

Abstract
Here we describe a novel caged form of the highly reactive bioeffector molecule, nitroxyl (HNO). Reacting the labile nitric oxide (NO)- and HNO-generating salt of structure iPrHN−N(O)=NO−Na+ (1, IPA/NO) with BrCH2OAc produced a stable derivative of structure iPrHN-N(O)=NO−CH2OAc (2, AcOM-IPA/NO), which hydrolyzed an order of magnitude more slowly than 1 at pH 7.4 and 37 °C. Hydrolysis of 2 to generate HNO proceeded by at least two mechanisms. In the presence of esterase, straightforward dissociation to acetate, formaldehyde, and 1 was the dominant path. In the absence of enzyme, free 1 was not observed as an intermediate and the ratio of NO to HNO among the products approached zero. To account for this surprising result, we propose a mechanism in which base-induced removal of the N−H proton of 2 leads to acetyl group migration from oxygen to the neighboring nitrogen, followed by cleavage of the resulting rearrangement product to isopropanediazoate ion and the known HNO precursor, CH3−C(O)−NO. The trappable yield of HNO from 2 was significantly enhanced over 1 at physiological pH, in part because the slower rate of hydrolysis for 2 generated a correspondingly lower steady-state concentration of HNO, thus, minimizing self-consumption and enhancing trapping by biological targets such as metmyoglobin and glutathione. Consistent with the chemical trapping efficiency data, micromolar concentrations of prodrug 2 displayed significantly more potent sarcomere shortening effects relative to 1 on ventricular myocytes isolated from wild-type mouse hearts, suggesting that 2 may be a promising lead compound for the development of heart failure therapies.
Introduction
Nitroxyl (HNO) has been shown to possess intriguing biological properties.1−21 For instance, HNO has been implicated in the mechanism of the inhibitory effect of cyanamide on aldehyde dehydrogenase in treating alcohol abuse(4) and has been demonstrated to enhance contractility in experimental models of heart failure.(9) That HNO rapidly and irreversibly dimerizes and dehydrates to produce nitrous oxide (eq 1),22,23 making it impossible to isolate in pure form, has been a major barrier to achieving a quantitative understanding of its chemical and bioeffector behavior.(24)
As an approach to studying the chemistry and biology of HNO, considerable effort has been devoted to designing compounds capable of serving as caged HNO donors, or prodrugs.1−5,20,21,25−33 We are among those working to develop a platform for systematically generating reliable, controlled fluxes of HNO at programmable rates in physiological media.
Historically, Angeli’s salt (Scheme 1) has been the reagent of choice for generating HNO in aqueous solution.(31) Recently, the related compound IPA/NO (1, the sodium salt of diazeniumdiolated isopropylamine; Scheme 1) has been shown to mimic the chemical and biological properties of Angeli’s salt.(34) Unfortunately, neither of these salts is itself ideal from the drug development perspective, as they are prone to deterioration on storage and they are difficult to purify. Additionally, their similar half-lives of ∼2 min for spontaneous hydrolysis at physiological pH and temperature do not allow for prolonged exposure of target tissues.
Scheme 1. Structures of the Two Related, Spontaneously HNO-Generating Salts, Angeli’s Salt and IPA/NO (1).

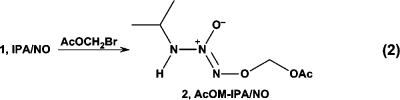
A possible approach to circumventing the limitations but harnessing the advantages of such ions is to alkylate the salts to produce stable neutral species that can be rigorously purified and repurified as necessary, then induced to release HNO by way of enzymatic, hydrolytic, or other tunable activation pathways. To our knowledge, alkylating or otherwise suitably derivatizing Angeli’s salt has been unsuccessful to date. In contrast, 1 has proven amenable to alkylation.(35) Here, we report that acetoxymethylation of 1 as shown in eq 2 leads to prodrug 2 (AcOM-IPA/NO), which is easily purifiable by column chromatography and functions as an advantageous HNO donor with an unexpected mechanism of activation.
 |
Results and Discussion
Compound 2 Hydrolyzes to HNO More Slowly and Efficiently than 1
A key goal of this research is to introduce an array of agents that display a broad range of half-lives for HNO release while minimizing the simultaneous generation of NO that accompanies the spontaneous dissociation of salt 1 at physiological pH. Ideally, this would come in the form of a series of water-soluble compounds that produce reliable fluxes of pure HNO at rates that are tunable for any given experimental application. Compound 2 appears to represent a step in that direction.
