuraminidase, influenza entry using the pseudotyped virus expressing influenza hemagglutinin, HApp (17), and the cell-based reporter assay for the influenza RdRP (18). Except for the known neuraminidase inhibitors Oseltamivir and Zanamivir, no other compounds are inhibitory to the influenza neuraminidase activity. Similarly, we found none of the new antiinfluenza compounds inhibiting influenza entry at 5 μM or below using the HApp reporting system. The RdRP inhibitory activities of the antiinfluenza compounds were tested using the RdRP reporter assay employing 293 cells transfected with plasmids for the expression of influenza NP, PA, PB1, and PB2 proteins and the luciferase RNA in negative-sense orientation. The presence of functional influenza RNA-dependent RNA polymerase consisting of NP, PA, PB1, and PB2 can transcribe the negative sensed luciferase RNA into mRNA for the synthesis of the reporter luciferase. Fig. 1C shows that compound 581 inhibited the RDRP function almost completely at 10 and 3 μM and was less active at 1 μM. Compound 1061 that is structurally similar to 581 and a weaker antiinfluenza inhibitor is also a weaker inhibitor in the RdRP reporter assay. Compound 788 (Nucleozin), being the most active inhibitor, inhibited the RdRP activity at about 1 μM. Compound 367 is also inhibitory to the RdRP activity, although it is weaker. Finally, 1075 is a potent antiinfluenza compound; however, it is not an inhibitor of RdRP. To gain insights to the mode of action of these inhibitors, we selected inhibitor-resistant WSN viruses by propagating parental WSN virus in media containing increasing contents of these inhibitors. Fit and inhibitor-resistant WSN variants were obtained that are resistant to 788 (Nucleozin), 1075, and 367 (SI Text and Fig. S4) suggesting that these three compounds most likely target influenza encoded gene products.
Antiinfluenza Properties of the 788 (Nucleozin) Analogs with Substituted Isoxazolyl Carbonyl Piperazine Structures.
Since 788 (Nucleozin) is the most potent antiinfluenza compound, more analogs were collected from commercial sources for studies. Table 1 summarizes the antiviral assay results of these analogs against influenza viruses derived from WSN and several other laboratory influenza strains. Among the analogs, 3061 (FA-2) was found to be the most potent compound. The antiinfluenza activities of 3061 (FA-2) and other active analogs are roughly equal when tested against either Oseltamivir sensitive or the resistant WSN viruses that are different at the 274th amino acid of the neuraminidase protein as either the parental 274H or the Oseltamivir-resistant 274Y. Compound 3061 (FA-2) is also active in inhibiting several other tested influenza A strains with varying IC50 values (Table 1). Moreover, we tested ten Taiwan clinical H1N1 isolates that are either sensitive or resistant to Oseltamivir and found that 3061 (FA-2) at 5 μM completely block the replication of these H1N1 strains. In contrast, at similar concentrations, noticeable influenza yield reduction was not observed in the treatment using ribavirin (SI Text and Fig. S5). The results that both Oseltamivir sensitive and Oseltamivir-resistant stains are susceptible to 3061 (FA-2) are consistent with its proposed mode of action at the influenza RNA polymerase. In addition, we showed the in vivo efficacy of 3061 (FA-2) at 2.5 mg/kg for partial protection (P < 0.05) of mice infected with WSN viruses (SI Text and Fig. S6).
Table 1.
