

Atherosclerosis is the major cause of death and disability in the United States, Europe, and much of Asia. The lesions of atherosclerosis have a nonuniform distribution in the vasculature and are prone to develop at bends, branches, and bifurcations of the aorta and its conduit arteries. At these sites in the vasculature laminar flow is disturbed by recirculation, which resembles an eddy at a river branch. Here the endothelium of the blood vessel is exposed to reduced and oscillating flow, a hemodynamic condition that produces physicochemical and biological alterations predisposing to atherogenesis (Fig. 1). Recirculation causes an increase in particle residence time, so that lipoproteins and monocytes (the anlagen of atherosclerosis) have greater contact with the endothelium in these regions (1). The endothelium itself is morphologically altered. In relatively straight segments of a conduit vessel, endothelial cells are regularly aligned, each cell with its longitudinal axis oriented in the direction of flow. By contrast, in areas of disturbed flow, the usual orientation of endothelial cells is lost, and the cells become more polygonal in appearance. Intriguingly, endothelial cells at branch points appear to age faster. Endothelial cells exposed to disturbed flow in vitro turn over more rapidly (2). In human iliac arteries, endothelial cells at the bifurcation have shorter telomeres, consistent with a focal acceleration of senescence (3). Senescent human endothelial cells produce less nitric oxide (NO⋅), generate more superoxide anion (O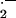 ), and are more adhesive for monocytes, effects that can be reversed by transfection with the gene encoding telomerase (4).
), and are more adhesive for monocytes, effects that can be reversed by transfection with the gene encoding telomerase (4).
Figure 1.

Vascular effects of laminar or disturbed flow. The tractive force of fluid flow, i.e., shear stress, is sensed by the endothelium; the signal transduction apparatus is incompletely characterized but involves activation of specific kinases and ion channels that induce the release of vasoactive factors and the expression of genes affecting vascular function and structure. The response to physiological laminar flow and disturbed flow (such as that observed at bends, branches, and bifurcations) is quite different. The response activated by disturbed flow predisposes to atherogenesis. KCa, calcium-activated potassium channel; BK, bradykinin; Ach, acetylcholine; PGI2, prostacyclin; EDHF, endothelium-dependent hyperpolarizing factor; SOD, superoxide dismutase; COX, cyclooxygenase; CAM, cell adhesion molecule (e.g., vascular cell adhesion molecule); tPA, tissue plasminogen activator; VSM, vascular smooth muscle; TGFβ, transforming growth factor β; PDGF, platelet-derived growth factor; SMAD, family of flow-regulated transcriptional factors; ET-1, endothelin; EC, endothelial cell; NFκB, Elk-1, and p-CREB, redox-activated transcriptional proteins.
Laminar flow in a straight segment of the conduit vessel exposes the endothelium to the tractive force of shear stress. Shear stress in a physiological range of 10–40 dynes/cm2 (1 dyne = 10 μN) activates signaling pathways that induce endothelial elaboration of a number of vasoactive factors such as NO⋅, prostacyclin, tissue plasminogen activator, and transforming growth factor β (5–8). Nitric oxide is paradigmatic of these factors that promote vasodilatation, inhibit adherence of circulating blood elements, and suppress the proliferation and migration of vascular smooth muscle cells (9). In addition to triggering the immediate release of NO⋅, laminar shear stress increases the expression of NO synthase (NOS) (10), and that of superoxide dismutase (which reduces oxidative degradation of NO⋅; ref. 11). Thus, in a physiological range, laminar shear stress maintains vascular homeostasis. By contrast, disturbed flow (causing low and oscillating shear stress) inhibits the release of these factors. At the branches of coronary arteries in humans, endothelium-dependent vasodilatation is impaired (12). Similarly, when cultured endothelial cells are exposed to disturbed flow, NOS expression is not up-regulated as when endothelial cells are exposed to laminar flow (13). This hemodynamic alteration of endothelial biology predisposes to processes involved in atherogenesis. This theme is further explored by de Nigris et al. (14) in studies that document the interaction of shear stress, NOS, and proatherogenic transcriptional factors. They find that in the aorta of the hypercholesterolemic low-density lipoprotein receptor-deficient mouse, the expression of NOS is down-regulated, whereas expression of the proatherogenic transcriptional factors Elk-1 and p-CREB is increased. This alteration in protein expression is particularly evident at lesion-prone sites in the aorta, i.e., areas of disturbed flow. The reduction in NOS expression was reversed by administration of l-arginine or the antioxidant vitamins C and E. There was a synergistic interaction of l-arginine and the antioxidant vitamins to increase NOS expression and reduce Elk-1 and p-CREB. Moreover, the administration of l-arginine or the antioxidant vitamins suppressed lesion formation, again with a synergistic interaction. They observed a similar interaction between antioxidant vitamins and l-arginine on the expression of NOS, Elk-1, and p-CREB in cultured endothelial cells exposed to variable levels of shear stress by using a cone-plate viscometer.
