

Abstract
The polycomb repressive complex 2 (PRC2) is the major methyltransferase for H3K27 methylation, a modification critical for maintaining repressed gene expression programs throughout development. It has been previously shown that PRC2 maintains histone methylation patterns during DNA replication in part through its ability to bind to H3K27me3. However, the mechanism by which PRC2 recognizes H3K27me3 is unclear. Here we show that the WD40 domain of EED, a PRC2 component, is a methyllysine histone-binding domain. The crystal structures of apo-EED and EED in complex respectively with five different trimethyllysine histone peptides reveal that EED binds these peptides via the top face of its β-propeller architecture. The ammonium group of the trimethyllysine is accommodated by an aromatic cage formed by three aromatic residues, while its aliphatic chain is flanked by a fourth aromatic residue. Our structural data provide an explanation for the preferential recognition of the Ala-Arg-Lys-Ser motif-containing trimethylated H3K27, H3K9, and H1K26 marks by EED over lower methylation states and other histone methyllysine marks. More importantly, we found that binding of different histone marks by EED differentially regulates the activity and specificity of PRC2. Whereas the H3K27me3 mark stimulates the histone methyltransferase activity of PRC2, the H1K26me3 mark inhibits PRC2 methyltransferase activity on the nucleosome. Moreover, H1K26me3 binding switches the specificity of PRC2 from methylating H3K27 to EED. In addition to determining the molecular basis of EED-methyllysine recognition, our work provides the biochemical characterization of how the activity of a histone methyltransferase is oppositely regulated by two histone marks.
Keywords: methyllysine-binding domain, WD40 repeat-containing protein, X-ray crystallography
The polycomb (PcG) and trithorax groups of proteins act in concert to maintain the transcriptional state of developmental control genes such as the Hox genes. These patterns are established during early embryonic development and are heritable through many generations of cell division (1–3). Polycomb group proteins are transcriptional repressors that maintain target genes in an inactive state, whereas trithorax group proteins are transcriptional activators that maintain their target genes in an active state. This system of cellular memory is conserved in nearly all multicellular eukaryotes (4).
PcG proteins often function as complexes, and the two best-characterized complexes are the polycomb repressive complex 1 (PRC1) and 2 (PRC2). PRC2 exhibits histone methyltransferase activity on H3K27 (2, 5–7) and weakly on H1K26 (8). Studies in Drosophila have shown that PRC1 contains a chromodomain protein, polycomb, which preferentially recognizes the histone H3K27me3 mark (9, 10). One hypothesis is that H3K27 methylation by PRC2 creates a binding site recognized by the chromodomain of polycomb of PRC1, which in turn maintains transcriptional silencing through an unknown mechanism (11). In addition to silencing Hox genes, the polycomb group complexes are also involved in X-inactivation, germ-line development, stem cell pluripotency and differentiation, and cancer metastasis (2).
PRC2 complex contains four core components: EZH2, EED, SUZ12, and RbAp46/48. The catalytic activity of PRC2 is conferred by the suppressor of variegation [Su(var)3-9], enhancer of zeste [E(z)], and trithorax (SET) domain in EZH2, but PRC2 exhibits robust methylation activity only as a complex because in vitro enzymatic assays indicate that EZH2 has virtually no histone methylation activity on its own (6). Biochemical and genetic studies suggest that both EED and SUZ12 are required for the integrity of PRC2 and PRC2-mediated H3K27 methylation because mutations in either protein lead to EZH2 to destabilization and deficient methylation of H3K27 (12–14).
EED contains seven WD40 repeats at its C terminus (residues 81–441) preceded by a small N-terminal domain (residues 1–80). Studies of the Drosophila EED orthologue extra sex combs (ESC) revealed that the N-terminal region interacts directly with the core domain of histone H3 and that this interaction is essential for E(Z)-dependent trimethylation of H3K27 (15). Furthermore, although an N-terminally truncated ESC can form a complex with E(Z) when expressed in stable S2 cell lines, this complex cannot carry out trimethylation of histone H3 (15). A similar mechanism may regulate PRC2 function in vertebrates because the ESC/EED–histone H3 interaction is evolutionarily conserved (15).
