

Abstract
Despite increasing importance of protein glycosylation, most of the large-scale glycoproteomics have been limited to profiling the sites of N-glycosylation. However, in-depth knowledge of protein glycosylation to uncover functions and their clinical applications requires quantitative glycoproteomics eliciting both peptide and glycan sequences concurrently. Here we describe a novel strategy for the multiplexed quantitative mouse serum glycoproteomics based on a specific chemical ligation, namely, reverse glycoblotting technique, focusing sialic acids and multiple reaction monitoring (MRM). LC-MS/MS analysis of de-glycosylated peptides identified 270 mouse serum peptides (95 glycoproteins) as sialylated glycopeptides, of which 67 glycopeptides were fully characterized by MS/MS analyses in a straightforward manner. We revealed the importance of a fragment ion containing innermost N-acetylglucosamine (GlcNAc) residue as MRM transitions regardless the sequence of the peptides. Versatility of the reverse glycoblotting-assisted MRM assays was demonstrated by quantitative comparison of 25 targeted glycopeptides from 16 proteins between mice with homo and hetero types of diabetes disease model.
Clinical proteomics focusing on the identification and validation of biomarkers and the discovery of proteins as therapeutic targets is an emerging and highly important area of proteomics. Biomarkers are measurable indicators of a specific biological state (particularly one relevant to the risk of contraction) and the presence or the stage of disease, and are thus expected to be useful for the prediction, detection, and diagnosis of disease as well as to follow the efficacy, toxicology, and side effects of drug treatment, and to provide new functional insights into biological processes.
At present, proteomics methods based on mass spectrometry (MS) have emerged as the preferred strategy for discovery of diagnostic, prognostic, and therapeutic protein biomarkers. Most biomarker discovery studies use unbiased, “identified-based” approaches that rely on high performance mass spectrometers and extensive sample processing. Semiquantitative comparisons of protein relative abundance between disease and control patient samples are used to identify proteins that are differentially expressed and, thus, to populate lists of potential biomarkers. De novo proteomics discovery experiments often result in tens to hundreds of candidate biomarkers that must be subsequently verified in serum. However, despite the large numbers of putative biomarkers, only a small number of them are passed through the development and validation process into clinical practice, and their rate of introduction is declining. The first non-standard abbreviation (MS above is standard) must be footnoted the same as the abbreviation footnote, and MRM must be the first abbreviation in the list because it is the one footnoted. After that the order does not matter.
Targeted proteomics using multiple reaction monitoring (MRM)1 is emerging as a technology that complements the discovery capabilities of shotgun strategies as well as an alternative powerful novel MS-based approach to measure a series of candidate biomarkers (1–7). Therefore, MRM is expected to provide a powerful high throughput platform for biomarker validation, although clinical validation of novel biomarkers has been traditionally relying on immunoassays (8, 9). MRM exploits the unique capabilities of triple quadrupoles (QQQ) MS for quantitative analysis. In MRM, the first and the third quadrupoles act as filters to specifically select predefined m/z values corresponding to the peptide precursor ion and specific fragment ion of the peptide, whereas the second quadrupole serves as collision cell. Several such transitions (precursor/fragment ion pairs) are monitored over time, yielding a set of chromatographic traces with retention time and signal intensity for a specific transition as coordinates. These measurements have been multiplexed to provide 30 or more specific assays in one run. Such methods are slowly gaining acceptance in the clinical laboratory for the routine measurement of endogenous metabolites (10) (e.g. in screening newborns for a panel of inborn errors of metabolism) some drugs (11) (e.g. immunosuppressants), and the component analysis of sugars (12).
One of the profound challenges in clinical proteomics is the need to handle highly complex biological mixtures. This complexity presents unique analytical challenges that are further magnified with the use of clinical serum/plasma samples to search for novel biomarkers of human disease. The serum proteome is composed of tens of thousands of unique proteins, of which concentrations may exceed 10 orders of magnitude. Protein glycosylation, one of the most common post-translational modifications, generates tremendous diversity, complexity, and heterogeneity of gene products. It changes the biological and physical properties of proteins, which include functions as signals or ligands to control their distribution, antigenicity, metabolic fate, stability, and solubility. Protein glycosylation, in particular by N-linked glycans, is prevalent in proteins destined for extracellular environments. These include proteins on the extracellular side of the plasma membrane, secreted proteins, and proteins contained in body fluids (such as blood serum, cerebrospinal fluid, urine, breast milk, saliva, lung lavage fluid, or pancreatic juice). Considering that such body fluids are most easily accessible for diagnostic and therapeutic purposes, it is not surprising that many clinical biomarkers and therapeutic targets are glycoproteins. These include, for example, cancer antigen 125 (CA125) in ovarian cancer, human epidermal growth factor receptor 2 (Her2/neu) in breast cancer, and prostate-specific antigen (PSA) in prostate cancer. In addition, changes in the extent of glycosylation and the structure of N-glycans or O-glycans attached to proteins on the cell surface and in body fluids have been shown to correlate with cancer and other disease states, highlighting the clinical importance of this modification as an indicator or effector of pathologic mechanisms (13–16). Thus, clinical proteomic platforms should have capability to provide protein glycosylation information as well as sufficient analytical depth to reliably detect and quantify specific proteins with sufficient accuracy and throughput.