Under physiological conditions, Angeli’s salt is primarily an HNO donor (<0.1% NO in the presence of a metal chelator such as 50 μM diethylenetriaminepentaacetic acid (DTPA)).(36) In contrast, 1 generates mixtures of NO and HNO.(34) Although 1 decomposes with nearly the same half-life as Angeli’s salt (2.3 min in pH 7.4 phosphate buffer at 37 °C),(37) conversion of 1 to 2 increased the half-life to 41 min (pH 7.4, 37 °C), which offers a useful addition to the otherwise short half-lives available for spontaneous generation of HNO in aqueous media.
The methods available to detect NO are quite varied and adaptable, but quantitative analysis of HNO is limited by its rapid and irreversible conversion to N2O (eq 1). The most common method for identifying HNO and establishing a lower limit for the amount formed from a given donor is to trap HNO with metmyoglobin (metMb)38,39 according to eq 3. This method was used earlier to suggest that 1 is a donor of both NO and HNO while Angeli’s salt is a donor of only HNO under physiological conditions.(34) Here, we employed this established method to determine whether the products of hydrolyzing 2 include HNO, as would be expected for a prodrug form of 1. For longer-lived donors of HNO such as 2, the rate of autoxidation of MbNO back to metMb (eq 4) requires use of anaerobic conditions to detect HNO.
Both 1 and 2 reductively nitrosylated metMb (Figure 1), but 2 did so more efficiently. We conclude that the slower hydrolysis rate of 2 decreases the steady-state concentration of HNO to impede self-consumption such that MbNO is formed in higher yield. The rates of MbNO formation were comparable to the decomposition rates of 1 and 2, respectively.
Figure 1.
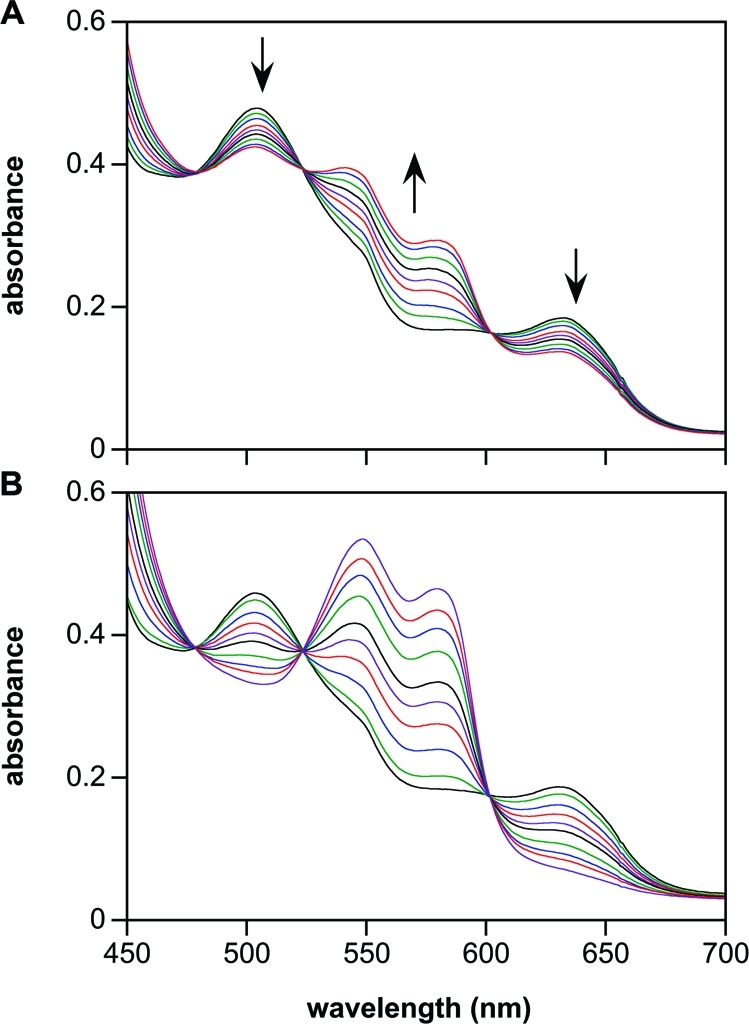
Reductive nitrosylation of metMb (50 μM) by (A) 1 (100 μM), or (B) 2 (100 μM). The assay was performed in phosphate-buffered saline (PBS) (pH 7.4) containing 50 μM DTPA at 37 °C under deaerated conditions until there were no further spectral changes at 543 and 575 nm (1, 2, 4, 6, 9, 14, 23, and 46 min for 1; 3−107 min in 4- to 10-min intervals shown for 2). Concentrations of MbNO produced under these conditions by 1 and 2 were ∼18 and 45 μM, respectively.