The balance between vascular generation of two free radicals, NO⋅ and O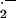 , is a major determinant of endothelial adhesiveness and lesion formation. Cultured endothelial cells exposed to physiological laminar flow generate more NO and less superoxide anion; consequently, activation of the transcriptional protein NF-κB is attenuated, expression of the endothelial adhesion molecule VCAM-1 is reduced, and endothelial adhesiveness for monocytes is diminished (15). As confirmed by de Nigris et al. (14), the balance between vascular NO⋅ and O
, is a major determinant of endothelial adhesiveness and lesion formation. Cultured endothelial cells exposed to physiological laminar flow generate more NO and less superoxide anion; consequently, activation of the transcriptional protein NF-κB is attenuated, expression of the endothelial adhesion molecule VCAM-1 is reduced, and endothelial adhesiveness for monocytes is diminished (15). As confirmed by de Nigris et al. (14), the balance between vascular NO⋅ and O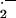 can also be shifted by administration of antioxidants or the NO precursor l-arginine. In cell culture and animal models of hypercholesterolemia and atherosclerosis, l-arginine becomes rate limiting for NO production (16, 17). This may be caused by increased tissue or plasma levels of the endogenous inhibitor of NOS, asymmetric dimethylarginine; increased expression of vascular arginase; reduced l-arginine transport; and/or increased local utilization of l-arginine by immune or vascular cells expressing inducible NOS (18–21). In the setting of reduced availability of l-arginine, NOS transfers electrons to oxygen to create superoxide anion (22). Superoxide anion generated by NOS, NADPH oxidase, mitochondrial oxidases, or other enzymes oxidizes NO to the highly reactive and cytotoxic free radical peroxynitrite anion (23). Furthermore, oxidative stress attenuates the expression of endothelial NOS, in part by reducing mRNA stability (24). Finally, oxidant-responsive genes such as VCAM-1 or MCP-1 are induced, increasing monocyte adhesion, infiltration, and lesion formation (15, 25). Under these conditions, l-arginine and/or antioxidants can reverse the pathobiology induced by oxidative stress (18, 26).
can also be shifted by administration of antioxidants or the NO precursor l-arginine. In cell culture and animal models of hypercholesterolemia and atherosclerosis, l-arginine becomes rate limiting for NO production (16, 17). This may be caused by increased tissue or plasma levels of the endogenous inhibitor of NOS, asymmetric dimethylarginine; increased expression of vascular arginase; reduced l-arginine transport; and/or increased local utilization of l-arginine by immune or vascular cells expressing inducible NOS (18–21). In the setting of reduced availability of l-arginine, NOS transfers electrons to oxygen to create superoxide anion (22). Superoxide anion generated by NOS, NADPH oxidase, mitochondrial oxidases, or other enzymes oxidizes NO to the highly reactive and cytotoxic free radical peroxynitrite anion (23). Furthermore, oxidative stress attenuates the expression of endothelial NOS, in part by reducing mRNA stability (24). Finally, oxidant-responsive genes such as VCAM-1 or MCP-1 are induced, increasing monocyte adhesion, infiltration, and lesion formation (15, 25). Under these conditions, l-arginine and/or antioxidants can reverse the pathobiology induced by oxidative stress (18, 26).
Endothelial dysfunction is an independent predictor of the severity of cardiovascular disease.
Whether interventions to enhance endothelial NOS and/or reduce oxidative stress will be clinically useful remain to be determined. l-arginine supplementation has been shown to improve endothelial function, enhance coronary blood flow, reduce angina, and improve exercise tolerance in patients with coronary artery disease (CAD; refs. 27 and 28); however, there are no data regarding its effects on progression of CAD or cardiovascular mortality. Although antioxidants C and E can improve endothelial function in humans with CAD or with risk factors for CAD (29), large randomized clinical trials with antioxidant vitamin therapy have been for the most part disappointingly negative (30). In fact, high doses of antioxidant vitamins blunt the beneficial effects of statins to increase high-density lipoprotein cholesterol and reduce progression of CAD (31). By contrast, statins or angiotensin-converting enzyme inhibitors reduce the progression of CAD and total mortality (32, 33). Some of the benefit of these agents has been ascribed to their effect of improving endothelial function (e.g., increasing NOS activity and expression, reducing the generation of O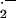 ), but this remains conjecture. However, endothelial dysfunction (as manifested by reduced vasodilatation to endothelial agonists, or by increased plasma levels of the NOS inhibitor ADMA) has been shown in multiple studies to be an independent predictor of the severity and sequelae of cardiovascular disease (34–39). Thus endothelial-targeted diagnostics and therapy are likely to be important tools in the future management of cardiovascular diseases.