Antiinfluenza IC50 values (μM) of 788 (nucleozin) analogs against tested influenza viruses 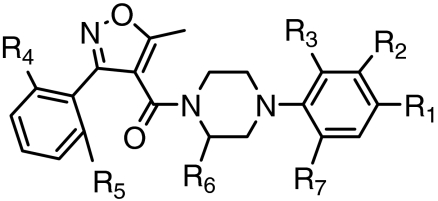
| IC50 against influenza viruses (μM)* |
|||||||||||||||||
| Name |
R1 |
R2 |
R3 |
R4 |
R5 |
R6 |
R7 |
WSN (274H) |
WSN (274Y) |
rWSN (52Y) |
rWSN (52H) |
H1N1 (Ca/07/09) |
H1N1 (Br/59/09) |
H3N2 (Br/10/09) |
H3N2 (Udorn/72) |
H5N1 (RG14) |
CC50†(μM) |
| 788 | NO2 | H | Cl | H | H | H | H | 0.14 | 0.21 | 0.25 | > 30 | 16.90 | 7.80 | 12.50 | 3.50 | 1.2 | > 100 |
| 3061 | NO2 | H | Cl | Cl | H | H | H | 0.07 | 0.14 | 0.13 | > 30 | 4.20 | 1.20 | 8.10 | 0.70 | 0.32 | > 100 |
| 4332 | NO2 | H | Cl | H | H | CH3 | H | 1.30 | 1.30 | 1.64 | > 30 | 8.70 | 3.30 | > 30 | 11.6 | 3.4 | 71 |
| 2130 | Cl | H | NO2 | Cl | H | H | H | 3.50 | 3.50 | 3.40 | > 30 | 3.20 | 1.30 | 28.90 | 9.20 | 18.7 | 77 |
| 3822 | NO2 | H | H | H | H | H | H | 1.20 | 2.60 | 3.00 | > 30 | 24.50 | > 30 | > 30 | > 30 | 4.6 | > 100 |
| 6074 | NO2 | H | H | Cl | Cl | H | H | 5.00 | 5.10 | 3.20 | > 30 | 15.80 | 6.70 | > 30 | > 30 | 18.6 | > 100 |
| 0927 | H | H | Cl | Cl | H | H | NO2 | 14.00 | 13.1 | > 30 | > 30 | 9.20 | > 30 | 21.70 | > 30 | > 30 | 25 |
| 0131 | NO2 | pyrrolidine | H | Cl | H | H | H | 17.50 | 30.00 | > 30 | > 30 | 4.00 | 0.90 | > 30 | > 30 | > 30 | 71 |
| 5614 | Cl | H | NO2 | H | H | H | H | 25.40 | > 30.0 | > 30 | > 30 | 5.50 | 3.00 | > 30 | > 30 | > 30 | 87 |
| 4427 | NO2 | H | H | Cl | H | H | H | 21.00 | 28.70 | > 30 | > 30 | 25.90 | 4.30 | > 30 | > 30 | > 30 | > 100 |
| 9168 | H | H | NO2 | H | H | H | Cl | 19.50 | 24.40 | > 30 | > 30 | 15.40 | 20.80 | > 30 | > 30 | > 30 | 58 |
| 4812 | NO2 | H | H | H | H | H | morpholine | > 30 | > 30 | > 30 | > 30 | 28.00 | 27.30 | > 30 | 28.50 | > 30 | > 100 |
*WSN(274H): A/WSN/1933 (H1N1) with Tamiflu sensitive neuraminidase (274H); WSN(274Y): A/WSN/1933 (H1N1) with Tamiflu resistant neuraminidase (274Y); rWSN (52Y): rWSN virus with wild-type NP (52Y); rWSN (52H): rWSN virus with mutant NP (52H); H1N1(Ca/07/09): A/California/07/2009 H1N1 strain; H1N1(Br/59/07): A/Brisbane/10/2007 H1N1 strains; H3N2(Br/10/09): A/Brisbane/10/2007 H3N2 strains; H3N2(Udorn/72): A/Udorn/1972 H3N2 strain; H5N1 (RG14): the NIBRG14 reassortant strains that harbors the hemagglutinin and neuraminidase genes from A/VietNam/1194/2004, and other influenza genes from PR8 viruses.
†CC50 indicated the concentration needed to inhibit 50% growth of human 293T cells in 48 h.
Antiinfluenza Compound 3061 (FA-2) and 788 (Nucleozin) Target Influenza NP, and the NP Mutation at Y52H Causes Resistance.