Another biological target known to react rapidly with HNO is glutathione (GSH). To confirm the production of HNO in the spontaneous hydrolysis of 2, the metMb assay was repeated in the presence of excess GSH, which reacts with HNO more rapidly than metMb (eqs 3 vs 5) but does not interact appreciably with low concentrations of NO. GSH inhibited reductive nitrosylation by 2 under deaerated conditions, as expected based on the respective rate constants and concentrations employed (Figure 2A, compare Figure 1B). Interestingly, under deaerated conditions, GSH also enhanced MbNO formation during the hydrolysis of 1 (Figure 2B), which hydrolyzes to produce both HNO and NO.(34) In deaerated solution, GSH has been shown to enhance the interaction of NO with metMb (eqs 6 and 7).42,43 Note that the spectral changes for 1 were comparable to those observed when the NO donor DEA/NO(37) was hydrolyzed in the presence of both metMb and GSH (Figure 2C, compare Figure 2B). Quenching by GSH of the reaction of 2 with metMb (Figure 2A) was similar to that of the HNO donor Angeli’s salt (Figure 2D), confirming that HNO generation by 2 under these conditions is comparable to that of Angeli’s salt but enhanced relative to that of 1.
Figure 2.

Effect of GSH on the reductive nitrosylation of metMb (50 μM) by (A) 2 (100 μM), (B) 1 (100 μM), (C) DEA/NO (50 μM), or (D) Angeli’s salt (100 μM). The assay was performed in PBS (pH 7.4) containing 50 μM DTPA and 250 μM GSH at 37 °C under deaerated conditions. Scans are plotted at 0.5 and 60 min in panel A, 2, 4, 6, 9, 14, 23, and 56 min in panel B, and at 1-min intervals to 8 min and then at 11, 14, 18, 23, 28, and 40 min in panel C. Panel D includes spectra at 500 s (green trace), where the decomposition of Angeli’s salt was complete, and at 60 min (blue trace). The effect of GSH appears to be dependent on the batch of myoglobin, indicating the possible interaction with a contaminating protein such as Cu, Zn superoxide dismutase (data not shown; product formation is variable).
Compound 2 Generates Negligible NO on Spontaneous Hydrolysis, in Contrast to 1
As mentioned above, 1 produces both NO and HNO on spontaneous hydrolysis at neutral pH.(34) Surprisingly, the total amount of NO detected on hydrolyzing 2 under the same conditions while purging gases formed in the reaction into an NO-specific chemiluminescence detector was <1% of theoretical (data not shown), suggesting a different hydrolysis mechanism.
To gain insight into the mechanistic origins of this unexpected finding, we explored the pH/rate profiles of the two diazeniumdiolates. Hydrolysis of 1 slowed as pH was increased from 3 to 10 (Figure 3A). This is quite the opposite of the results for 2, whose hydrolysis rate was essentially first order in hydroxide ion concentration between pH 6 and 12 (Figure 3B). Below pH 6, the long half-life of 2 (≥2 days) precluded quantitation.
Figure 3.

The pH-dependence of the first-order rate constant of decomposition measured at 250 nm (mean ± SEM, n ≥ 3, all R2 values >0.995 for 1 and >0.98 for 2) of 1 (A, 100 μM) and 2 (B, 100 μM) at 37 °C in PBS containing 50 μM DTPA and adjusted to the indicated pHs as necessary by adding NaOH or HCl; measured pH was the same at the end of each kinetic run as it was at time zero.
Interestingly, at and above pH 8, the rate of decomposition of 2 exceeded that of 1 (Figure 3), further indicating a mechanism more complicated than simple hydrolysis of 2 to 1. Correspondingly, absorbance at 250 nm due to 1 was not observed in the spontaneous hydrolysis of 2 at pH 7.4 (Figure 4A). Of further importance, as the pH was raised to 12, the higher energy shoulder resolved into a peak at 217 nm of significant intensity, and there was a shift in the 236-nm peak to 278 nm, consistent with ionization of the N−H bond (Figure 4B).(35)
Figure 4.