The WD40 repeat is a structural motif of approximately 40 amino acids, which forms a four-stranded antiparallel β-sheet and often contains a Gly-His dipeptide at the end of the fourth strand and a Trp-Asp dipeptide at the end of the third strand (16). Most WD40 repeat proteins contain seven or eight WD40 repeats and adopt a β-propeller architecture. WD40 proteins are involved in diverse functions such as transcription regulation, signal transduction, vesicular trafficking, cytoskeletal assembly, cell cycle control, and apoptosis (16). A common function of these WD40 repeat proteins is to serve as a scaffold for protein–protein or protein–DNA interactions and to coordinate downstream events. Since the first WD40 repeat structure Gβ was solved (17, 18), tens of WD40 structures have been reported, which, together, show that the WD40 β-propeller is capable of recognizing a diverse group of ligands, including proteins (17, 18), phosphopeptides (19–21), and nucleic acids (22), mainly through the small top surface. For example, WDR5 uses the top surface of its β-propeller to recognize histone H3K4 peptide (23–26) or a mixed lineage leukemia (MLL) fragment (27–29). WDR5 recognizes a peptide through an arginine residue that inserts into the central pore of the β-propeller and anchors the peptides into a shallow groove at its top face.
WD40 proteins can also bind their ligands through the larger bottom surface. For example, the structure of the WD40 repeat domain of EED in complex with an N-terminal fragment of EZH2 revealed that the peptide bound to the WD40 repeat domain of EED on the larger bottom surface (Fig. 1A) (30). Mutations in the WD40 region of the ESC perturbed its binding to E(Z) in vitro and abolished its silencing function in vivo (31).
Fig. 1.

Preferential binding of EED to trimethylated lysine histone peptides. (A) The top surface of EED harbors an aromatic cage consisting of Phe97, Tyr148, Trp364, and Tyr365, distinct from the EZH2-binding site shown in the crystal structure of EED in complex with an EZH2 fragment (30). The aromatic cage residues on the top surface of EED are displayed in a stick model. EED and the EZH2 peptide are colored in green and blue, respectively. (B) Binding of different trimethylated histone peptides to EED measured by fluorescence polarization. (C) Tabulated binding affinities (Kd) of different histone peptides for EED, measured by fluorescence polarization binding assays. The target lysines are colored in red. NB, no detectable binding; ND, not determined.
Recently, it was shown that the EZH2–EED–SUZ12 trimeric PRC2 complex binds to and colocalizes with the H3K27me3 mark at sites of active DNA replication and in the G1 phase of a cell cycle (32). However, it is unclear which component is responsible for recognizing the H3K27me3 mark. An examination of the top surface of the EED structure revealed an aromatic cage consisting of residues Phe97, Tyr148, Trp364, and Tyr365 (Fig. 1A). An aromatic cage is a common feature of methyllysine-binding motifs (33) such as the chromodomain (9, 10, 34, 35) and the malignant brain tumor repeats (36, 37). This finding led us to propose that EED may possess a methyllysine-binding activity that is important for PRC2 function. Here, we provide evidence in support of this notion. Specifically, we show that the WD40 domain of EED is a histone methyllysine-binding motif that preferentially recognizes Ala-Arg-Lys-Ser (ARKS) motif-containing trimethylated H3K27, H3K9, and H1K26 peptides and that the nucleosome methylation activity of PRC2 is enhanced by binding to the H3K27me3 histone peptide via EED but inhibited by H1K26me3. We further investigated the molecular basis for the selective binding of EED to trimethylated lysine histones by determining the crystal structure of apo-EED and its complexes with five trimethylated histone peptides, respectively.
Results and Discussion
The WD40 Repeat Domain of EED Selectively Recognizes Trimethylated Histone Peptides.
PRC2 methylates histone lysine residues, mainly on H3K27 (2, 5–7), and also binds to the H3K27me3 mark (32). Efficient binding requires a ternary complex of EZH2, EED, and SUZ12 but is independent of the catalytic SET domain of EZH2 (32). However, which component in PRC2 is responsible for binding to H3K27me3 is unclear. Aided by the crystal structure of EED in complex with a short N-terminal fragment of EZH2 (30), we identified an aromatic cage on the top face of the EED β-propeller structure (Fig. 1A), raising the possibility that EED may mediate direct binding of PRC2 to H3K27me3 through this aromatic cage. To test this hypothesis, we measured the binding of EED to a panel of peptides that represented the histone marks H3K4, H3K9, H3K27, H3K36, H3K79, H4K20, and H1K26 in different methylation states (Fig. 1 B and C). We found that EED preferentially bound trimethylated histone peptides, with higher affinities to the H3K9me3, H1K26me3, and H3K27me3 peptides than the other trimethylated histone peptides. Our EED-histone peptide binding data agree with the result of Margueron et al. that was published while this manuscript was under review (38).
H3K27me3 Stimulates Whereas H1K26me3 Inhibits the Methyltransferase Activity of PRC2 on Nucleosomal Histone H3K27.