We obtained seven 3061 (FA-2)-resistant WSN variants by selections using either 788 (Nucleozin) or 3061 (FA-2) (SI Text and Fig. S4) to study the mode of action of these compounds. All seven independently isolated 3061 (FA-2)-resistant WSN strains carry the same Y52H mutation of NP suggesting that NP may be the target of these compounds. Using reverse genetics, we rescued recombinant influenza viruses, rWSN(52Y) from transfected cells using plasmid constructs expressing all eight parental WSN genes and also rescued its isogenic recombinant virus, rWSN(52H), from similarly transfected cells except the NP construct was replaced with a plasmid for the expression of histidine at the 52nd residue of NP. Unlike the parental recombinant strain, rWSN(52Y), that failed to replicate in the presence of 3061 (FA-2), rWSN(52H) grew equally well with or without the presence of 3061 (FA-2) (Fig. 2A). Our genetic results thus strongly suggest that the antiinfluenza activity of 3061 (FA-2) is most likely targeting the influenza NP. While the resistant mutations found by Kao et al., are at Y289H (16), our WSN resistant mutants carry the Y52H NP mutation, as confirmed by studies on isogenic recombinant viruses. We observed that treatment of influenza infected cells with 3061 (FA-2) blocked the NP synthesis (Fig. 2B), consistent with the efficient 3061 (FA-2) inhibition in the RdRP reporter assay (Fig. 1C). To correlate the role of 3061 (FA-2) in inhibiting influenza replication, we compared the 3061 (FA-2) susceptibility of the reconstituted RdRP activities consisting of either the parental NP (52Y) or the mutant NP (52H) in the assay. Table 2 shows that both 788 (Nucleozin) and 3061 (FA-2) inhibited parental NP reconstituted RdRP with low IC50 values of 0.3 and 0.1 μM respectively. The antiviral IC50 values of the recombinant RdRP reconstituted with the mutant NP (52H) are about 100-fold greater. Parallel to the much increased RdRP resistance due to the mutant NP of 52H, recombinant WSN expressing the mutant NP (52H), are highly resistant to the antiinfluenza activities of either 788 (Nucleozin) or 3061 (FA-2). Consistent with this result is the observation that NP synthesis is completely blocked in infected MDCK cells if 3061 (FA-2) is added at the beginning of influenza infection (Fig. 2B). To examine the effects of 3061 (FA-2) on the NP protein during infection, we treated WSN-infected MDCK cells with 3061 (FA-2) for 2 h, beginning at 6 h postinfection. For the rWSN(52Y) infected cells, 3061 (FA-2) treatment at 6–8 h postinfection resulted in the appearance of distinct NP aggregates in the cytoplasm. In contrast, 3061 (FA-2) treatments of MDCK cells infected with the 3061 (FA-2)-resistant rWSN(52H) virus did not inhibit the NP synthesis nor induce the aggregation of NP protein (Fig. 2B). The distinct responses of the 3061 (FA-2) treatments to the cells that were infected with the isogenic recombinant WSN viruses are striking. NP is a multifunction protein playing several roles in the life cycle of influenza infection. The apparent structural change of the parental NP protein by 3061 (FA-2) treatment could be very destructive to many NP involved functions resulting in the inhibition of the influenza replication. While both results by Kao et al. (16) and this report suggested that NP is the target of nucleozin or 3061 (FA-2), the conclusions were deduced from different NP mutations. We prepared additional NP mutant constructs and showed that similar to the mutational effects of Y52H, reconstituted RdRP with either Y289H or Y52H or double NP mutations are resistant to 3061 (FA-2) (Table 2 and Fig. S7).
Fig. 2.

Inhibition of 3061 (FA-2) to the isogenic recombinant influenza viruses expressing parental and mutant NP proteins. (A) The growth of the recombinant isogenic NP viruses on agar without (top) or with 3 μM 3061 (FA-2) (bottom). (B) Effects of 3061 (FA-2) treatment on NP expressions in MDCK cells were examined at 8 h postinfection with rWSN(52Y) or rWSN(52H) viruses. Treatment in the course of the infection with 3061 (FA-2) blocked the NP synthesis of the rWSN(52Y) infected but not the rWSN(52H) infected MDCK cells (center). Treatment with 3061 (FA-2) at 6–8 h postinfection caused the cytoplasmic aggregation of NP protein of the rWSN(52Y) infected but not the rWSN(52H) infected MDCK cells (right).