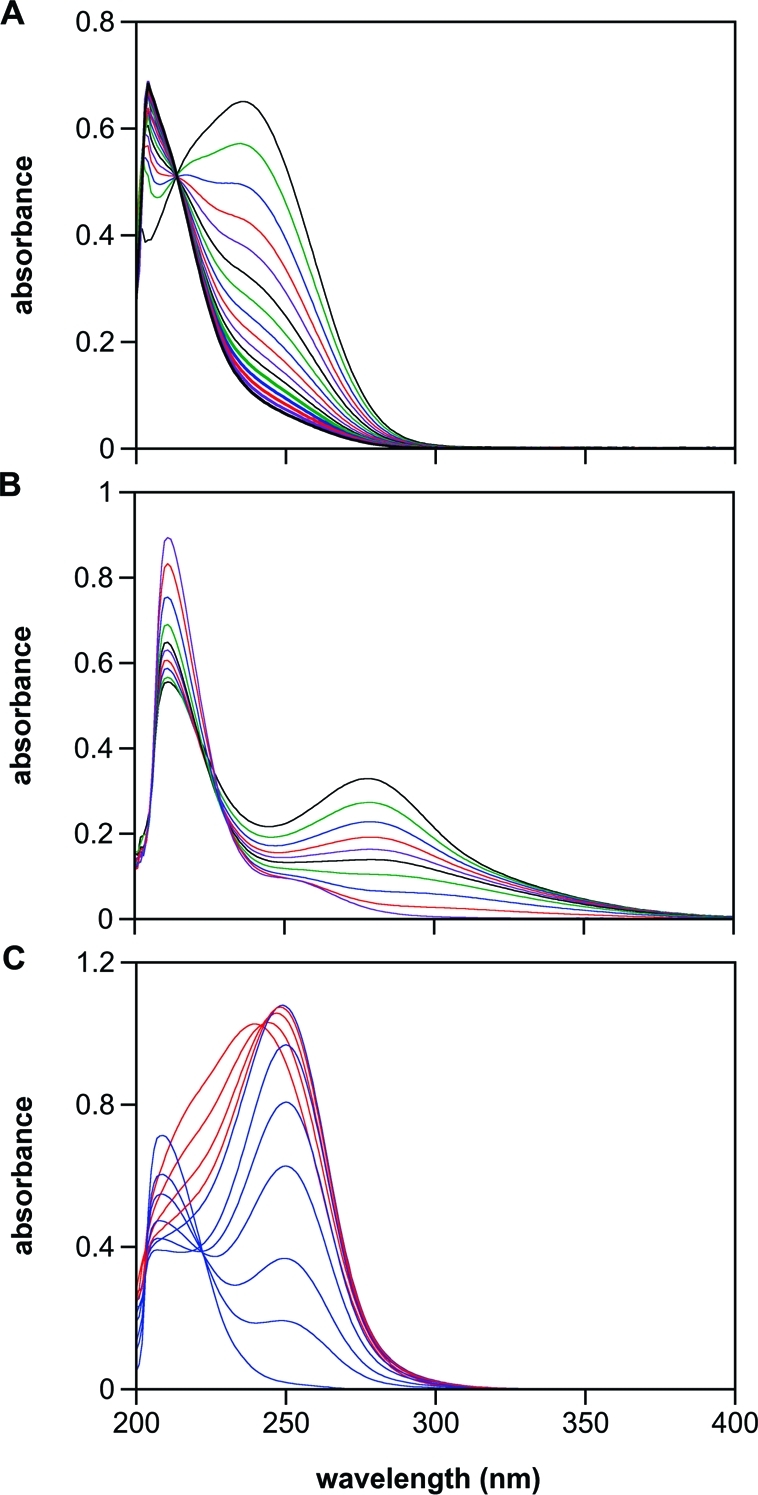
Spontaneous (A and B), and esterase-catalyzed (C) hydrolysis, of 2. In panel A, spectra were collected every 60 s (10-min intervals shown) after dissolving 2 in PBS (pH 7.4) at 37 °C, and only loss of the parent compound was observed. In panel B, the pH was elevated to 12, and scans were collected every 0.5 s (through 2.5 s and 3.5, 5.5, 9.5, and 54.5 s shown). In panel C, porcine liver esterase (1.4 U/mL) was added (pH 7.4), and spectra were collected every 10 s. Hydrolysis of 2 (λmax 236 nm) to 1 (λmax 250 nm) was complete within 40 s (0.067 s−1; red spectra) under these conditions. The rate of decay of 1 in panel C (blue spectra at 40, 100, 160, 240, 400, 600, and 2460 s) was comparable to that shown in Figure 3A (0.0033 s−1 vs 0.0012 s−1, respectively). Elevation of the intensity near 200 nm indicates formation of autoxidation products such as nitrite. Deaeration inhibited this peak (data not shown).
In contrast to the results in simple buffer solutions, 1 was an observable intermediate in the hydrolysis of 2 when active esterase was present. As shown in Figure 4C, the 250-nm peak for 1 dominated the spectrum within a short time after adding the enzyme to 2 in PBS at pH 7.4. Additional studies confirmed that the esterase does not significantly affect the stability of 1 or the ability of metMb and GSH to trap the hydrolytically produced HNO (Figure S2 in Supporting Information).