The binding of EED to trimethylated lysine marks on histones suggests a mechanism for PRC2 to propagate and spread the H3K27me3 mark to daughter strands during cell division. To investigate how methylated histones may regulate the function of PRC2, we measured its histone methyltransferase activity on nucleosome in the presence of different histone peptides. Our results showed that the H3K27me3 peptide stimulated the enzymatic activity of PRC2 by approximately three folds (Fig. 2A). These data are consistent with a recent report showing that H3K27me3 peptide stimulates the PRC2 activity (38). With the exception of H1K26 peptide, we did not observe any significant effects of other peptides on the activity of PRC2. Addition of the H1K26me3 peptide into the reaction inhibited nucleosomal histone H3K27 methylation (Fig. 2A). Although histone H1 inhibition of PRC2 methyltransferase activity on nucleosome was previously shown (8), the strong inhibitory effect of a methyllysine peptide on PRC2 activity has not been reported to date. To investigate whether the methylation status of H1K26 affects the inhibitory activity, we carried out the in vitro methylation assay in the presence of the H1K26 peptide with varying methylation states. Interestingly, H1K26 peptides of lower methylation states exhibited a weaker inhibitory effect compared to the H1K26me3 peptide (Fig. 2B). PRC2 has been shown to methylate H1K26 weakly (8). Presumably, H1K26 will exhibit full inhibition effect only when it is trimethylated because lower methylated H1K26 peptides have much weaker binding affinity for EED (38).
Fig. 2.

The H3K27me3 peptide specifically stimulates, but the H1K26me3 peptide inhibits, the activity of PRC2. (A) Effects of different histone peptides on the activity of PRC2 on histone H3 methylation. Top is an autoradiograph of the Middle (Coomassie blue staining). Quantification of two independent experiments is shown in Bottom. Error bars are standard deviations of the two experiments. (B) Effects of different methylation states of the H1K26 peptide on the methylation activity of PRC2. (C) The H1K26me3 peptide containing an A24E mutation lost its ability to inhibit PRC2 HMT activity on nucleosome.
To further investigate whether the inhibitory effect of H1K26me3 is mediated by its direct binding to EED, we carried out a peptide competition assay. Our results show that the H1K26me3 peptide binds to EED directly and that the binding can be competed off by the H3K27me3 peptide, suggesting that H1K26me3 and H3K27me3 bind to the same site on EED (Fig. 1C and Fig. S1). We next investigated whether impairment of H1K26me3 binding to EED affects the inhibitory effect of PRC2 complex on nucleosome. On the basis of our EED–histone peptide complex structures, which will be discussed later, the residue at the P-2 position relative to the methyllysine at the histone peptides is important for complex formation. Thus, an Ala to Glu mutation at the P-2 position in the conserved ARKS motif of H3K27me3, H3K9me3, and H1K26me3 would impair its binding to EED, which is confirmed by our isothermal titration calorimetry (ITC) and peptide competition assays (Fig. S1 B and C). Consistent with its loss of binding, the H1K26me3A24E peptide was unable to inhibit PRC2 activity (Fig. 2C). Taken together, these data clearly demonstrate that H1K26me3 inhibition of PRC2 activity on nucleosome is conferred by its direct binding to EED.
It is therefore likely that, in vivo, the activity of PRC2 is regulated by both the H3K27me3 and the H1K26me3 marks that play opposing roles. This finding suggests a mechanism for dynamic control of histone methylation in that the recruitment of PRC2 by H3K27me3 enhances its activity and allows for propagation of the H3K27me3 mark to sister chromatins during a cell division, whereas recruitment of the PRC2 complex to the H1K26me3 mark shuts down the methylation activity toward core histones.
Whereas the addition of the H1K26me3 peptide into the in vitro methylation reaction inhibited nucleosomal H3 methylation, it promoted methylation of another protein of approximately 50 kDa (Fig. S2A). This protein was subsequently identified as EED by liquid chromatography MS/MS. To further characterize the methylation of EED, we used multiple reaction monitoring mass spectrometry to identify the specific sites that are methylated. We checked all 32 lysines in EED and obtained high quality data on 23 lysines, of which K66, K197, K268, and K284 were methylated (Fig. S3). Moreover, all three methylation states were detected for these four sites, and the most abundant methylation states are shown in Fig. S3. The identities of these methyl peptides were confirmed by both multiple reaction monitoring and the corresponding MS/MS spectra collected in information-dependent acquisition mode. Of these methylation sites identified in EED, the K66 site is located in a sequence motif of Gly-Arg-Lys-Ser that resembles the H3K27 sequence, ARKS. However, mutation of K66 to an alanine did not abolish the methylation of EED (Fig. S2B). Given the multiple sites that could be methylated in EED, this result is not unexpected. Therefore, our data indicate that, in contrast to H3K27me3 and other histone peptides, the H1K26me3 peptide changes the substrate preference of PRC2.