To improve the detection limits to the required sensitivities, one needs to dramatically reduce the complexity of the sera samples. For focused glycoproteomics, several techniques using lectins or antibodies enabling the large-scale identification of glycoproteins have recently been developed (17–19). Notably, Zhang et al. reported a method for the selective isolation of peptides based on chemical oxidation of the carbohydrate moiety and subsequent conjugation to a solid support using hydrazide chemistry (20–26). However, it is not possible to provide any structural information about N-glycans because the MS analysis is performed on peptides of which N-glycans are removed preferentially by treating with peptide N-glycanase (PNGase). In 2007, we developed a method for rapid enrichment analysis of peptides bearing sialylated N-glycans on the MALDI-TOF-MS platform (27). The method involves highly selective oxidation of sialic acid residues of glycopeptides to elaborate terminal aldehyde group and subsequent enrichment by chemical ligation with a polymer reagent, namely, reverse glycoblotting technique inspired from an original concept of glycoblotting method (28). This method, in principle, is capable identifying both glycan and peptide sequences concurrently. Recently, Nilsson et al. reported that glycopeptides from human cerebrospinal fluid can be enriched on the basis of the same principle as the reverse glycoblotting protocol, and captured glycopeptides were analyzed with ESI FT-ICR MS (29). Because it is well known that sialic acids play important roles in various biological processes including cell differentiation, immune response, and oncogenesis (30–34), our attention has been directed toward feasibility of the reverse glycoblotting technique in quantitative analysis of the specific glycopeptides carrying sialic acid(s) by combining with multiplexed MRM-based MS.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Sodium periodate (NaIO4), ammonium bicarbonate (NH4HCO3), dithiothreitol (DTT), iodoacetamide, trifluoroacetic acid (TFA), sodium metabisulfite (Na2S2O5), and 2-aminopyridine (2-AP) were obtained from Wako Pure Chemical (Osaka, Japan). Yolk sialylated glycopeptide used as an internal standard was obtained from Tokyo Kasei Co. N-osyl-L-phenylalanine chloromethyl ketone–treated trypsin was purchased from Thermo Fisher Scientific, Inc. (Rockford, IL). Peptide N-glycosidase F (PNGF) was obtained from Roche Applied Science. Sephadex G-25 was purchased from Pharmacia Biotech Inc. (Piscataway, NJ). Hydrazide resin (Affi-Gel Hz) was obtained from Bio-Rad 2-Picoline borane (2-Pic-BH3) was purchased from Junsei Chemical Co. Ltd. (Tokyo, Japan). Mouse serum was purchased from Sigma-Aldrich. Sera of male and female C57BL/KsJ db/db mice (disease model) and 7 weeks age-matched db/ + mice (control) were purchased from Charles River Japan (Yokohama, Japan).
A General Protocol for Reverse Glycoblotting of Sialyl Glycopeptides from Serum
Mouse serum (50 μl, an initial protein concentration (60 mg/ml) determined by the Bradford method (Bio-Rad)) was mixed with a final concentration of 50 mm DTT in 100 mm NH4HCO3, and subjected to the reduction reaction at 80 °C for 15 min, then the mixture was alkylated with final concentration of 125 mm iodoacetamide in 100 mm NH4HCO3 at room temperature for 15 min. Then, the alkylated proteins were digested with trypsin (100 μg) in 50 mm NH4HCO3 at 37 °C overnight (protease was added with 1:30 (w/w) of protein). For removal of excess small molecules, such as DTT and acetamide derivatives, the mixture was fractionated with Sephadex G-25 column (1.0 × 40 cm, 32 ml), and the digested peptides and glycopeptides were further treated with trypsin (50 μg) to cleavage incomplete peptide fragment at 37 °C overnight. After heating at 80 °C for 15 min, the tryptic peptides was added with yolk sialyl glycopeptide (250 pmol) as an internal standard. The mixture was mixed with NaIO4 (final concentration: 30 mm), and reacted at 0 °C for 60 min (35–37). The oxidized glycopeptides mixture was added with Affi-Gel Hz resin (wet vol. 200 μl), and subsequently quenched with Na2S2O5 (the equivalent mol of NaIO4 added) to prevent over-oxidation, and reacted at 37 °C for 2 h. For removal of peptides, asialoglycopeptides, and other components, the resin was washed thoroughly with water (1 ml), 1 m sodium chloride (NaCl; 1 ml), 30% methyl alcohol (MeOH; 1 ml), 0.1% acetic acid (1 ml), and water (1 ml). The resin was then treated with ice-cold 1 m hydrochloride (HCl; 500 μl) under cooling in an ice bath, and filtered quickly. To the solution was then added 2-AP (50 mg) to make the solution pH 7.0, and the mixture was reacted with 2-Pic-BH3 (10 mg) in methanol (100 μl) at room temperature overnight. The reaction mixture was subjected to the purification by Sephadex G-25 column (1.0 × 40 cm, 32 ml) to remove excess reductive reagents and 2-AP. Finally, pyridyl aminated (PA)–labeled sialyl glycopeptides enriched from mouse serum (50 μl) were used directly for further LC-MS and MS/MS analyses. To optimize a condition for achieving a complete oxidation, the reaction using a standard glycopeptide (100 pmol) was monitored by MALDI-TOF-MS (supplemental Fig. 1).
Characterizations of PA-labeled Sialyl Glycopeptides
Profiling by HPLC
PA-labeled sialyl glycopeptides with and without PNGF digestion were analyzed by HPLC (Hitachi d-7000 HPLC system, Hitachi High-Technologies Co. Ltd., Tokyo, Japan) equipped with fluorescence detection (Ex. 320 nm, Em. 400 nm) on a reverse-phase column (Inertsil ODS-3, 3 μm, 2.1 × 150 mm, GL sciences Inc.). The samples (1 μl of PA-labeled sialyl glycopeptides or deglycosylated peptides) were eluted with gradients of 0∼42% acetonitrile with 0.1% trifluoroacetic acid (0∼90 min), washed with 60% acetonitrile with 0.1% TFA (90–105 min), and equilibrated with 0.1% TFA (105–130 min) at a constant flow of 200 μl/min (column oven at 40 °C).