Quantitative Insight into the NO/HNO Partition by Analysis of the Organic Products
Two limiting mechanisms of hydrolysis for an ionic primary amine diazeniumdiolate are depicted in Scheme 2.(44) Path a is simply the reverse of the synthesis reaction, with the ion dissociating on N-protonation to yield the parent amine and 2 equiv of NO. Path b also begins with a protonation step, but at a different nitrogen, yielding an intermediate that dissociates to form 1 equiv of HNO and a primary nitrosamine; the latter species then rearranges to a diazoate that, in a protonating medium, loses a molecule of water to form a highly reactive alkyldiazonium ion. This intermediate can then react rapidly with available nucleophiles (represented in Scheme 2 as water) to displace a dinitrogen molecule and produce a nucleophile/carbocation adduct (shown as the alcohol in Scheme 2); if the R+ group bears a hydrogen atom in the β-position, loss of that proton would produce an olefin.
Scheme 2. Two Limiting Hydrolysis Mechanisms for Generation of NO versus HNO from a Diazeniumdiolated Primary Amine Anion.

With the assumption that paths a and b are the only routes by which 1 can dissociate, the yield of amine observed in such a dissociation can be taken as a quantitative measure of the preponderance of path a, while the combined yields of olefin and RX species quantitatively reflect the extent of competing route b. With this in mind, we followed the course of hydrolysis for both 1 and 2 by NMR and quantified the organic products. Consistent with the HNO/NO yield data reported above, complete dissociation of 1 at pH 7.4 generated similar amounts of isopropylamine and isopropanol, with essentially saturating concentrations of propene also being observed (Figure 5A). In contrast, methyl peaks attributable to the amine bordered on undetectable when 2 was hydrolyzed under the same conditions, pointing to a hydrolysis mechanism for 2 that does not involve 1 as an intermediate (Figure 5B).
Figure 5.

NMR spectra of products generated on exhaustive hydrolysis of 1 (A) and 2 (B) in 0.1 M phosphate buffer containing 50 μM DTPA at pH 7.4 and 37 °C. Note the near-absence of isopropylamine, a byproduct of the NO-forming pathway, in the hydrolysis of 2.
Differences in time course as well as stoichiometry in the alkaline hydrolysis of 1 and 2, illustrated in Figure 6, also matched expectation based on the HNO/NO yield and rate data. Ionic 1 hydrolyzed slowly at pH 11. Compound 2, on the other hand, completely dissociated within 5 min at 37 °C in pH 11 carbonate buffer. Peaks for formaldehyde and acetate were seen in addition to those for the carbocation-derived alcohol and alkene, but the amine was undetectable.
Figure 6.

Distribution of products in the hydrolysis at 37 °C of 1 (left side panels) and 2 (right side panels) in pH 11 aqueous 0.05 M sodium carbonate containing 10% deuterium oxide. Note that hydrolysis of 1 to isopropanol and propene proceeded to the extent of only ∼15% during 1 h while dissociation of 2 was complete within 5 min. Since 1 is more stable than 2 at this pH, the absence of a peak for it in the spectrum of fully dissociated 2 indicates that free 1 is not significantly produced in the hydrolysis of 2 under these conditions. An expanded view of the spectrum at the lower right is included as Figure S1 in Supporting Information. It provides integrals for all the organic products including formaldehyde, and shows that the combined yield of propene plus isopropanol essentially matches that of acetate, as expected for a hydrolysis mechanism for 2 that produces an NO/HNO ratio approaching zero.
Mechanism of Base-Induced Hydrolysis
Although 1 hydrolyzed 2 orders of magnitude more slowly than 2 at pH 11 (Figure 3), the characteristic 250-nm wavelength maximum of 1 was detected only at low intensity during the rapid hydrolysis of 2 at this pH (Figure 4B), and the corresponding NMR signals for 1 could not be separated from the noise (Figure 6). One way to rationalize the near absence of free 1 as a product of hydrolyzing 2 is to postulate that base-induced removal of the N−H proton of 2 generates 3, as in Scheme 3. This intermediate might be expected to undergo 1−4 acyl migration via cyclic intermediate 4. Expulsion of formaldehyde from 4 could give 5, which could then fragment with N−N bond cleavage to produce diazoate ion 6 and acylnitroso derivative 7. Aliphatic diazoates are well-known progenitors of unstable carbenium ions that can indiscriminately alkylate ambient nucleophiles (such as water to produce the corresponding alcohol) with cogeneration of an equivalent of dinitrogen,(45) and acylnitroso compounds such as 7 are known to hydrolyze to HNO with the derived carboxylate as byproduct.21,46
Scheme 3. Proposed Mechanism of Hydrolytic HNO Generation from 2 in the Absence of Esterase.