Table 2.
The NP mutations results in increased 788/3061 resistance in both RdRP inhibition and antiinfluenza activities
| RdRP IC50 (μM)* | Anti-rWSN IC50 (μM)† | |||||||
| parental | NP 52H | NP 289H | NP 52H/289H | parental | NP 52H | NP 289H | NP 52H/289H | |
| 788 (nucleozin) | 0.3 | 20 | N.D.‡ | N.D. | 0.25 | > 100 | > 100 | > 100 |
| 3061 (FA-2) | 0.1 | 9.3 | > 30 | > 30 | 0.13 | > 30 | > 30 | > 30 |
*Reporter assay using reconstituted RdRP of parental PB1, PB2, PA, and NP. NP can be the parental protein or the one with Y52H or Y289H or double mutants.
†Antiinfluenza activities using recombinant virus in WSN background. NP can be the parental protein or the one with Y52H or Y289H or double mutants.
‡N.D.: Not Determined.
Possible Molecular Mechanism for the Interaction of 788 (Nucleozin) and 3061 (FA-2) with NP.
We then used analytical ultracentrifuge to verify the 3061 (FA-2) induced NP aggregation observed in infected cells (Fig. 2B). As shown in Fig. S8A and SI Text, both compounds cause severe aggregation to the wild-type NP protein. These results clearly suggest that binding of 788 (Nucleozin) or 3061 (FA-2) causes aggregation of NP, and that the binding and its effect probably involve a key residue Tyr52.
To provide a molecular explanation, we modeled the N-terminal region of the NP protein based on the reported NP structure (19). It appears that the regions of the α2 loop, β1, and the β3 sheets are stabilized by the pi-stacking interactions involving Y52, R99, and Y313 residues. We surmise that compounds such as 788 (Nucleozin) or 3061 (FA-2) could insert between the residues R99 and Y52 or Y52 and Y313, disrupt the loop stability, and lead to the formation of oligomeric aggregates (SI Text and Fig. S8B). It is conceivable that inhibitor induced NP structural change could have profound effects in the influenza life cycle. For example, the NP in aggregated form may be unable to enter the nuclei and may prevent the RNP formation for the production of viral particles (16). In addition to having defective nuclear entry, structurally altered NP may have aberrant interaction with PB1, PB2, RNA, and several of its cellular binding partners affecting the mRNA synthetic activity of the RdRP, leading to 3061 (FA-2) inhibition of the RdRP reporter assay and prevention of the NP synthesis in infected cells.
We noticed that the antiviral activities of 3061 (FA-2) and 788 (Nucleozin) are not very potent to several influenza strains, particularly to A/Brisbane/10/2007 (H3N2) and A/California/07/2009 (H1N1) (Table 1). Looking into the NP sequences, we noticed that NP for A/Brisbane/10/2007 (H3N2) has a histidine at the 52nd residue, and that for A/California/07/2009 (H1N1) is 289H NP. The presence of a histidine at either the amino acid 52 or 289 may contribute the greater IC50 values for 3061 (FA-2) or 788 (Nucleozin) against these two influenza strains. We also looked into the NCBI Influenza research database (http://www.fludb.org) on the reported NP sequences at these two amino acid residues. Among the reported 7,757 influenza NP sequences for strains reported in the periods of 2001 to 2010, the majority of the strains (58%) have tyrosine at both AA52 and AA289. About 13% of the strains carry 52H, and 27% of the collected influenza strains have NP sequences with 289H. Only 0.05% of these influenza strains have histidines in both amino acid residues. We recently prepared four recombinant influenza strains in WSN background different only at these two NP residues. All four viruses appear to grow well on MDCK cells (SI Text and Fig. S9) suggesting that replacement of either one or both tyrosines to histidines will not affect the NP functions but will reduce the susceptibility to nucleozin or 3061 (FA-2).
Antiinfluenza Activity of Compound 367 Targeting the Influenza PB1.