Reverse-phase Capillary L-MS/MS and MRM Assays
PA-labeled sialyl glycopeptides and deglycosylated peptides (5 μl) were subjected to the analysis by electrospray LC-MS/MS using LC systems (Dionex, Sunnyvale, CA) with C-18 reversed-phase column (Inertsil ODS-3 0.5 × 150 mm, GL sciences Inc.) and eluted with gradients of 0–42% acetonitrile with 0.1% TFA (0–90 min), washed with 60% acetonitrile with 0.1% TFA (90–105 min) and equilibrated with 0.1% TFA (105–130 min) at a constant flow of 30 μl/min (column oven at 40 °C). Electrospray MS data were collected using the TurboIonSpray® source on a 4000 QTRAP hybrid triple quadrupole/linear ion trap instrument (Applied Biosystems/MDS Sciex, Foster City, CA), and the peaks and masses were integrated using annotation procedures in the Analyst software 1.4.1 (intelli-Quan algorithm). The source temperature was set at 250 °C, with a voltage of 5.5 kV, and gases 1 and 2 were set at 30 and 20, respectively. MRM transitions acquired at unit resolution in both the Q1 and Q3 quadrupoles to maximize specificity and the scan rate on Q3 was 4000 amu/sec. The acquired MS/MS spectra of PA-labeled sialyl glycopeptides from mouse serum were searched from identified deglycosylated peptides after the treatment with PNGF, which can be identified from the ion sets of specific peptide fragments containing single N-acetylglucosamine (GlcNAc) residue and the corresponding nonglycosylated (naked) peptide fragment. The fact that retention time of glycopeptide is closely related to that of deglycosylated peptides (core peptides) should greatly assist the above searching analysis. Search parameters included carbamidomethylation of cysteines (and oxidized methionines; trypsin digestion with partial site cleavages (semitrypsin); variable modifications including pyroglutamic acid and aspartic acid to identify a number of peptides (38, 39). A mass tolerance for the precursor ions and the fragments ions were 1.2 Da and 0.6 Da, respectively. De-N-glycosylated peptides derived by PNGF digestion were used for these modifications and the conversion of asparagines into aspartic acid and searched by an in-house MASCOT server (Version 2.1, Matrix Science) using Mass Spectrometry Protein Sequence Database (MSDB database, updated 31st August, 2006, 3,239,079 proteins), which was modified for a consensus sequence of N-glycosylated peptides O-X-S/T from N-X-S/T to distinguish between asparagine of N-glycosylation and other asparagine and Protein PilotTM software 3.0 (mouse_KBMS5.0 database, updated March 2, 2005, 231,316 proteins). The obtained candidates from these searches were estimated from the presence of at least one NXS/T motif, in which the probability-based matching scores should exceed their thresholds (p < 0.05). Data analysis of MRM assays and quantification based on the MRM channels for individual glycopeptides were performed using MultiQuant 1.0 software from Applied Biosystems. In the MRM assay on the basis of the channels established in the present experiments, 5 μl of mouse serum samples were proved to be sufficient for further quantitative analysis in the presence of 25 pmol of PA-labeled standard glycopeptide as an internal standard (supplemental Figs. 4–8).
RESULTS
Principle of Reverse Glycoblotting Technique Designated for Sialyl Glycopeptides-focused MRM Assays—The basic strategy for multiplexed quantitative glycoproteomics of mouse serum samples (50 μl) based on the reverse glycoblotting method and LC-MRM assays was outlined in Figure 1. The objective of this approach is the generation of reproducible glycopeptide patterns representing the serum glycoproteome, leading to the quantitative detection of specific glycopeptides that discriminate between related groups of glycoproteomes and the subsequent identification of these discriminatory glycopeptides. We herein established a novel method for the enrichment analysis of serum glycopeptides bearing sialic acids, in which nonreducing sugar residues are labeled selectively by reductive amination of the aldehyde group generated at C7 position of sialic acid with 2-AP (Scheme 1). This improved process greatly facilitated further LC-MS and MS/MS characterization involving MRM-based measurements of mouse serum glycopeptides because PA-labeled glycopeptides allowed for general fluorescence detection. In our previous report (27), we used an aminooxy-functionalized polyacrylamide for the enrichment of glycopeptides having at least one terminal sialic acid. This protocol is based on the formation of stable oxime bond between aldehyde groups generated by selective oxidation at glycerol moiety of sialic acid residues and aminooxy groups of polymer supports. Glycopeptides captured are therefore recovered as their asialo forms due to this stable oxime linkage because the treatment with 3% TFA at 100 °C for 1 h, a common condition to cleave oxime bonds, was proved to digest predominantly the glycoside linkage between sialic acid and galactose (Gal) residues. Thus, our attention was directed toward chemical flexibility of hydrazone linkage (40–42) derived from hydrazide-functionalized polymers. We considered that hydrazone linkage between glycopeptides and polymer supports appears to be much more sensitive to general conditions of acid hydrolysis than α2,3/2,6 sialosides. As anticipated, an improved protocol allowed for the enrichment of glycopeptides bearing sialic acids safely. Upon rapid, selective, and quantitative oxidation of the tryptic digests of whole glycoproteins derived from 50 μl of mouse serum by treating with 30 mm NaIO4 (final concentration) at 4 °C for 60 min (supplemental Fig.1), aldehydes generated at terminal sialic acid residues were selectively captured at 37 °C for 2 h by chemical ligation with commercially available hydrazide modified polymer (Affi-Gel Hz). After thorough washing to remove nonspecifically bound molecules (e.g. nonsialylated glycopeptides and other peptides), glycopeptides covalently enriched were released by treating with ice-cold aqueous 1 m HCl as free aldehydes. One-pot reductive amination of regenerated aldehydes with 2-AP was performed in the presence of 2-Pic-BH3 (pH 7.0) at room temperature. Advantage of PA-labeling with sialyl glycopeptides is evident because the glycopeptides can be monitored and quantified on HPLC by common fluorescence detector independent from the peptide sequence. Moreover, it is expected that enhanced ionization potency by PA-derivatization should greatly facilitate the structural characterization of sialyl glycopeptides through ESI-MS and MS/MS analysis of ideal fragment ions (supplemental Fig. 2). When a known egg yolk glycopeptide was tested as a control for the validation of the present protocol, it seems that the general MRM assay requires at least approximately 10 pmol of unknown glycopeptides, whereas we usually spiked 25 pmol of PA-labeled glycopeptide as an internal standard. In the present results, no significant signals due to the derivatives from N-terminal Ser/Thr was detected, although it is considered that the peptides having such terminal might also be oxidized by treatment with periodate (43).
Fig. 1.

Strategy for comprehensive approach to multiplexed quantitative analysis of sialyl glycoproteins on the basis of reverse glycoblotting-assisted MRM assays.
Scheme 1.

An improved protocol for reverse glycoblotting technique.
