

Abstract
Bacillus subtilis, a Gram-positive, endospore-forming soil bacterium, was grown in media made with water of varying oxygen (δ18O) and hydrogen (δD) stable isotope ratios. Logarithmically growing cells and spores were each harvested from the cultures and their δ18O and δD values determined. Oxygen and hydrogen stable isotope ratios of organic matter were linearly related with those of the media water. We used the relationships determined in these experiments to calculate the effective whole-cell fractionation factors between water and organic matter for B. subtilis. We then predicted the δ18O and δD values of spores produced in nutritionally identical media and local water sources for five different locations around the United States. Each of the measured δ18O and δD values of the spores matched the predicted values within a 95% confidence interval, indicating that stable isotope ratio analyses may be a powerful tool for tracing the geographic point-of-origin for microbial products.
Both plants and animals record aspects of their environment and physiology through the stable isotope ratios of elements (e.g., 2H/1H, 13C/12C, 15N/14N, and 18O/16O) that form the organic matter of those organisms. For example, isotope ratios have been used to trace the origins of migratory butterflies (1), birds (2, 3), and elephants (4, 5). Point-of-origin information can span several spatial scales. As an example, the physiological differences between C3 and C4 photosynthetic pathways, which result in large 13C/12C differences, allow one to trace the flow of this organic carbon as differential dietary inputs to animals (6), the transport of carbon across ecosystems (7), and, ultimately, the movement of region-specific carbon back into the atmosphere (8–10). This sourcing aspect of stable isotopes has also been applied to forensic issues, such as determining the point-of-origin of illicit drugs (11, 12) and adulteration of foods and alcoholic beverages (13–15).
Continentality, storm-track trajectories, and moisture origins result in substantial geographic gradients in the δ18O and δD values of precipitation and, therefore, of local waters (16). Several maps have been produced that show the extent of isotopic variations in waters at the continental and global scales (17, 18). Again, organisms often record the isotopic composition of these source waters in their organic compounds. In trees, for example, the isotopic composition of cellulose is correlated with that of source water (19, 20). Similar examples are found in animals where oxygen isotope ratios of bone and blood are closely related to that of the local water (21).
We hypothesized that microorganisms, too, should show a record of their growth environment in their cellular components. If relationships between growth environment and cellular isotope composition could be elucidated, it might become possible to draw conclusions about the waters in which the microbes had been grown, particularly for genetically identical organisms cultured in different geographic regions. We used the growth of Bacillus subtilis in laboratory culture as a model system. B. subtilis is a nonpathogenic, endospore-forming, Gram-positive soil bacterium. Because B. subtilis can grow on a variety of nutrients, we focused first on the relationship between the δ18O and δD values in the microbial products (cells or spores) and those ratios in the water used to make the culture media. We then tested the values predicted by laboratory model with cultures grown in different geographical regions across the United States.
Methods
Our experimental organism was B. subtilis strain 6051 (American Type Culture Collection). Cultures were grown in Schaeffer's sporulation medium (SSM) (22), which contained 8 g of Difco nutrient broth powder per liter plus the following volumes of solutions: 10% KCl, 10 ml; 1.2% MgSO4 heptahydrate, 10 ml; 1M NaOH, 1 ml; 1 M Ca(NO3)2, 1 ml; 0.01 M MnCl2, 1 ml; and 1 mM FeSO4, 1 ml. To make the media, the nutrient broth powder was dissolved in 976 ml of water, the KCl, MgSO4, and NaOH solutions were added, and the medium was autoclaved. The autoclaved solution was cooled to ≈50°C, and the remaining sterile solutions were added. The same salt solutions were used to make all of the media, even when the water was varied.
We prepared four containers of water of different oxygen and hydrogen isotope ratios by adding various amounts of D2O and H218O to local deionized water. We used waters from these containers throughout our experiments. The containers had airtight lids and were kept in the cold room. Isotope ratios of the four waters are listed in Table 1.
Table 1.