Precedent for ionization of O2-substituted IPA/NO derivatives such as 2 with an accompanying shift in absorbance maximum from ∼236 to ∼278 nm (see Figure 4B) has been reported for methylated IPA/NO,(47) with a measured pKa of 12.3. For 2, with increasing alkalinity this shift became apparent at pH 11, indicating a comparable pKa. The importance of anionic resonance form 3 as a nucleophilic site is supported by the finding that glucosylated IPA/NO can be methylated at that site in basic media.(48) Examples of 1−4 acyl migrations reminiscent of that shown in Scheme 3 for conversion of 3 to 5 have been reported,(49) and cleavage of 5 to the two well established reactive intermediates 6 and 7 would not be surprising. Consistent with the view that the base-catalyzed dissociation of 2 according to Scheme 3 is the only pathway by which it decomposes in enzyme-free media, product ratios were the same in the slow pH 7.4 reaction (Figure 5) and its faster pH 11 counterpart (Figure 6), except that propene appeared to be present in somewhat higher concentration at the higher pH. The observed integral for acetate corresponded to a yield (taken as 100%) essentially matching that expected for the sum of propene plus isopropanol (26% + 79%, respectively; see Figure S1 in the Supporting Information), suggesting that analysis of the organic products in these reactions can provide reliable quantitative insight into the partition between the NO- and HNO-forming pathways.
Effects of 1 and 2 on Isolated Cardiac Myocytes
To determine whether the slower and more efficient generation of HNO by 2 relative to 1 translates to enhanced bioactivity, we turned to a model for one of the most exciting applications of HNO donors, the treatment of heart failure. Donors of HNO enhance contractility in failing hearts.(9) As might be expected for an agent that generates substantially more HNO than 1, prodrug 2 proved more potent at micromolar concentrations in ventricular myocytes isolated from wild-type mice. As shown in Figure 7, 2 elicited a roughly 4-fold higher change than 1 in sarcomere shortening relative to baseline at 3 μM. While this finding suggests potential pharmacological utility on the part of 2, it is important to note its apparently negative inotropic effect at 50 μM, a possible reflection of toxicity not seen with 1 (Figure 7). We are currently exploring the origin of this undesirable effect in an attempt to infer a means of circumventing it.
Figure 7.

Relative increases in contractility in ventricular myocytes isolated from wild-type mouse hearts. Dose−response effects of 1 and 2 on cell shortening. ***P < 0.001 vs control. Number of cells = 6 at 1 μM, 5 at 2 μM, and 15 for 3 and 50 μM.
Summary and Significance
Our results suggest that diazeniumdiolate chemistry may offer a versatile platform for development of advantageous HNO sources for fundamental research and biomedical applications. The present comparison shows that suitable derivatization of 1 can dramatically slow the rate of spontaneous HNO generation, with concomitant improvement in the efficiency of its capture by biological targets. Furthermore, conversion of relatively unstable 1 to 2 provides a rigorously purifiable derivative with alternate mechanisms of hydrolysis to HNO and improved ability to strengthen contraction of beating cardiac myocytes at low micromolar concentrations. Work currently in progress is focused on further exploring the structure−activity and −reactivity relationships in the primary amine diazeniumdiolate series, with the aim of developing a variety of improved tools for probing the chemistry and pharmacology of the intriguing bioeffector molecule HNO.
Acknowledgments
This work was supported by: the Marie Curie Intra European Fellowship within the 7th European Community Framework Programme (PIEF-GA-2008-221666 to S.D.); the Forschungsförderungsfonds (NWF-08/04 to S.D.); the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research; NCI contract HHSN261200800001E; National Institutes of Health grants R01-GM076247 to K.M.M. and 1F31AA018069-01A1 (National Institute on Alcohol Abuse and Alcoholism) to D.J.S.