We compared the susceptibilities of the 3061 (FA-2)-resistant mutants to 367 and the 367-resistant mutants to 3061 (FA-2) and found that they are not cross-resistant to each other, suggesting that 367 and 3061 (FA-2) probably target different gene products (Fig. 3 A and B). Sequence analysis of a 367 resistant isolate showed the H456P alteration in the PB1 gene. We used reverse genetics to construct a pair of isogenic recombinant influenza viruses differing only at the 456th codon of the PB1 protein. The recombinant WSN with parental PB1 sequence, rWSN(456H), is sensitive to 367 with measured IC50 values between 0.3 to 1 μM. The IC50 value of the recombinant isogenic virus, rWSN(456P), is greater than 100 μM (Fig. 3C). The antiinfluenza activity of the 367 analog, 715, was measured against these two viral strains, and the determined IC50 values were 3 μM and > 100 μm, respectively. The inhibition of 367 to the RdRP reporter assay was also determined and found that the IC50 value of the reconstituted RdRP consisting of the parental 456H PB1 subunit is 5.8 μM. Reconstituted RdRP with the mutant H456P PB1 subunit contributes a sixfold increase in the IC50 value at 36 μM (Fig. 3D). The genetic studies of the 367 resistant viruses suggest that the antiviral activity of 367 is targeting the PB1 gene product, and the inhibition to the viral mRNA transcription contributes to the observed inhibition to influenza replication. We also referenced the NCBI data base for the identified PB1 mutations at the 456th codon. In contrast to the more frequent alterations at the 52nd and the 289th codons of the NP protein, H456P mutation was not found in the 7,653 influenza strains identified in the 2001 to 2010 period.
Fig. 3.

Properties of the antiinfluenza inhibitor 367. (A) The 788 (Nucleozin)-selected WSN variant virus is also resistant to 3061 (FA-2) but is still susceptible to 367. (B) The 367-selected WSN variant is resistant to 367 but does not grow in the presence of 3061 (FA-2). (C) The antiviral dose response curves of 367 for rWSN(456H) influenza with parental PB1 (circle) and the isogenic rWSN(456P) virus with mutant PB1 (triangle) (N = 3). (D) Inhibition measurements at varied 367 concentrations against RdRP activities reconstituted using the parental 456H PB1 (circle) or the mutant 456P PB1 (triangle) (N = 3).
In conclusion, we developed an antiinfluenza screening strategy for a HTS campaign against a large compound library. We further established a high throughout virus yield reduction methodology to confirm hits that are inhibitory to influenza replication. The screening of a large library resulted in the identification of several classes of unique compounds that appear to inhibit influenza at different targets. Two of the identified antiinfluenza compounds inhibit influenza RdRP activities by targeting different subunit proteins. The compound 3061 (FA-2), an analog of the recently reported nucleozin, interacts with the NP, while the other antiinfluenza inhibitor, 367, prevents PB1 functions. Influenza RdRP is an enzyme with multiple enzymatic functions and it interacts with other viral and cellular proteins for the replication and expression of influenza genes (14, 20). It is conceivable that RdRP could be targeted at many different sites. Our HTS campaign has resulted in the identification of two classes of inhibitors that are potent antivirals by targeting the NP and PB1 proteins. Optimization of these compounds could result in the development of new antiinfluenza agents.
Materials and Methods
Compounds.
Compounds 3061 (FA-2) and 788 (Nucleozin) were purchased from ChemDiv. The purity was measured by analytical HPLC and the spectra were recorded at 260 nm. The purities of 3061 and 788 were 96% and 95%, respectively.
Viruses, Cells, and Reagents.