Overall Structure of Apo-EED and EED-H3K27me3.
To investigate the molecular basis for the selective binding of EED to trimethylated histone peptides, we determined the crystal structure of apo-EED and its complex with the H3K27me3 peptide (Fig. 3). As shown previously (30), the WD40 repeat domain of EED folds into a seven-blade β-propeller structure (Fig. 3A). The seven blades are symmetrically arranged around a pseudosymmetry axis, and a narrow channel runs through the middle of the β-propeller structure. The crystal structures of apo-EED and the EED–H3K27me3 peptide complex were almost identical, with an rmsd of ∼0.2 Å for all aligned  atoms. As predicted from the structural analysis, EED utilizes the smaller top surface of the β-propeller to recognize H3K27me3. Since the first WD40 repeat structure Gβ was solved (17, 18), many WD40–ligand complex structures have been determined, most of which use the smaller top surface to accommodate a ligand with a small interface area (Figs. S4 and S5).
atoms. As predicted from the structural analysis, EED utilizes the smaller top surface of the β-propeller to recognize H3K27me3. Since the first WD40 repeat structure Gβ was solved (17, 18), many WD40–ligand complex structures have been determined, most of which use the smaller top surface to accommodate a ligand with a small interface area (Figs. S4 and S5).
Fig. 3.

Structure of EED in complex with a trimethylated H3K27 peptide. (A) Top view of the EED WD40 domain in complex with a trimethylated H3K27 peptide. EED is shown in a cartoon representation and colored green, and the H3K27me3 peptide is shown in a stick model. (B) The complex structure of EED and H3K27me3. EED is shown in a surface representation with positive electrostatic potential denoted in blue and negative potential by red. The H3K27me3 peptide is depicted in a stick model. (C) Detailed view of intermolecular interactions between EED and H3K27me3. EED residues in contact with the H3K27me3 peptide are shown in sticks. Hydrogen bonds are marked with black dashed lines. (D) Superposition of the crystal structures of EED in complex with different trimethylated histone peptides. Methyllysines are shown in a stick model. (E) Top view of WDR5 in complex with an H3K4me2 peptide (23). The H3K4me2 peptide and the H3R2-binding residues in WDR5 are shown in stick models. Hydrogen bonds are shown in red dashed lines.
The histone-binding mode of EED is reminiscent of WDR5, which also binds histone and other arginine-containing peptides through its top surface (23–29). WDR5 binds histone and MLL peptides mainly via an arginine residue of the peptide that acts like a pin and inserts into the central pore of the β-propeller to form a large network of hydrogen bonds and cation-π interactions with WDR5 (23) (Fig. 3E). In the EED–H3K27me3 peptide complex structure, the histone peptide is anchored to EED through the trimethyllysine, which is accommodated by a binding pocket formed by four aromatic residues (Fig. 3 B and C). The side chain of the trimethylated lysine is inserted into an aromatic cage formed by F97, Y148, and Y365. The aromatic rings of these three residues make close contact and are oriented roughly perpendicular to each other, forming the three sides of the methyllysine-binding pocket and establishing van der Waals and cation-π interactions with the trimethylated ammonium group. The extended aliphatic chain of the methyllysine lies against the planar ring of W364, further enhancing the hydrophobic interaction between the protein and the methylated lysine. This interaction mode is a unique methyllysine-binding feature for EED. The four aromatic residues identified in the EED–H3K27me3 complex are highly conserved in EED orthologues (Fig. S6). The trimethyllysine-binding mode of EED is similar to that of other classical trimethylated histone-binding proteins, such as HP1 and polycomb (9, 10, 34, 35).
The importance of the methyllysine-binding residues in EED was verified by subsequent mutagenesis experiments. Fluorescence polarization measurements showed that mutating F97, Y148, W364, or Y365 to alanine, or W364 to leucine, completely abolished binding of the resulting EED mutant to the H3K27me3 peptide (Fig. S7A). We next reconstituted the PRC2 complex with the EED mutant W364A or Y365A and examined its histone lysine methyltransferase (HKMT) activity. The activity of the reconstituted PRC2 was severely diminished when these methyllysine-binding residues in EED were mutated (Fig. S7B). Besides providing biochemical evidence to support the structure, these data suggest that the function of PRC2 is dependent on EED, in particular its WD40 repeats. Our data agree with that obtained for the Drosophila EED orthologue, ESC (31), in which certain point mutations in the WD40 repeats of ESC perturbs its binding to E(Z) (31).