Supporting Information Available
Complete ref (34); experimental procedures including synthesis and characterization of 2; details of the analytical and biological methodologies employed; and additional spectral data from studies of the esterase-mediated hydrolyses of 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lee M. J. C.; Nagasawa H. T.; Elberling J. A.; DeMaster E. G. J. Med. Chem. 1992, 35, 3648–3652. [DOI] [PubMed] [Google Scholar]
- Fukuto J. M.; Hszieh R.; Gulati P.; Chiang K. T.; Nagasawa H. T. Biochem. Biophys. Res. Commun. 1992, 187, 1367–1373. [DOI] [PubMed] [Google Scholar]
- Nagasawa H. T.; Yost Y.; Elberling J. A.; Shirota F. N.; DeMaster E. G. Biochem. Pharmacol. 1993, 45, 2129–2134. [DOI] [PubMed] [Google Scholar]
- Nagasawa H. T.; Kawle S. P.; Elberling J. A.; DeMaster E. G.; Fukuto J. M. J. Med. Chem. 1995, 38, 1865–1871. [DOI] [PubMed] [Google Scholar]
- Lee M. J. C.; Shoeman D. W.; Goon D. J. W.; Nagasawa H. T. Nitric Oxide 2001, 5, 278–287. [DOI] [PubMed] [Google Scholar]
- Wink D. A.; Miranda K. M.; Katori T.; Mancardi D.; Thomas D. D.; Ridnour L.; Espey M. G.; Feelisch M.; Colton C. A.; Fukuto J. M.; Pagliaro P.; Kass D. A.; Paolocci N. Am. J. Physiol.: Heart Circ. Physiol. 2003, 285, H2264–H2276. [DOI] [PubMed] [Google Scholar]
- Feelisch M. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 4978–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda K. M.; Paolocci N.; Katori T.; Thomas D. D.; Ford E.; Bartberger M. D.; Espey M. G.; Kass D. A.; Feelisch M.; Fukuto J. M.; Wink D. A. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 9196–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolocci N.; Katori T.; Champion H. C.; St. John M. E.; Miranda K. M.; Fukuto J. M.; Wink D. A.; Kass D. A. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 5537–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong E.; Tumbev V.; Abramson J.; Salama G.; Stoyanovsky D. A. Cell Calcium 2005, 37, 87–96. [DOI] [PubMed] [Google Scholar]
- Miranda K. M.; Ridnour L.; Espey M.; Citrin D.; Thomas D.; Mancardi D.; Donzelli S.; Wink D. A.; Katori T.; Tocchetti C. G.; Ferlito M.; Paolocci N.; Fukuto J. M. Prog. Inorg. Chem. 2005, 54, 349–384. [Google Scholar]
- Miranda K. M. Coord. Chem. Rev. 2005, 249, 433–455. [Google Scholar]
- Paolocci N.; Jackson M. I.; Lopez B. E.; Miranda K.; Tocchetti C. G.; Wink D. A.; Hobbs A. J.; Fukuto J. M. Pharmacol. Ther. 2007, 113, 442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáenz D. A.; Bari S. E.; Salido E.; Chianelli M.; Rosenstein R. E. Neurochem. Int. 2007, 51, 424–432. [DOI] [PubMed] [Google Scholar]
- Landino L. M.; Koumas M. T.; Mason C. E.; Alston J. A. Chem. Res. Toxicol. 2007, 20, 1693–1700. [DOI] [PubMed] [Google Scholar]
- Favaloro J. L.; Kemp-Harper B. K. Cardiovasc. Res. 2007, 73, 587–596. [DOI] [PubMed] [Google Scholar]
- Irvine J. C.; Favaloro J. L.; Widdop R. E.; Kemp-Harper B. K. Hypertension 2007, 49, 885–892. [DOI] [PubMed] [Google Scholar]
- Fukuto J. M.; Jackson M. I.; Kaludercic N.; Paolocci N. Methods Enzymol. 2008, 440, 411–431. [DOI] [PubMed] [Google Scholar]
- Lancel S.; Zhang J.; Evangelista A.; Trucillo M. P.; Tong X.; Siwik D. A.; Cohen R. A.; Colucci W. S. Circ. Res. 2009, 104, 720–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisz J. A.; Bechtold E.; King S. B. Dalton Trans. 2010, 39, 5203–5212. [DOI] [PubMed] [Google Scholar]
- Kovacic P.; Edwards C. L.. J. Recept. Signal Transduct. [Early Online]. DOI: 10.3109/10799893.2010.497152. Published online: Jun 30, 2010. [Google Scholar]
- Smith P. A. S.; Hein G. E. J. Am. Chem. Soc. 1960, 82, 5731–5740. [Google Scholar]