Identification of Mouse Serum Sialyl Glycopeptides by LC-MS and MS/MS
Mouse serum PA-labeled sialyl glycopeptides enriched by reverse glycoblotting protocol were subjected to LC-MS analysis according to the systematic strategy shown in Figure 1. It is clear that the digestion of PA-labeled N-glycans from glycopeptides proceeded smoothly by treating with PNGF because LC profiles monitored by fluorescence detector changed dramatically to converge at early retention times after de–N-glycosylation (Fig. 2A). Interestingly, the retention times due to original glycopeptides and the corresponding naked peptides were found to be in close proximity on the reversed-phased chromatography while peak numbers were reduced by PNGase digestion, indicating the existence of significant heterogeneity in glycan structures at individual N-glycosylation sites (Fig. 2B). For example, MS/MS analysis revealed that a glycopeptide from alpha-1-acid glycoprotein 1 and its de–N-glycosylated peptide were eluted at 45.9 and 49.1 min, respectively (Fig. 2C and 2D). De–N-glycosylated peptides were subjected to the pattern matching analysis based on the MS/MS analysis and the mass of discriminatory peptides was determined using in-house MASCOT server and Protein PilotTM software. Eventually, we identified 270 unique sialylated glycopeptides mapping to 95 unique sialylated glycoproteins by reverse glycoblotting method as shown in Table I and Fig. 3. It should be noted that two peptide search algorithms significantly influence the fragment scoring calculation of the number of matching between the row data and the theoretical fragment ions to afford differences in the identified glycopeptides/glycoproteins (44–46).
Fig. 2.

LC profiles of PA-labeled sialyl glycopeptides enriched from mouse serum before (top) and after (bottom) the treatment with PNGase F. A, LC pattern monitored by fluorescence detector. B, LC pattern monitored by MS detector. C, MS/MS spectrum of PA-labeled sialyl glycopeptide from α1-acid glycoprotein 1 (NLINDTIELR) detected at 45.9 min. D, MS/MS spectrum of the corresponding de-N-glycosylated peptide identified as NLIDDTIELR detected at 49.1 min.
Table I. Identified mouse glycopeptides from de–N-glycosylated peptides using reverse glycoblotting (Mascot score from in-house MASCOT server, Confidence (Conf) from Protein PilotTM, O residue means N-glycosylated asparagine site).



Fig. 3.
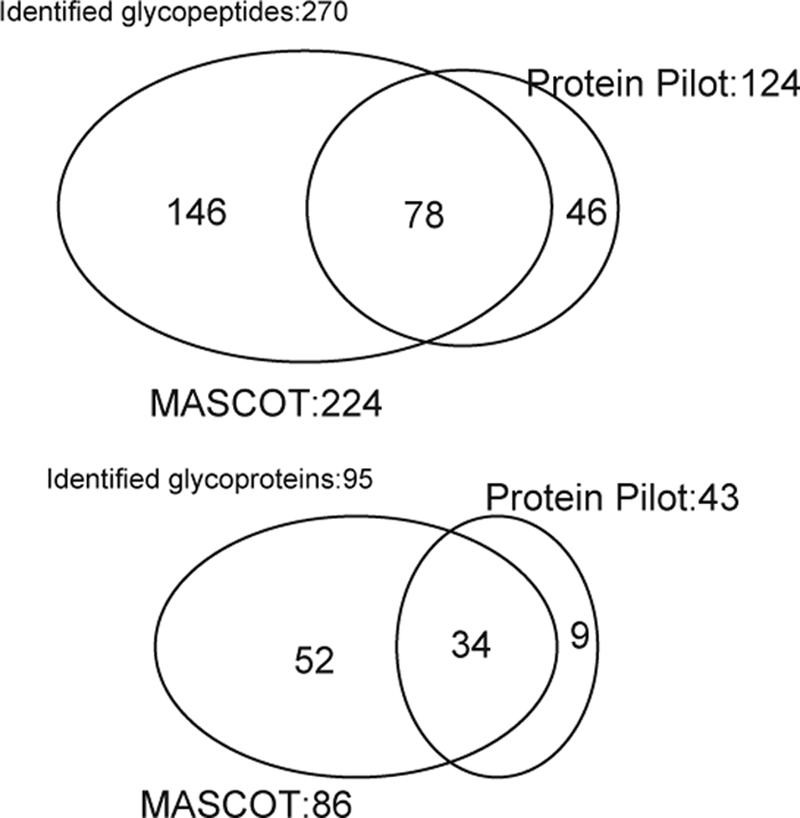
Diagram of glycopeptides and glycoproteins identified by MASCOT® and ProteinPilotTM peptide search.
Next, our attention was focused on assignment of the enriched glycopeptides having sialic acid–containing glycans by direct LC-MS/MS analysis of PA-labeled sialyl glycopeptides without PNGase treatment. In the typical MS/MS spectra of PA-labeled sialyl glycopeptides, we found the generation of specific carbohydrate fragment ions (i.e. m/z 324; PA-N-glycolyl neuraminic acid (NeuGc), m/z 486; PA-NeuGc-Gal, m/z 689; PA[-NeuGc-Gal-GlcNAc, etc.) as well as peptide fragments with and without an innermost GlcNAc residue. It seems likely that these signature signals in MS/MS spectra can be used for rapid identification of PA-labeled sialyl glycopeptide. In fact, MS/MS analysis of the glycopeptide (m/z 868.6 (+4)) derived from alpha-1-acid glycoprotein 1 gave the following signature signals (m/z 324; PA-NeuGc, m/z 366; Gal-GlcNAc, m/z 486; PA–NeuGc-Gal, m/z 689; PA–NeuGc-Gal-GlcNAc, and m/z 851; PA–NeuGc-Gal-GlcNAc-mannose (Man)) as well as fragment ions from peptide moiety (m/z 1201; NLINDTIELR and m/z 1404; NLIN(GlcNAc)DTIELR) as indicated in Fig. 2C. Feasibility of this simple procedure was demonstrated by uncovering glycoform microheterogeneity at each N-glycosylation site. For instance, it was revealed that a peptide, NSTLCDLCIGPLK from serotransferrin, bears three types glycan chains such as NeuGc2Gal2GlcNAc2Man3GlcNAc2 (m/z 941.0 (+4)), NeuGc2Gal2GlcNAc2Man3GlcNAc2Fuc (m/z 977.5 (+4)), and NeuGc3Gal2GlcNAc2Man3GlcNAc2 (m/z 1021.8 (+4)). In the present study, we identified 67 mouse serum glycopeptides modified with at least one PA-labeled sialic acid residue as listed in Table II. It is also noteworthy that PA-labeling of terminal sialic acids facilitated the differentiation of two important species, NeuAc and NeuGc, as PA-NeuAc (m/z 308) and PA-NeuGc (m/z 324) existing on a single glycopeptide (No.45 in Table II) from alpha-2-macroglobulin (NYLNETQQLTEAIK).