In addition to the interactions of methyllysine with the aromatic cage, other interactions may also help stabilize the complex. These include a backbone hydrogen bond between Trp364 in EED and H3R26 in the peptide, a hydrogen bond between the side chain of EED Trp364 and the backbone of H3A25, and the side chain of EED Arg414 and the backbone of methyllysine (Fig. 3C). Interestingly, Arg414 is absolutely conserved in all orthologues of EED and forms a salt bridge with another conserved residue Asp430 (Fig. 3C). The Arg414–Asp430 pair may therefore play an important role in binding methyllysine. Residues C-terminal to the methyllysine in the peptide form virtually no interactions with the protein, which explains why EED binds its ligands more weakly (Kd: 108 μM with H3K27me3) than do the HP1 chromodomain (Kd: 4 μM with H3K9me3) and the JMJD2A tudor domains (Kd: 0.4 μM with H3K4me3) to their respective peptide ligands (10, 39).
Preferential Recognition of the ARKme3S Motif by EED.
Besides the complex structure of EED with the H3K27me3 peptide, we also determined the crystal structures of EED complexed with the H3K4me3, H3K9me3, H3K79me3, and H4K20me3 peptides, respectively. In all five complex structures, the backbone orientations of the peptides are almost identical, suggesting that the interaction of methyllysine with the aromatic cage drives the complex formation (Fig. 3D). However, the five histone peptides bind to EED with varying affinities, and EED preferentially binds H3K9me3, H1K26me3, and H3K27me3, which share a conserved ARKS sequence motif.
We compared the five EED complex structures to understand the structural basis for the preferential binding of the H3K27me3, H1K26me3, and H3K9me3 marks by EED. This comparative analysis identified two residues preceding the methyllysine in a peptide (referred to as the P-1 and P-2 positions) as key determinants of such specificity. In the EED-H3K27me3 complex, the P-2 residue alanine (H3A25) is accommodated in a relatively hydrophobic and shallow pocket consisting of Y308, C324, and W364 (Fig. 3C). H3K9me3 also contains an alanine residue (H3A7) at the same position, so this binding mode is conserved in the EED–H3K9me3 complex (Fig. S8A). In contrast, a histidine residue is found at the corresponding position in the H4K20me3 peptide. The histidine side chain would be too bulky to be inserted into the P-2 pocket without causing significant steric hindrance. Indeed, in the corresponding crystal structure, this histidine residue (H3H18) points to the solvent and makes a hydrogen bond with the backbone carbonyl oxygen of D362 in EED (Fig. S8B). This alternative mode of P-2 recognition could explain why the H4K20me3 peptide has comparable binding affinity to EED. In the EED–H3K79me3 complex structure, the corresponding residue Asp (H3D77) is disordered, suggesting that it did not contribute significantly to binding (Fig. S8C). In the case of the H3K4me3 peptide, the long side chain of the H3R2 could not be accommodated by the shallow binding pocket and is also disordered (Fig. S8D).
In all the EED–peptide complexes, the peptide residue immediately preceding the methyllysine (P-1 position) protrudes out of the binding interface and points to the solvent. Therefore, a hydrophobic residue at this position would not be favored energetically, which is the case for H3K36me3 (V35 at P-1) and H3K79me3 (F78 at P-1). Together, these structures explain why EED preferentially binds H3K9me3, H3K27me3, and H1K26me3 marks.
Implication of EED-H3K27me3 Binding in the Transmission of the H3K27me3 Epigenetic Mark.
On the basis of our binding assays and structural studies, we conclude that (i) EED binds to multiple trimethylated histone peptides, but preferably to repressive marks, (ii) the recognition of the methyllysine residue by the aromatic cage plays a key role in the formation of an EED–methyllysine peptide complex, (iii) residues at the P-1 and P-2 positions relative to the methyllysine in a peptide are responsible for the differential binding affinities, and (iv) the H3K27me3, H3K9me3, and H1K26me3 peptides all bind EED with comparable affinities but exhibit different outcomes: The H3K27me3 peptide stimulates PRC2 HKMT activity, H1K26me3 inhibits PRC2 HKMT activity, and H3K9me3 has no effect.
It has been proposed that H3K27me3 binding to PRC2 introduces a conformational change in PRC2 and activates the PRC2 HKMT activity (38). It is likely that H1K26me3 changes the conformation of PRC2 in a different way that makes EED a preferred substrate for PRC2, whereas H3K9me3 binding to PRC2 does not change its conformation significantly. Furthermore, H3K9 is not a substrate of PRC2, and, even though H3K9me3 can localize with PRC2 in cells, it does not form a feedback loop that helps spread the H3K9me3 mark. The structures of the holoenzyme in complex with these peptides would help clarify the distinct effects of different histone peptides on PRC2.