The influenza viruses of A/WSN/1933 (H1N1), A/California/07/2009 (H1N1), A/brisbane/59/2007 (H1N1), A/Brisbane/10/2007 (H3N2), A/Udorn/1972 (H3N2), and reassortant viruses RG14 harboring the hemagglutinin and neuraminidase from A/Viet Nam/1194/2004 (H5N1) were provided by J.-T. Jan (GRC, Academic Sinica). The clinical H1N1 viruses that are either Oseltamivir sensitive or resistant were provided by Centers for Disease Control, Taiwan. The influenza viruses A/WSN/1933 (H1N1) and A/California/07/2009 (H1N1) were cultured in the allantoic cavities of 10-day-old embryonated chicken eggs for 72 h, harvested, and purified by sucrose gradient centrifugation. All other influenza viruses were cultured in MDCK cells. The influenza virus titers were determined by conventional plaque assay and represented as plaque forming units (pfu) (21). MDCK cells were obtained from American Type Culture Collection and were grown in DMEM (Dulbecco’s modified Eagle medium) containing 10% FBS and penicillin-streptomycin at 37 °C under 5% CO2 unless stated otherwise. All cell culture reagents were obtained from Invitrogen Inc.. The antibodies against influenza NP were purchased from Chemicon Inc. and the fluorescein-labeled secondary antibodies were from Sigma. Anti-WSN rabbit antibody was purified from sera of rabbits that were immunized with two injections of formalin-inactivated WSN33 and was used for immuno-detection and entry neutralization studies.
Indirect Immunofluorescence Staining and Confocal Microscopy.
MDCK cells were grown on coverslips and incubated in DMEM containing 2% FBS followed by the infection with influenza virus [A/WSN/1933 (H1N1), moi = 5] for 8 h. Compound 3061 (FA-2) was added to medium at different times: 0 and 6 h after infection. The infected cells were fixed at 8 h postinfection by 4% paraformaldehyde at RT for 60 min and permeability by 0.1% Triton-X100 at RT for 10 min. After fixation, the cells were incubated with NP antibody (Chemicon Inc.) at RT for 1 h and then the DyLight 488-conjugated secondary antibody (Jackson ImmunoReseaarch) in PBS at RT for 1 h. Nuclei were visualized by DAPI. Immunofluorescence images were obtained by using a Leica TCS-SP5 laser scanning confocal microscope (Leica Microsystems GmbH).
Cell-Based Influenza Polymerase Assay.
The plasmids for virus-inducible luciferase pLKOHArFlu and expression plasmid constructs pFluNP, pFluPB1, pFluPB2, pFluPA, for NP, PB1, PB2, and PA expressions (18) were used to transfect 293 cells. For the evaluation of the effects of the NP 52H mutation or the PB1 456P mutation on the RdRP activities, both the NP and the PB1 expressing plasmids were mutagenized employing QuikChange® II XL Site-Directed Mutagenesis Kit from Agilent Technologies using two primers: 5′-caccgaacttaaactcagtgatcatgagggacggc-3′ and 5′-gccgtccctcatgatcactgagtttaagttcggtg-3′ for NP mutagenesis and two primers 5′-tgtgaatgcacccaatcctgaagggattcaagccg-3′ and 5′-cggcttgaatcccttcaggattgggtgcattcaca-3′ for PB1 modification to generate pFluNP52H and pFluPB1456P that were used for the expression of mutant RdRP activities. After being cotransfected with five plasmids, each at 2 μg plasmid DNA, 293T cells were harvested 6 h later by trypsin and reseeded to 96-well plates at 104 cell per well in media added with inhibitors. After incubation for 24 h, the treated cells were assayed for the luciferase activities with BrightGlo® (Promega). Statistical analysis was performed using one-way ANOVA (Prism, Graphpad Software).
Selection, Isolation, and Characterization of Inhibitor-Resistant Influenza Viruses.