Table II. Identified glycopeptides in mouse serum.

Design of MRM Assays for PA-labeled Sialyl Glycopeptides
In MRM experiments, two mass analyzers (Q1 and Q3) are used as static mass filters to monitor a particular fragment ion of a selected precursor ion (Fig. 4A). Unlike conventional shotgun proteomic studies, MRM measurements are strictly targeting a predetermined set of peptides and depend strongly on specific MRM transitions (specific pair of m/z values associated with the precursor and fragment ions selected) for each targeted peptides. Because the intensities of individual fragments derived from one precursor ion differ substantially, it is essential to select transitions specific for the most intense fragments to obtain a hig-sensitivity assay. For this purpose, 67 MS/MS spectra of glycopeptides from 26 mouse serum glycoproteins were actually obtained, and information on peptide sequence, glycoform, observed parent mass, and retention time on reversed-phase chromatography were also summarized in Table II. The optimization of collision energy (CE) was performed for 38 glycopeptides to achieve high sensitivity and efficiency in the detection of specific fragment ions (supplemental Fig. 3).
Fig. 4.

Schematic diagram of MRM assays and typical procedure for MRM setting. A, a general concept of MRM. B, MS/MS spectrum of PA-labeled sialyl glycopeptide from egg yolk. C, MRM chromatogram under different collision energy (CE) at 22, 27, 32, 37, and 42 eV for 724.6/863.3 as Q1/Q3. D, relationship between fragment ion counts versus CE for MRM assay (the numerical value means each coefficient of variation (CV)). E, relationship between fragment ion counts versus the amount of PA-labeled glycopeptide.
Regardless of the peptide sequences, the MS/MS cleavage occurs preferentially between the two GlcNAc residues of the core chitobiose, resulting in a peptide fragment ion retaining one GlcNAc residue on the asparagine residue with high intensity (Fig. 4B). Therefore, we selected a fragment ion containing the innermost GlcNAc residue as key transitions. Basically, the choice of the transitions is observed with parents mass as Q1 filter, observed fragment ions as Q3 filter, and the CE is selected from optimal CE which is guided to several measurements with different CE on the basis of observed MS/MS analysis with rolling collision energy program (Fig. 4C and 4D). As anticipated, the obtained MRM setting named as MRM channel for PA-labeled yolk glycopeptide as an internal standard showed high sensitivity and reproducibility (Fig. 4D and 4E, supplemental Fig. 4). It was also demonstrated that the relationship between fragment ion counts and the amount of PA-labeled glycopeptide tested exhibited a good linearity at a range from 1 to 6 pmol.
Reproducibility of Quantitative MRM Assays of Mouse Serum Sialyl Glycopeptides Enriched by Reverse–Glycoblotting
To assess the reproducibility of quantitative MRM assays of mouse serum PA-labeled sialyl glycopeptides, MRM LC-MS/MS analysis was performed for 25 targeted glycopeptides known as high-to-medium abundance glycoproteins of mouse serum. (supplemental Fig. 5). Typically, triply or quadruply charged parent glycopeptide ions and singly or doubly charged fragment ions containing the innermost GlcNAc residue were selected in the first and third quadrupoles (Q1 and Q3), respectively. The retention time of each glycopeptide on the reversed-phase chromatography was found to be highly reproducible (CV < 1.5%, data not shown) and 25 targeted glycopeptides generated acceptable quantitative ion counts data (CV ≤ 19%). Because the optimal CE for individual target molecules gave lower CVs and higher sensitivities in MRM assays (Fig. 4 and supplemental Fig. 5), it was suggested that MRM assays of glycopeptides can be performed in a wide dynamic range of mouse serum glycoproteins (approximately 1,000-fold, from alpha 2-macroglobulin (86.4) to complement factor H (0.08) estimated from the concentration of the internal standard spiked).
It is not surprising that the condition for the preparation of tryptic peptides from serum samples often influences mass-based quantitative proteomics because a common biological sample consists of various factors affecting profiles of tryptic digests such as endogenous proteases and protease inhibitors (21). Thus, we examined the effect of this step during sample preparation on the reproducibility and concluded that repetitive tryptic digests provide highly stable and reliable MRM assay of PA-labeled sialyl glycopeptides enriched by the reverse glycoblotting method (supplemental Fig. 6). In addition, it was demonstrated that the use of an internal standard (PA-labeled sialyl glycopeptides derived from egg yolk) allows for calibration of the raw data to determine the relative concentrations of targeted glycopeptides in MRM assays (supplemental Fig. 7). However, these relative concentrations estimated by comparing with an internal standard spiked are not actual concentrations in mouse serum because of the different ionization potentials of individual glycopeptides (6).
Application of Reverse Glycoblotting–assisted Multiplex MRM Assays of Sialyl Glycopeptides for Searching Biomarkers in Disease-Model Mouse Serum
Finally, our interest was focused on the clinical importance of serum sialyl glycopeptides as potential disease-related biomarkers as well as the feasibility of the present strategy in quantitative glycoproteomics based on MRM assays. Merit of reverse glycoblotting-assisted multiplex MRM assays was demonstrated by an in-depth quantitative approach toward biomarker discovery using the sera of male and female C57BL/KsJ db/db mice known as diabetic models and 7-weeks age-matched db/+ control mice. In the present study, MRM channels for 25 glycopeptides from 16 glycoproteins were used for the quantitative glycoproteomics of PA-labeled sialyl glycopeptides enriched from serum samples by reverse glycoblotting protocol (Fig. 5 and supplemental Fig. 8). It was revealed that sialyl glycopeptides from immunoglobulin gamma-2B (Number. 26, 27, and 28) increased significantly in diabetic model males in comparison with that of control subjects (p < 0.05). This type-specific alteration in the relative expression levels of sialyl glycopeptides was observed in serotransferrin (Number. 2 and 3), murinoglobulin 1 (Number. 13 and 16), alpha-2-macroglobulin (Number. 48), and serine protease inhibitor A3K (Number. 6) enriched from the serum of a diabetic model male. Interestingly, we did not observe any significant differences in the concentration of these glycopeptides between the diabetes model female and the control serum. These results clearly indicate the differences in posttranslational modification processes during diabetic disease progression between male and female mice, although the effect of gender on the mechanism of these N-glycan alterations in targeted glycopeptides remains unclear. Judging from the increase of glycopeptide (write out no. in text and check N against PDF to see if it is underlined, if so use N No. 3, NSTLCDLCIGPLK +NeuGc2Gal2GlcNAc2Man3GlcNAc2Fuc) and the decrease of glycopeptide (No. 2, NSTLCDLCIGPLK +NeuGc2Gal2GlcNAc2Man3GlcNAc2) found in the case of serotransferrin, it seems that quantitative fucosylation at this N-glycosylation site occurs specifically in the diabetic model male. This finding suggests that altered N-glycosylation in the relatively abundant serum proteins may yield distinct differences between patients and healthy controls (47, 48).