The ability of EED to bind to the product of PRC2 (H3K27me3) provides an effective means for the propagation and/or spreading of the H3K27me3 repressive mark. In quiescent cells, recruitment of PRC2 via EED to an H3K27me3 mark may help propagate the mark to neighboring nucleosomes. In postmototic cells, targeting of PRC2 to the H3K27me3 mark associated with the parent DNA strand may help spread this mark to the nascent sister strand. This mode of dual function for a methyltransferase or coexistence of catalytic and effector functions in the same protein complex is not without precedent. For instance, Clr4 is a histone H3K9 methyltransferase (40, 41) that also contains a chromodomain at its N terminus, which binds specifically to the H3K9me3 mark it creates. The ability of Clr4 to generate and bind H3K9me3 is essential for the spreading of heterochromatin silencing (42). Similar modes of regulation may underlie the function of other histone-modifying enzymes.
In summary, our work identifies EED as a central player in PRC2 function. The unique ability of EED to bind multiple histone marks, which leads to distinct outcomes, may play a pivotal role in controlling the activity of PRC2 and its function in propagating and spreading repressive histone marks in the nucleus. By mediating a series of interactions—binding the histone H3 core domain via the N-terminal fragment, the N-terminal motif of EZH2 via the bottom surface, and the H3K27me3/H1K26me3 mark via the top surface of the EED WD40 domain—EED may function as an adaptor and/or conformational switch that controls the integrity, targeting, activity, and specificity of PRC2. It should be noted, however, that the affinity of EED for H3K27me3 is significantly weaker than that of HP1 and Pc chromodomains for the H3K9me3 or H3K27me3 peptide. The recruitment of the PRC2 complex to the correct chromatin location must therefore involve multiple sites of EED and, likely, multiple components of PRC2. Indeed, it has been shown that all components of PRC2 are required for optimal binding (32). In addition, it has been shown that RbAp46/46 is capable of binding the first helix of histone H4 (43–45).
Methods
Protein Expression and Purification.
Two fragments of human EED (amino acids 40–441 and 76–441) covering the WD40 repeats were subcloned into a modified pET28GST-LIC vector. The recombinant proteins were overexpressed at 16 °C in Escherichia coli BL21 (DE3) Codon plus RIL (Stratagene) as N-terminal GST-tagged fusion proteins and were purified by affinity chromatography on glutathione-sepharose (GE Healthcare). After cleavage using thrombin (Sigma Aldrich) on the column at room temperature for 4–5 h, the flow-through was collected and purified further by size exclusion chromatography (Superdex 200; GE Healthcare). The proteins were concentrated to 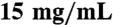 in a buffer containing 20 mM Tris-HCl, pH 7.5, 0.2 M NaCl, and 1 mM DTT. Mutated EED cDNAs were made by using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene); mutations were confirmed by sequencing complete cDNAs. Mutated proteins were expressed in bacterial cells and purified as above.
in a buffer containing 20 mM Tris-HCl, pH 7.5, 0.2 M NaCl, and 1 mM DTT. Mutated EED cDNAs were made by using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene); mutations were confirmed by sequencing complete cDNAs. Mutated proteins were expressed in bacterial cells and purified as above.
Histone Methyltransferase Assay.
Lentiviral vectors encoding wild-type and mutant FLAG-EED were transfected into a 293T cell by using Effectene reagents (Qiagen). Then 48 h after transfect ion, cells were harvested and lysed in nondenaturing lysis buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM EDTA) at 4 °C for 1 h. After brief centrifugation (16,100 × g for 10 min), cell lysates were subjected to immunoprecipitate with anti-FLAG M2 agarose beads (Sigma) at 4 °C for 3 h. PRC2 complexes were eluted from the beads with 0.4 mg/mL FLAG peptide (0.4 mg/mL) and dialysed against BC50 before used for the histone methyltransferase assay. The histone methyltransferase assay was performed as described previously (46). Briefly, affinity purified PRC2 samples were incubated with 10 μg of oligonucleosome or mononuclesomes prepared from a HeLa-S3 cell (47), 1 μL 3H-S-adenosylmethionine (NEN Life Science Products), and histone H3 peptides (40 mM) in histone methyltransferase buffer (20 mM Tris-HCl, pH 8.0, 4 mM EDTA, 1 mM PMSF, 0.5 mM DTT) at 30 °C for 1 h. The reaction products were resolved on 15% SDS-PAGE. After staining and destaining, gels were treated with 3H enhancer (En3hance; Perkin Elmer), dried, and exposed to X-film for 1–3 d.