- Kohout F. C.; Lampe F. W. J. Am. Chem. Soc. 1965, 87, 5795–5796. [Google Scholar]
- Shafirovich V.; Lymar S. V. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 7340–7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware R. W. Jr.; King S. B. J. Org. Chem. 2000, 65, 8725–8729. [DOI] [PubMed] [Google Scholar]
- Zeng B. B.; Huang J.; Wright M. W.; King S. B. Bioorg. Med. Chem. Lett. 2004, 14, 5565–5568. [DOI] [PubMed] [Google Scholar]
- King S. B. Free Radical Biol. Med. 2004, 37, 735–736. [DOI] [PubMed] [Google Scholar]
- Huang J.; Kim-Shapiro D. B.; King S. B. J. Med. Chem. 2004, 47, 3495–3501. [DOI] [PubMed] [Google Scholar]
- King S. B. Curr. Top. Med. Chem. 2005, 5, 665–673. [DOI] [PubMed] [Google Scholar]
- Pennington R. L.; Sha X.; King S. B. Bioorg. Med. Chem. Lett. 2005, 15, 2331–2334. [DOI] [PubMed] [Google Scholar]
- Miranda K. M.; Nagasawa H. T.; Toscano J. P. Curr. Top. Med. Chem. 2005, 5, 649–664. [DOI] [PubMed] [Google Scholar]
- Sha X.; Isbell T. S.; Patel R. P.; Day C. S.; King S. B. J. Am. Chem. Soc. 2006, 128, 9687–9692. [DOI] [PubMed] [Google Scholar]
- Kirsch M.; Büscher A. M.; Aker S.; Schulz R.; de Groot H. Org. Biomol. Chem. 2009, 7, 1954–1962. [DOI] [PubMed] [Google Scholar]
- Miranda K. M.; et al. J. Med. Chem. 2005, 48, 8220–8228. [DOI] [PubMed] [Google Scholar]
- Saavedra J. E.; Bohle D. S.; Smith K. N.; George C.; Deschamps J. R.; Parrish D.; Ivanic J.; Wang Y.-N.; Citro M. L.; Keefer L. K. J. Am. Chem. Soc. 2004, 126, 12880–12887. [DOI] [PubMed] [Google Scholar]
- Lopez B. E.; Shinyashiki M.; Han T. H.; Fukuto J. M. Free Radical Biol. Med. 2007, 42, 482–491. [DOI] [PubMed] [Google Scholar]
- Maragos C. M.; Morley D.; Wink D. A.; Dunams T. M.; Saavedra J. E.; Hoffman A.; Bove A. A.; Isaac L.; Hrabie J. A.; Keefer L. K. J. Med. Chem. 1991, 34, 3242–3247. [DOI] [PubMed] [Google Scholar]
- Bazylinski D. A.; Hollocher T. C. J. Am. Chem. Soc. 1985, 107, 7982–7986. [Google Scholar]
- Doyle M. P.; Mahapatro S. N.; Broene R. D.; Guy J. K. J. Am. Chem. Soc. 1988, 110, 593–599. [Google Scholar]
- Andersen H. J.; Skibsted L. H. J. Agric. Food. Chem. 1992, 40, 1741–1750. [Google Scholar]
- Laverman L. E.; Wanat A.; Oszajca J.; Stochel G.; Ford P. C.; van Eldik R. J. Am. Chem. Soc. 2001, 123, 285–293. [DOI] [PubMed] [Google Scholar]
- Reichenbach G.; Sabatini S.; Palombari R.; Palmerini C. A. Nitric Oxide 2001, 5, 395–401. [DOI] [PubMed] [Google Scholar]
- Ford P. C.; Fernandez B. O.; Lim M. D. Chem. Rev. 2005, 105, 2439–2456. [DOI] [PubMed] [Google Scholar]
- Dutton A. S.; Suhrada C. P.; Miranda K. M.; Wink D. A.; Fukuto J. M.; Houk K. N. Inorg. Chem. 2006, 45, 2448–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. B.; March J.. March’s Advanced Organic Chemistry. Reactions, Mechanisms, and Structure. Fifth ed.; John Wiley & Sons, Inc.: New York, 2001; pp 447−448. [Google Scholar]
- Cohen A. D.; Zeng B.-B.; King S. B.; Toscano J. P. J. Am. Chem. Soc. 2003, 125, 1444–1445. [DOI] [PubMed] [Google Scholar]
- Wang Y.-N.; Bohle D. S.; Bonifant C. L.; Chmurny G. N.; Collins J. R.; Davies K. M.; Deschamps J.; Flippen-Anderson J. L.; Keefer L. K.; Klose J. R.; Saavedra J. E.; Waterhouse D. J.; Ivanic J. J. Am. Chem. Soc. 2005, 127, 5388–5395. [DOI] [PubMed] [Google Scholar]
- Bohle D. S.; Keefer L. K.; Saavedra J. E. Tetrahedron Lett. 2009, 50, 5917–5919. [Google Scholar]
- Banthorpe D. V.Rearrangements involving amino groups. In The Chemistry of the Amino Group, Patai S., Ed.; Interscience Publishers: London, 1968; pp 651−654. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.