Fig. 5.

MRM assays of PA-labeled sialyl glycopeptides enriched from diabetic model mouse serum and control mouse. A, MRM chromatogram of male control. B, MRM chromatogram of male diabetic model. C, MRM chromatogram of female control. D, MRM chromatogram of female diabetic model. Quantification of 25 target sialyl glycopeptides by MRM assays of male control/diabetic mouse (E) and female control/diabetic mouse.
DISCUSSION
We describe a promising method for quantitative glycoproteomics by combining the reverse glycoblotting method and MRM-based MS. The method allows the identification and quantification of glycoproteins bearing sialylated oligosaccharides in a complex sample and the determination both of N-glycosylation sites and glycoforms concurrently. The improved reverse glycoblotting procedure made recovering enriched glycopeptides possible without loss of sialic acid residues, and the terminal sialic acid residues regenerated can be labeled directly using a fluorescence tag. It was demonstrated that the present protocol greatly facilitates highly sensitive monitoring and accurate quantitation of mouse serum sialyl glycopeptides on LC-ESI MS. We identified 270 glycopeptides from 95 mouse serum glycoproteins carrying sialyl N-glycan. As anticipated, we did not detect any sialylated O-glycopeptide though the experiments because of its extremely lower abundance in mouse serum than that of sialylated N-glycopeptides. However, the present protocol worked well to capture such sialylated O-glycopeptides when glycoprotein samples preferentially enriched by means of some specific affinity-ligand were used (data not shown). Glycoforms (glycan structures) of 67 PA-labeled sialyl glycopeptides from 26 glycoproteins were further characterized by MS/MS analysis.
Focused glycoproteomics based on the reverse glycoblotting technique revealed the existence of many irregularly cleaved peptides such as tryptic peptides. For instance, it was demonstrated that 10 redundant peptides involving the same N-glycosylation site were enriched and identified from serum alpha-1-antitrypsin 1–2 as shown in Table I. This result clearly suggests the presence of various endogenous proteases and peptidases in serum, as well as the appearance of different truncated glycoproteins from cellular and tissue “leakage” or the removal of signal peptides. Indeed, we identified the proteases, such as carboxypeptidase N subunit 2 and carboxypeptidase B2, as sialyl glycoproteins (Table I) from mouse serum in this work. Considering that many human serum endogenous proteases and peptidases are known to correlate with various diseases such as aminopeptidase N for acute myeloid leukemia, cathepsin D for breast cancer and possibly Alzheimer's disease, and cathepsin L for bone resorption, quantitative profiling of such irregular cleavage(s) in glycoproteins may contribute to biomarker discovery and the elucidation of the mechanical process of pathogenesis.
MS/MS data of 67 PA-labeled sialyl glycopeptides and their de–N-glycosylated species allowed us to generate MRM parameters in a straightforward manner. We demonstrated the importance of a fragment ion containing an innermost GlcNAc residue as a transition regardless of the sequence of the core peptides. For the first time, we constructed 25 MRM channels for PA-labeled sialyl glycopeptides enriched from mouse serum by setting this highly specific fragment ion as a Q3 filter, the precursor glycopeptide ion as a Q1 filter, and we optimized the CE. Versatility of MRM channels for 25 targeted glycopeptides was demonstrated by applying this assay to the multiplex quantitative measurements of targeted glycoproteins having the sialylated N-glycan of interest in diabetic model mice in comparison with normal mice. Reverse glycoblotting–assisted MRM assays uncovered changes in the expression level of some sialyl glycopeptides in mouse serum during disease development in the diabetic model male. Surprisingly, no significant alteration was observed in these 25 targeted glycoproteins between diabetic and normal female mice. MRM assays focusing on nonsialylated glycopeptides, Gal and/or GlcNAc terminated glycopeptides, remain unable, although glycopeptides bearing high mannose type N-glycans can be isolated by means of conventional lectin-based affinity chromatography.
MRM assays for detection and quantitation of human serum N-glycopeptides are of growing importance because changes in the posttranslational modification process have relevance to various diseases, including cancer (5, 48). However, it has been shown that in silico calculation indicates that on average 1/20 of the tryptic peptides have the N-X-S/T sequence known as the N-glycosylation consensus sequence (49). Considering the microheterogeneity of glycoforms in the redundant N-glycosylation sites, we believe that the glycoform-focused reverse glycoblotting technique greatly contributes to reducing the complexity of biological samples and accelerates discovery research of potential biomarkers on the basis of multiplex MRM MS(50, 51).
Supplementary Material
Footnotes
* This work was supported in part by grants for “Development of Systems and Technology for Advanced Measurement and Analysis (SENTAN)” and “The Matching Program for Innovations in Future Drug Discovery and Medical Care” from the Japan Science and Technology Agency (JST) and the Ministry of Education, Culture, Science, and Technology, Japan.
 This article contains supplemental Figs. 1–9.
This article contains supplemental Figs. 1–9.
1 The abbreviations used are:
- MRM
- multiple reaction monitoring
- PNGase
- peptide N-glycanase
- NaIO4
- sodium periodate
- NH4HCO3
- ammonium bicarbonate
- 2-AP
- 2-aminopyridine
- PNGF
- peptide N-glycosidase
- 2-Pic-BH3
- 2-picoline borane
- PA
- pyridyl aminated
- GlcNAc
- N[-acetylglucosamine
- NeuGc
- N-glycolyl neuraminic acid
- CE
- collision energy
- CV
- coefficient of variation.