Crystallization and Structure Determination.
Purified protein EED (40–441) was mixed with histone trimethylated peptides by directly adding a twofold molar excess of peptide to the protein solution and crystallized by using the sitting drop vapor diffusion method at 18 °C. The complex of GST-tag cleaved EED and H4K20me3 peptide was crystallized in a buffer containing 3.5 M sodium formate, 10 mM Tris(2-carboxyethyl)phosphine (TCEP) chloride. For the other complexes, EED (76–441) was mixed with the histone peptides by directly adding a twofold molar excess of peptides to the protein solution. All those complexes were crystallized by using the sitting drop vapor diffusion method at 18 °C and in a buffer containing 0.1 M 1,3-bis[tris(hydroxymethyl)methylamino]propane, 3.5 M sodium formate, 10 mM TCEP, and 15–20% glycerol. Prior to flash-freezing the crystals in liquid nitrogen, the crystals were soaked in a cryoprotectant consisting of 100% reservoir solution and 20% glycerol.
X-ray diffraction data were collected at 100 K by using the Rigaku FRE High Brilliance X-Ray Generator with R-AXIS IV detector and Canadian Light Source. Data were processed by using the HKL software package (48). All structures were solved by molecular replacement by using the program MOLREP (49). The crystal structure of EED and EZH2 peptide was used as the search model (Protein Data Bank ID code 2QXV). ARP/wARP was used for automatic model building (50). Graphics program COOT (51) was used for model building and visualization. Crystal diffraction data and refinement statistics for these structures are displayed in Table S1.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Alexey Bochkarev and Guillermo Senisterra for advice and technical assistance and Yanming Wang, Aled Edwards, and Cheryl Arrowsmith for critically discussing and reading the manuscript. This work was supported by the Structural Genomics Consortium, a registered charity (1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, The Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research, and the Wellcome Trust. H.W. is a Sidney Kimmel Scholar and is supported by National Institutes of Health Grant GM081489. S.S.-C.L. holds a Canada Research Chair in Functional Genomics and Cellular Proteomics. Open access publication costs were defrayed by the Ontario Genomics Institute Genomics Publication Fund.
Note.
While this manuscript was under consideration, R. Margueron and colleagues published similar work (38).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3JZN, 3K26, 3K27, 3JZG, 3JZH, and 3JPX).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1008937107/-/DCSupplemental.
References
- 1.Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet. 2007;8:9–22. doi: 10.1038/nrg1981. [DOI] [PubMed] [Google Scholar]
- 2.Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev. 2004;14:155–164. doi: 10.1016/j.gde.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Martin C, Zhang Y. Mechanisms of epigenetic inheritance. Curr Opin Cell Biol. 2007;19:266–272. doi: 10.1016/j.ceb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Pirrotta V, Gross DS. Epigenetic silencing mechanisms in budding yeast and fruit fly: Different paths, same destinations. Mol Cell. 2005;18:395–398. doi: 10.1016/j.molcel.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Czermin B, et al. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–196. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 6.Muller J, et al. Histone methyltransferase activity of a Drosophila polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 7.Cao R, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 8.Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell. 2004;14:183–193. doi: 10.1016/s1097-2765(04)00185-6. [DOI] [PubMed] [Google Scholar]
- 9.Min J, Zhang Y, Xu RM. Structural basis for specific binding of polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003;17:1823–1828. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischle W, et al. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, et al. Hierarchical recruitment of polycomb group silencing complexes. Mol Cell. 2004;14:637–646. doi: 10.1016/j.molcel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Montgomery ND, et al. The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation. Curr Biol. 2005;15:942–947. doi: 10.1016/j.cub.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 13.Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004;23:4061–4071. doi: 10.1038/sj.emboj.7600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. 2004;15:57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 15.Tie F, Stratton CA, Kurzhals RL, Harte PJ. The N terminusof Drosophila ESC binds directly to histone H3 and is required for E(Z)-dependent trimethylation of H3 lysine 27. Mol Cell Biol. 2007;27:2014–2026. doi: 10.1128/MCB.01822-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith TF, Gaitatzes C, Saxena K, Neer EJ. The WD repeat: A common architecture for diverse functions. Trends Biochem Sci. 1999;24:181–185. doi: 10.1016/s0968-0004(99)01384-5. [DOI] [PubMed] [Google Scholar]