), but this remains conjecture. However, endothelial dysfunction (as manifested by reduced vasodilatation to endothelial agonists, or by increased plasma levels of the NOS inhibitor ADMA) has been shown in multiple studies to be an independent predictor of the severity and sequelae of cardiovascular disease (34–39). Thus endothelial-targeted diagnostics and therapy are likely to be important tools in the future management of cardiovascular diseases.
Footnotes
See companion article on page 1420.
References
- 1.Glagov S, Zarins C, Giddens D P, Ku D N. Arch Pathol Lab Med. 1988;112:1018–1031. [PubMed] [Google Scholar]
- 2.Davies P F, Remuzzi A, Gordon E J, Dewey C F, Jr, Gimbrone M A., Jr Proc Natl Acad Sci USA. 1986;83:2114–2117. doi: 10.1073/pnas.83.7.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang E, Harley C B. Proc Natl Acad Sci USA. 1995;92:11190–11194. doi: 10.1073/pnas.92.24.11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsushita H, Chang E, Glassford A J, Cooke J P, Chiu C P, Tsao P S. Circ Res. 2001;89:793–798. doi: 10.1161/hh2101.098443. [DOI] [PubMed] [Google Scholar]
- 5.Davies P. Physiol Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooke J P, Rossitch E, Andon N, Loscalzo J, Dzau V J. J Clin Invest. 1991;88:1663–1671. doi: 10.1172/JCI115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhagyalakshmi A, Frangos J A. Biochem Biophys Res Commun. 1989;158:31–37. doi: 10.1016/s0006-291x(89)80172-x. [DOI] [PubMed] [Google Scholar]
- 8.Ohno M, Cooke J P, Dzau V J, Gibbons G H. J Clin Invest. 1995;95:1363–1369. doi: 10.1172/JCI117787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooke J P, Dzau V J. Annu Rev Med. 1997;48:489–509. doi: 10.1146/annurev.med.48.1.489. [DOI] [PubMed] [Google Scholar]
- 10.Uematsu M, Ohara Y, Navas J P, Nishida K, Murphy T J, Alexander R W, Nerem R M, Harrison D G. Am J Physiol. 1995;269:C1371–C1378. doi: 10.1152/ajpcell.1995.269.6.C1371. [DOI] [PubMed] [Google Scholar]
- 11.Topper J N, Cai J, Falb D, Gimbrone M A., Jr Proc Natl Acad Sci USA. 1996;93:10417–10422. doi: 10.1073/pnas.93.19.10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLenachan J M, Vita J, Fish D R, Treasure C B, Cox D A, Ganz P, Selwyn A P. Circulation. 1990;82:1169–1173. doi: 10.1161/01.cir.82.4.1169. [DOI] [PubMed] [Google Scholar]
- 13.Topper J N, Gimbrone M A., Jr Mol Med Today. 1999;5:40–46. doi: 10.1016/s1357-4310(98)01372-0. [DOI] [PubMed] [Google Scholar]
- 14.de Nigris F, Lerman L O, Ignarro S W, Sica G, Lerman A, Palinski W, Ignarro L J, Napoli C. Proc Natl Acad Sci USA. 2003;100:1420–1425. doi: 10.1073/pnas.0237367100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsao P S, Buitrago R, Chan J R, Cooke J P. Circulation. 1996;94:1682–1689. doi: 10.1161/01.cir.94.7.1682. [DOI] [PubMed] [Google Scholar]
- 16.Cooke J P, Singer A H, Tsao P, Zera P, Rowan R A, Billingham M E. J Clin Invest. 1992;90:1168–1172. doi: 10.1172/JCI115937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsao P, McEvoy L M, Drexler H, Butcher E C, Cooke J P. Circulation. 1994;89:2176–2182. doi: 10.1161/01.cir.89.5.2176. [DOI] [PubMed] [Google Scholar]
- 18.Cooke J P. Arterioscler Thromb Vasc Biol. 2000;20:2032–2037. doi: 10.1161/01.atv.20.9.2032. [DOI] [PubMed] [Google Scholar]