REFERENCES
- 1.Martin D. B., Holzman T., May D., Peterson A., Eastham A., Eng J., McIntosh M. (2008) MRMer, an interactive open source and cross-platform system for data extraction and visualization of multiple reaction monitoring experiments. Mol. Cell. Proteomics 7, 2270–2278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuzyk M. A., Smith D., Yang J., Cross T. J., Jackson A. M., Hardie D. B., Anderson N. L., Borchers C. H. (2009) Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics 8, 1860–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson L., Hunter C. L. (2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell. Proteomics 5, 573–588 [DOI] [PubMed] [Google Scholar]
- 4.Kay R. G., Gregory B., Grace P. B., Pleasance S. (2007) The application of ultra-performance liquid chromatography/tandem mass spectrometry to the detection and quantitation of apolipoproteins in human serum. Rapid Commun. Mass Spectrom. 21, 2585–2593 [DOI] [PubMed] [Google Scholar]
- 5.Stahl-Zeng J., Lange V., Ossola R., Eckhardt K., Krek W., Aebersold R., Domon B. (2007) High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol. Cell. Proteomics 6, 1809–1817 [DOI] [PubMed] [Google Scholar]
- 6.Keshishian H., Addona T., Burgess M., Kuhn E., Carr S. A. (2007) Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 6, 2212–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haab B. B., Geierstanger B. H., Michailidis G., Vitzthum F., Forrester S., Okon R., Saviranta P., Brinker A., Sorette M., Perlee L., Suresh S., Drwal G., Adkins J. N., Omenn G. S. (2005) Immunoassay and antibody microarray analysis of the HUPO plasma proteome project reference specimens: systematic variation between sample types and calibration of mass spectrometry data. Proteomics 5, 3278–3291 [DOI] [PubMed] [Google Scholar]
- 9.Anderson N. L., Anderson N. G. (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 1, 845–867 [DOI] [PubMed] [Google Scholar]
- 10.Röschinger W., Olgemöller B., Fingerhut R., Liebl B., Roscher A. A. (2003) Advances in analytical mass spectrometry to improve screening for inherited metabolic diseases. Eur. J. Pediatr. 162Suppl. 1S67–S76 [DOI] [PubMed] [Google Scholar]
- 11.Streit F., Armstrong V. W., Oellerich M. (2002) Rapid liquid chromatography-tandem mass spectrometry routine method for simultaneous determination of sirolimus, everolimus, tacrolimus, and cyclosporin A in whole blood. Clin. Chem. 48, 955–958 [PubMed] [Google Scholar]
- 12.Hammad L. A., Saleh M. M., Novotny M. V., Mechref Y. (2009) Multiple-reaction monitoring liquid chromatography mass spectrometry for monosaccharide compositional analysis of glycoproteins. J. Am. Soc. Mass Spectrom. 20, 1224–1234 [DOI] [PubMed] [Google Scholar]
- 13.Durand G., Seta N. (2000) Protein glycosylation and diseases: blood and urinary oligosaccharides as markers for diagnosis and therapeutic monitoring. Clin. Chem. 46, 795–805 [PubMed] [Google Scholar]
- 14.Kam R. K. T., Poon T. C. W. (2008) The potentials of glycomics in biomarker discovery. Clin. Proteomics 4, 67–79 [Google Scholar]
- 15.Freeze H. H. (2001) Update and perspectives on congenital disorders of glycosylation. Glycobiology 11, 129R–143R [PubMed] [Google Scholar]
- 16.Spiro R. G. (2002) Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12, 43R-56R [DOI] [PubMed] [Google Scholar]
- 17.Qiu R., Regnier F. E. (2005) Use of multidimensional lectin affinity chromatography in differential glycoproteomics. Anal. Chem. 77, 2802–2809 [DOI] [PubMed] [Google Scholar]
- 18.Calvano C. D., Zambonin C. G., Jensen O. N. (2008) Assessment of lectin and HILIC based enrichment protocols for characterization of serum glycoproteins by mass spectrometry. J. Proteomics. 71, 304–317 [DOI] [PubMed] [Google Scholar]
- 19.Cho W., Jung K., Regnier F. E. (2008) Use of glycan targeting antibodies to identify cancer-associated glycoproteins in plasma of breast cancer patients. Anal. Chem. 80, 5286–5292 [DOI] [PubMed] [Google Scholar]
- 20.Zhang H., Li X.-J., Martin D. B., Aerbersold R. (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotech. 21, 660–666 [DOI] [PubMed] [Google Scholar]
- 21.Zhang H., Yi E. C., Li X. J., Mallick P., Kelly-Spratt K. S., Masselon C. D., Camp D. G., II, Smith R. D., Kemp C. J., Aebersold R. (2005) High throughput quantitative analysis of serum proteins using glycopeptide capture and liquid chromatography mass spectrometry. Mol. Cell. Proteomics 4, 144–155 [DOI] [PubMed] [Google Scholar]
- 22.Liu T., Qian W.J., Gritsenko M. A., Camp D. G., II, Monroe M. E., Moore R. J., Smith R. D. (2005) Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J. Proteome Res. 4, 2070–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Y., Aebersold R., Zhang H. (2007) Isolation of N-linked glycopeptides from plasma. Anal. Chem. 79, 5826–5837 [DOI] [PubMed] [Google Scholar]
- 24.Chen R., Jiang X., Sun D., Han G., Wang F., Ye M., Wang L., Zou H. (2009) Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J. Proteome Res. 8, 651–661 [DOI] [PubMed] [Google Scholar]
- 25.Zhou L., Beuerman, Chew R. W., A. P., Koh S. K., Cafaro T.A., Urrets-Zavalia J. A., Li S. F., Serra H. M. (2009) Quantitative analysis of N-linked glycoproteins in tear fluid of climatic droplet keratopathy by glycopeptide capture and iTRAQ. J. Proteome Res. 8, 1992–2003 [DOI] [PubMed] [Google Scholar]
- 26.Sun S., Yang G., Wang T., Wang Q., Chen C., Li Z. (2010) Isolation of N-linked glycopeptides by hydrazine-functionalized magnetic particles. Anal. Bioanal. Chem. 396, 3071–3078 [DOI] [PubMed] [Google Scholar]