- 17.Lambright DG, et al. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 18.Wall MA, et al. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 19.Nash P, et al. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 20.Wu G, et al. Structure of a beta-TrCP1-Skp1-beta-catenin complex: Destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell. 2003;11:1445–1456. doi: 10.1016/s1097-2765(03)00234-x. [DOI] [PubMed] [Google Scholar]
- 21.Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell. 2003;112:243–256. doi: 10.1016/s0092-8674(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 22.Scrima A, et al. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135:1213–1223. doi: 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuetz A, et al. Structural basis for molecular recognition and presentation of histone H3 by WDR5. EMBO J. 2006;25:4245–4252. doi: 10.1038/sj.emboj.7601316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han Z, et al. Structural basis for the specific recognition of methylated histone H3 lysine 4 by the WD-40 protein WDR5. Mol Cell. 2006;22:137–144. doi: 10.1016/j.molcel.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 25.Ruthenburg AJ, et al. Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat Struct Mol Biol. 2006;13:704–712. doi: 10.1038/nsmb1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Couture JF, Collazo E, Trievel RC. Molecular recognition of histone H3 by the WD40 protein WDR5. Nat Struct Mol Biol. 2006;13:698–703. doi: 10.1038/nsmb1116. [DOI] [PubMed] [Google Scholar]
- 27.Song JJ, Kingston RE. WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J Biol Chem. 2008;283:35258–35264. doi: 10.1074/jbc.M806900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel A, Dharmarajan V, Cosgrove MS. Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J Biol Chem. 2008;283:32158–32161. doi: 10.1074/jbc.C800164200. [DOI] [PubMed] [Google Scholar]
- 29.Patel A, Vought VE, Dharmarajan V, Cosgrove MS. A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J Biol Chem. 2008;283:32162–32175. doi: 10.1074/jbc.M806317200. [DOI] [PubMed] [Google Scholar]
- 30.Han Z, et al. Structural basis of EZH2 recognition by EED. Structure. 2007;15:1306–1315. doi: 10.1016/j.str.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Tie F, Furuyama T, Harte PJ. The Drosophila polycomb group proteins ESC and E(Z) bind directly to each other and co-localize at multiple chromosomal sites. Development. 1998;125:3483–3496. doi: 10.1242/dev.125.17.3483. [DOI] [PubMed] [Google Scholar]
- 32.Hansen KH, et al. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008;10:1291–1300. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- 33.Adams-Cioaba MA, Min J. Structure and function of histone methylation binding proteins. Biochem Cell Biol. 2009;87:93–105. doi: 10.1139/O08-129. [DOI] [PubMed] [Google Scholar]
- 34.Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 35.Nielsen PR, et al. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature. 2002;416:103–107. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- 36.Min J, et al. L3MBTL1 recognition of mono- and dimethylated histones. Nat Struct Mol Biol. 2007;14:1229–1230. doi: 10.1038/nsmb1340. [DOI] [PubMed] [Google Scholar]
- 37.Li H, et al. Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Mol Cell. 2007;28:677–691. doi: 10.1016/j.molcel.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Margueron R, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Botuyan MV, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- 41.Min J, Zhang X, Cheng X, Grewal SI, Xu RM. Structure of the SET domain histone lysine methyltransferase Clr4. Nat Struct Biol. 2002;9:828–832. doi: 10.1038/nsb860. [DOI] [PubMed] [Google Scholar]
- 42.Zhang K, Mosch K, Fischle W, Grewal SI. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat Struct Mol Biol. 2008;15:381–388. doi: 10.1038/nsmb.1406. [DOI] [PubMed] [Google Scholar]
- 43.Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosomal DNA regulates the core-histone-binding subunit of the human Hat1 acetyltransferase. Curr Biol. 1998;8:96–108. doi: 10.1016/s0960-9822(98)70040-5. [DOI] [PubMed] [Google Scholar]
- 44.Song JJ, Garlick JD, Kingston RE. Structural basis of histone H4 recognition by p55. Genes Dev. 2008;22:1313–1318. doi: 10.1101/gad.1653308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murzina NV, et al. Structural basis for the recognition of histone H4 by the histone-chaperone RbAp46. Structure. 2008;16:1077–1085. doi: 10.1016/j.str.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang H, et al. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol Cell. 2003;12:475–487. doi: 10.1016/j.molcel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 47.Fang J, Wang H, Zhang Y. Purification of histone methyltransferases from HeLa cells. Methods Enzymol. 2004;377:213–226. doi: 10.1016/S0076-6879(03)77012-8. [DOI] [PubMed] [Google Scholar]
- 48.Minor ZOaW. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 49.Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr D. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- 50.Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS. ARP/wARP and molecular replacement. Acta Crystallogr D. 2001;57:1445–1450. doi: 10.1107/s0907444901014007. [DOI] [PubMed] [Google Scholar]
- 51.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.