MDCK cells were seeded in 6-well plates and infected with 500–1,000 pfu of parental WSN viruses. For the first selection cultures, the infected cells were treated with 0.3 μM of 788 (Nucleozin), 1075, 3061 (FA-2), or 1 μM of 367. After the appearance of cytopathic effects in MDCK cells, 3 μL of the conditioned media were taken to infect fresh MDCK cells for second selection cycle using media containing 1 μM of 788 (Nucleozin), 1075, 3061 (FA-2), or 3 μM of 367. Similarly, threefold higher compound concentrations were used for the third round of selection. Finally, at the fourth selection, 10 μM of 788 (Nucleozin), 1075, 3061 (FA-2) or 30 μM, or 367 were used for the selection of mutants resistant to these inhibitors. The resistant mutant viruses were plaque purified and the inhibitor resistances were confirmed by growth on agars containing varied inhibitor contents. The identified mutants resistant to 788 (Nucleozin), 3061 (FA-2), or 367 were sequenced to determine the presence of sequence alterations at the PA, PB1, PB2, and NP genes. The sequencing primers used cover the sequences of the cDNAs for PB1 at 17-41, 855-836; 741-762, 1824-1785; and 1568-1589, 2288-2264, for PB2 at 55-74, 936-917; 728-747, 1528-1509; 1304-1323, 2296-2272; for PA at 56-77, 838-876; 741-761, 1590-1566; 1368-1391, 2052-2028; 1558-1577, 2178-2154; and for NP at 23-44, 894-870; and 723-743, 1507-1488.
Preparation of Isogenic Recombinant Influenza Viruses.
The procedure to generate recombinant influenza viruses using eight cDNA plasmids (22) was used to produce recombinant influenza pairs isogenic either at the 52nd amino acid of NP or at the 456th amino acid of PB1. Briefly, 5 x 106 human 293 cells were seeded on 30% confluent MDCK cells in Opti-MEM (GIBCO). After an overnight incubation, the cocultured cells were transfected with 10 μg each of the eight plasmid DNA samples and in the presence of TransIT®-LT1 transfection reagent (Mirus Bio Corporation). The transfected cells were replaced with fresh medium after a day and further incubated for 4–5 d at which time the conditioned media were added into fresh MDCK cell cultures for 2 d followed by cloning of the rescued recombinant influenza viruses.
Other Assays Used for Inhibitor Evaluations.
Standard antiviral assay using virus-induced cytopathic effects of MDCK cells was used to evaluate antiviral activities of compounds (23) unless otherwise described. The neuraminidase assay and quantitation of neuraminidase inhibitor strength were done as described (23). The assay to measure the influenza entry was done using pseudotype virus HApp that was produced and assayed as described previously (24). Cytotoxicity assay was performed with human 293T cells and analyzed using Cell-Titer Glo® (Promega) (23).
Supplementary Material
Acknowledgments.
We thank Dr. Jim-Min Fang for neuraminidase inhibitors, Dr. Tamio Saito at RIKEN for natural product compounds, Dr. King-Song Jeng for suggestions to rescue recombinant influenza strains, and Dr. Evan Cromwell for technical supports using Isocyte. We also thank research supports from the National Science Council and Genomics Research Center, Academia Sinica, Taiwan.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1013592107/-/DCSupplemental.
References
- 1.Monto AS. The risk of seasonal and pandemic influenza: prospects for control. Clin Infect Dis. 2009;48(Suppl 1):S20–25. doi: 10.1086/591853. [DOI] [PubMed] [Google Scholar]
- 2.Oxford JS, et al. Treatment of epidemic and pandemic influenza with neuraminidase and M2 proton channel inhibitors. Clin Microbiol Infect. 2003;9:1–14. doi: 10.1046/j.1469-0691.2003.00564.x. [DOI] [PubMed] [Google Scholar]
- 3.Tillack TW, Wong M, Allietta M, Thompson TE. Organization of the glycosphingolipid asialo-GM1 in phosphatidylcholine bilayers. Biochim Biophys Acta. 1982;691:261–273. doi: 10.1016/0005-2736(82)90415-1. [DOI] [PubMed] [Google Scholar]
- 4.Margo KL, Shaughnessy AF. Antiviral drugs in healthy children. Am Fam Physician. 1998;57:1073–1077. [PubMed] [Google Scholar]