- 19.Wei L H, Wu G, Morris S M, Jr, Ignarro L J. Proc Natl Acad Sci USA. 2001;98:9260–9264. doi: 10.1073/pnas.161294898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jay M T, Chirico S, Siow R C, Bruckdorfer K R, Jacobs M, Leake D S, Pearson J D, Mann G E. Exp Physiol. 1997;82:349–360. doi: 10.1113/expphysiol.1997.sp004030. [DOI] [PubMed] [Google Scholar]
- 21.Baker C S, Hall R J, Evans T J, Pomerance A, Maclouf J, Creminon C, Yacoub M H, Polak J M. Arterioscler Thromb Vasc Biol. 1999;19:646–655. doi: 10.1161/01.atv.19.3.646. [DOI] [PubMed] [Google Scholar]
- 22.Pritchard K A, Jr, Groszek L, Smalley D M, Sessa W C, Wu M, Villalon P, Wolin M S, Stemerman M B. Circ Res. 1995;77:510–518. doi: 10.1161/01.res.77.3.510. [DOI] [PubMed] [Google Scholar]
- 23.Beckman J S, Koppenol W H. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 24.Liao J K, Shin W S, Lee W Y, Clark S L. J Biol Chem. 1995;270:319–324. doi: 10.1074/jbc.270.1.319. [DOI] [PubMed] [Google Scholar]
- 25.Tsao P S, Wang B, Buitrago R, Shyy J Y, Cooke J P. Circulation. 1997;96:934–940. doi: 10.1161/01.cir.96.3.934. [DOI] [PubMed] [Google Scholar]
- 26.Witztum J L, Steinberg D. Trends Cardiovasc Med. 2001;11:93–102. doi: 10.1016/s1050-1738(01)00111-6. [DOI] [PubMed] [Google Scholar]
- 27.Lerman A, Burnett J C, Jr, Higano S T, McKinley L J, Holmes D R., Jr Circulation. 1998;97:2123–2128. doi: 10.1161/01.cir.97.21.2123. [DOI] [PubMed] [Google Scholar]
- 28.Maxwell A J, Zapien M P, Pearce G L, MacCallum G, Stone P H. J Am Coll Cardiol. 2002;39:37–45. doi: 10.1016/s0735-1097(01)01708-9. [DOI] [PubMed] [Google Scholar]
- 29.Diaz M N, Frei B, Vita J A, Keaney J F., Jr N Engl J Med. 1997;337:408–416. doi: 10.1056/NEJM199708073370607. [DOI] [PubMed] [Google Scholar]
- 30.Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. N Engl J Med. 2000;342:154–160. doi: 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- 31.Brown B G, Xue-Qiao Z, Chait A, Fisher L D, Cheung M C, Morse J S, Dowdy A A, Marino E K, Bolson E L, Alaupovic P, et al. N Engl J Med. 2001;345:1583–1592. doi: 10.1056/NEJMoa011090. [DOI] [PubMed] [Google Scholar]
- 32.Dzau V J, Bernstein K, Celermajer D, Cohen J, Dahlof B, Deanfield J, Diez J, Drexler H, Ferrari R, Van Gilst W, et al. Cardiovasc Drugs Ther. 2002;16:149–160. doi: 10.1023/a:1015709617405. [DOI] [PubMed] [Google Scholar]
- 33.Takemoto M, Liao J K. Arterioscler Thromb Vasc Biol. 2001;11:1712–1719. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 34.Miyazaki H, Matsuoka H, Cooke J P, Usui M, Ueda S, Okuda S, Imaizumi T. Circulation. 1999;99:1141–1146. doi: 10.1161/01.cir.99.9.1141. [DOI] [PubMed] [Google Scholar]
- 35.Schächinger V, Britten M B, Zeiher A M. Circulation. 2000;101:1899–1906. doi: 10.1161/01.cir.101.16.1899. [DOI] [PubMed] [Google Scholar]
- 36.Suwaidi J A, Hamasaki S, Higano S T, Nishimura R A, Holmes D R, Jr, Lerman A. Circulation. 2000;101:948–954. doi: 10.1161/01.cir.101.9.948. [DOI] [PubMed] [Google Scholar]
- 37.Gokce N, Keaney J F, Jr, Hunter L M, Watkins M T, Menzoian J O, Vita J A. Circulation. 2002;105:1567–1572. doi: 10.1161/01.cir.0000012543.55874.47. [DOI] [PubMed] [Google Scholar]
- 38.Zoccali C, Bode-Böger S M, Mallamaci F, Benedetto F, Tripepi G, Malatino L, Cataliotti A, Bellanuova I, Fermo I, Frolich J, Boger R. Lancet. 2001;358:2113–2117. doi: 10.1016/s0140-6736(01)07217-8. [DOI] [PubMed] [Google Scholar]
- 39.Valkonen V P, Paiva H, Salonen J T, Lakka T A, Lehtimaki T, Laakso J, Laaksonen R. Lancet. 2001;358:2127–2128. doi: 10.1016/S0140-6736(01)07184-7. [DOI] [PubMed] [Google Scholar]