- 27.Kurogochi M., Amano M., Fumoto M., Takimoto A., Kondo H., Nishimura S. I. (2007) Reverse glycoblotting allows rapid enrichment glycoproteomics of biopharmaceuticals and disease-related biomarkers. Angew. Chem. Int. Ed.Engl. 46, 8808–8813 [DOI] [PubMed] [Google Scholar]
- 28.Nishimura S. I., Niikura K., Kurogochi M., Matsushita T., Fumoto M., Hinou H., Kamitani R., Nakagawa H., Deguchi K., Miura N., Monde K., Kondo H. (2005) High-throughput protein glycomics: combined use of chemoselective glycoblotting and MALDI-TOF/TOF mass spectrometry. Angew. Chem. Int. Ed.Engl 44, 91–96 [DOI] [PubMed] [Google Scholar]
- 29.Nilsson J., Rüetschi U., Halim A., Hesse C., Carlsohn E., Brinkmalm G., Larson G. (2009) Enrichment of glycopeptides for glycan structure and attachment site identification. Nat. Methods 6, 809–811 [DOI] [PubMed] [Google Scholar]
- 30.Dwek R. A. (1996) Glycobiology: Toward understanding the function of sugars. Chem. Rev. 96, 683–720 [DOI] [PubMed] [Google Scholar]
- 31.Lis H., Sharon N. (1998) Lectins: carbohydrate-specific proteins that mediate cellular recognition. Chem. Rev. 98, 637–674 [DOI] [PubMed] [Google Scholar]
- 32.Schauer R. (2009) Sialic acids as regulators of molecular and cellular interactions. Curr. Opin. Struct. Biol. 19, 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varki A. (2008) Sialic acids in human health and disease. Trends Mol. Med. 14, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varki A. (2009) Multiple changes in sialic acid biology during human evolution. Glycoconj. J. 26, 231–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Lenten L., Ashewell G. (1971) Studies on the chemical and enzymatic modification of glycoproteins. J. Biol. Chem. 246, 1889–1894 [PubMed] [Google Scholar]
- 36.Quesenberry M. S., Lee Y. C. (1996) A rapid formaldehyde assay using purpald reagent: Application under periodation conditions. Anal. Biochem. 234, 50–55 [DOI] [PubMed] [Google Scholar]
- 37.Lee K. B., Lee Y. C. (1996) Conformational studies of glycopeptides by energy transfer. J. Biol. Chem. 271, 1462–1469 [DOI] [PubMed] [Google Scholar]
- 38.Tanner S., Pevzner P. A., Bafna V. (2006) Unrestrictive identification of post-translational modifications through peptide mass spectrometry. Nat. Protoc. 1, 67–72 [DOI] [PubMed] [Google Scholar]
- 39.Tanner S., Payne S. H., Dasari S., Shen Z., Wilmarth P. A., David L. L., Loomis W. F., Briggs S. P., Bafna V. (2008) Accurate annotation of peptide modifications through unrestrictive database search. J. Proteome Res. 7, 170–181 [DOI] [PubMed] [Google Scholar]
- 40.Shimaoka H., Kuramoto H., Furukawa J., Miura Y., Kurogochi M., Kita Y., Hinou H., Shinohara Y., Nishimura S.I. (2007) One-pot solid-phase glycoblotting and probing by transoximization for high-throughput glycomics and glycoproteomics. Chemistry Eur. J. 13, 1664–1673 [DOI] [PubMed] [Google Scholar]
- 41.Furukawa J., Shinohara Y., Kuramoto H., Miura Y., Shimaoka H., Kurogochi M., Nakano M., Nishimura S. (2008) Comprehensive approach to structural and functional glycomics based on chemoselective glycoblotting and sequential tag conversion. Anal. Chem. 80, 1094–1101 [DOI] [PubMed] [Google Scholar]
- 42.Amano M., Yamaguchi M., Takegawa Y., Yamashita T., Terashima M., Furukawa J., Miura Y., Shinohara Y., Iwasaki N., Minami A., Nishimura S. (2010) Threshold in stage-specific embryonic glycotypes uncovered by a full portrait of dynamic N-glycan expression during cell differentiation. Mol. Cell. Proteomics 9, 523–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chelius D., Shaler T. A. (2003) Capture of peptides with N-terminal serine and threonine: a sequence-specific chemical method for peptide mixture simplification. Bionconjugate. Chem. 14, 205–211 [DOI] [PubMed] [Google Scholar]
- 44.Baliban R. C., DiMaggio P. A., Plazas-Mayorca M. D., Young N. L., Garcia B. A., Floudas C. A. (2010) A novel approach for untargeted post-translational modification identification using integer linear optimization and tandem mass spectrometry. Mol. Cell. Proteomics 9, 764–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renard B. Y., Kirchner M., Monigatti F., Ivanov A. R., Rappsilber J., Winter D., Steen J. A., Hamprecht F. A., Steen H., (2009) When less can yield more: computational preprocessing of MS/MS spectra for peptide identification. Proteomics 9, 4978–4984 [DOI] [PubMed] [Google Scholar]
- 46.Brosch M., Swamy S., Hubbard T., Choudhary J., (2008) Comparison of mascot and X!Tandem performance for low and high accuracy mass spectrometry and the development of an adjusted Mascot threshold. Mol. Cell. Proteomics 7, 962–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miura Y., Hato M., Shinohara Y., Kuramoto H., Furukawa J., Kurogochi M., Shimaoka H., Tada M., Nakanishi K., Ozaki M., Todo S., Nishimura S.-I. (2008) BlotGlycoABCTM, an integrated glycoblotting technique for rapid and large scale clinical glycomics. Mol. Cell. Proteomics 7, 370–377 [DOI] [PubMed] [Google Scholar]
- 48.Hanash S.M., Pitteri S. J., Faca V. M. (2008) Mining the plasma proteome for cancer biomarkers. Nature 452, 571–579 [DOI] [PubMed] [Google Scholar]
- 49.Domon B. (2009) Glycosylation as means of reducing sample complexity to enable quantitative proteomics. Proteomics 9, 1488–1491 [DOI] [PubMed] [Google Scholar]
- 50.Hülsmeier A. J., Paesold-Burda P., Hennet T. (2007) N-Glycosylation site occupancy in serum glycoproteins using multiple reaction monitoring liquid chromatography-mass spectrometry. Mol. Cell. Proteomics 6, 2132–2138 [DOI] [PubMed] [Google Scholar]
- 51.Whiteaker J. R., Zhao L., Anderson L., Paulovich A. G., (2010) An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell. Proteomics 9, 184–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.