- 5.Oxford JS, Mann A, Lambkin R. A designer drug against influenza: the NA inhibitor oseltamivir (Tamiflu) Expert Rev Anti Infe. 2003;1:337–342. [PubMed] [Google Scholar]
- 6.Oxford J, Balasingam S, Lambkin R. A new millennium conundrum: how to use a powerful class of influenza antineuraminidase drugs (NAIs) in the community. J Antimicrob Chemother. 2004;53:133–136. doi: 10.1093/jac/dkh037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monto AS, Arden NH. Implications of viral resistance to amantadine in control of influenza A. Clin Infect Dis. 1992;15:362–367. doi: 10.1093/clinids/15.2.362. discussion 368–369. [DOI] [PubMed] [Google Scholar]
- 8.Deyde VM, et al. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis. 2007;196:249–257. doi: 10.1086/518936. [DOI] [PubMed] [Google Scholar]
- 9.de Jong MD, et al. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N Engl J Med. 2005;353:2667–2672. doi: 10.1056/NEJMoa054512. [DOI] [PubMed] [Google Scholar]
- 10.Lackenby A, et al. Emergence of resistance to oseltamivir among influenza A(H1N1) viruses in Europe. Eurosurveillance. 2008;13:1–2. doi: 10.2807/ese.13.05.08026-en. [DOI] [PubMed] [Google Scholar]
- 11.Hauge SH, Dudman S, Borgen K, Lackenby A, Hungnes O. Oseltamivir-resistant influenza viruses A (H1N1), Norway, 2007–08. Emerg Infect Dis. 2009;15:155–162. doi: 10.3201/eid1502.081031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang JJ, Liao CL, Liao JT, Lee YL, Lin YL. A Japanese encephalitis virus vaccine candidate strain is attenuated by decreasing its interferon antagonistic ability. Vaccine. 2009;27:2746–2754. doi: 10.1016/j.vaccine.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Scalera NM, Mossad SB. The first pandemic of the 21st century: a review of the 2009 pandemic variant influenza A (H1N1) virus. Postgrad Med J. 2009;121:43–47. doi: 10.3810/pgm.2009.09.2051. [DOI] [PubMed] [Google Scholar]
- 14.Das K, Aramini JM, Ma LC, Krug RM, Arnold E. Structures of influenza A proteins and insights into antiviral drug targets. Nat Struct Mol Biol. 2010;17:530–538. doi: 10.1038/nsmb.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong J, et al. Potential targets and their relevant inhibitors in antiinfluenza fields. Curr Med Chem. 2009;16:3716–3739. doi: 10.2174/092986709789104984. [DOI] [PubMed] [Google Scholar]
- 16.Kao RY, et al. Identification of influenza A nucleoprotein as an antiviral target. Nat Biotechnol. 2010;28:600–605. doi: 10.1038/nbt.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang SY, et al. HA-pseudotyped retroviral vectors for influenza antagonist screening. J Biomol Screen. 2009;14:294–302. doi: 10.1177/1087057108330786. [DOI] [PubMed] [Google Scholar]
- 18.Lutz A, Dyall J, Olivo PD, Pekosz A. Virus-inducible reporter genes as a tool for detecting and quantifying influenza A virus replication. J Virol Methods. 2005;126:13–20. doi: 10.1016/j.jviromet.2005.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye Q, Krug RM, Tao YJ. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature. 2006;444:1078–1082. doi: 10.1038/nature05379. [DOI] [PubMed] [Google Scholar]
- 20.Ruigrok RW, Crepin T, Hart DJ, Cusack S. Towards an atomic resolution understanding of the influenza virus replication machinery. Curr Opin Struct Biol. 2010;20:104–113. doi: 10.1016/j.sbi.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 21.Palese P, Tobita K, Ueda M, Compans RW. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology. 1974;61:397–410. doi: 10.1016/0042-6822(74)90276-1. [DOI] [PubMed] [Google Scholar]
- 22.Neumann G, et al. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci USA. 1999;96:9345–9350. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shie JJ, et al. Synthesis of tamiflu and its phosphonate congeners possessing potent anti-influenza activity. J Am Chem Soc. 2007;129:11892–11893. doi: 10.1021/ja073992i. [DOI] [PubMed] [Google Scholar]
- 24.Su CY, et al. In vitro evaluation of neuraminidase inhibitors using the neuraminidase-dependent release assay of hemagglutinin-pseudotyped viruses. Antiviral Res. 2008;79:199–205. doi: 10.1016/j.antiviral.2008.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.