δD and δ18O of media before and after preparation
| Sample | δD of solids, ‰ ± 2 | δ18O of solids, ‰ ± 0.2 | δD of water, ‰ ± 2 | δ18O of water, ‰ ± 0.2 |
|---|---|---|---|---|
| Nutrient broth powder | −97 | 15.5 | ||
| Salt Lake City lab deionized water (autoclave system water) | −122 | −15.3 | ||
| Water 1 | −124 | −15.8 | ||
| SSM in water 1 before autoclaving | −98 | 14.5 | −123 | −16.0 |
| SSM in water 1 after autoclaving | −96 | 14.7 | −123 | −15.8 |
| SSM in water 1 after final salt additions | −94 | 15.5 | −123 | −15.9 |
| Water 2 | 17 | −5.8 | ||
| SSM in water 2 before autoclaving | −98 | 14.8 | 14 | −6.0 |
| SSM in water 2 after autoclaving | −98 | 15.0 | 12 | −6.1 |
| SSM in water 2 after final salt additions | −94 | 14.9 | 14 | −6.1 |
| Water 3 | 145 | 3.8 | ||
| SSM in water 3 before autoclaving | −95 | 15.7 | 138 | 3.0 |
| SSM in water 3 after autoclaving | −96 | 14.7 | 138 | 2.9 |
| SSM in water 3 after final salt additions | −95 | 15.2 | 137 | 3.3 |
| Water 4 | 271 | 12.1 | ||
| SSM in water 4 before autoclaving | −95 | 14.9 | 266 | 13.0 |
| SSM in water 4 after autoclaving | −92 | 14.9 | 271 | 12.1 |
| SSM in water 4 after final salt additions | −96 | 15.2 | 264 | 12.8 |
Cultures were grown in 100 ml of media in 1-liter flasks at 37°C and aerated by shaking at 200–225 rpm. Growth of cells was monitored by measuring the OD of the culture at 560 nm. Samples of mid-log-phase cells were harvested when the OD reached 0.5. Cells were harvested by chilling the culture on ice, then centrifuging the culture at 8,000 × g for 10 min. The pellets were resuspended in cold 0.9% NaCl made with the same water as was the culture medium. Cells were spun down again and then resuspended in 1 ml of cold 0.9% NaCl, frozen, and lyophilized for analysis.
Spore cultures were grown for 48 h, then centrifuged for 10 min at 8,000 × g, resuspended in one-fourth volume of culture water, recentrifuged, and resuspended in one-fifth volume of culture water (20 ml for a 100-ml culture). Spores were purified by shaking in water for at least 1 week, centrifuging each day for 20 min at 20,000 × g, and resuspending the resulting pellet (23). Finally, the spore pellet was resuspended in 1 ml of water, frozen, and lyophilized for analysis.
Stable isotope contents are expressed in “delta” notation as δ values in ‰, where δ‰ = (RA/RStd − 1) · 1,000‰, and RA and RStd are the ratios of the rare to abundant isotope (e.g., 18O/16O) in the sample and the standard. The standard used for both O and H is Vienna Standard Mean Ocean Water (VSMOW).
Organic samples including growth media, cells, and spores were weighed and placed into silver capsules, which had been treated to remove silver oxide. Oxygen and hydrogen isotopic composition of each sample was determined on a Delta Plus XL isotope ratio mass spectrometer (IRMS, Thermo Finnigan, Bremen, Germany) equipped with a Thermo Chemical Elemental Analyzer (Thermo Finnigan) and a zero-blank autosampler (Costech Analytical, Valencia, CA).
Hydrogen isotope ratios of water samples were obtained by reducing the hydrogen in 2 μl of water to H2 with 100 mg of Zn reagent in a Pyrex tube at 500°C. The resulting hydrogen gas was analyzed on a Thermo Finnigan Delta S IRMS equipped with a dual inlet.
Water samples for oxygen isotopes were prepared by equilibration with CO2 as described (24). Isotopic analysis was done on a Thermo Finnigan Delta S IRMS equipped with an elemental analyzer (1108, Carlo Erba, Milan).
Results and Discussion
Media Preparation Had Little to No Effect on the Isotopic Content of Media Solids or Water.
The O and H atoms available for incorporation into cellular material originate from either the powdered media components (nutrient broth mix and salts) or water. To determine the effect of media preparation on the isotopic content of the media solids and water, we prepared media in waters of four different isotopic compositions (waters 1–4; see Table 1) made in our laboratory by adding various amounts of D2O and H218O to local deionized water. We took samples of the media (i) after dissolving the nutrient broth powder in the water and adding some of the salt solutions, (ii) after autoclaving the solution, and then (iii) after the addition of the remaining sterile salt solutions. We lyophilized a portion of each sample to recover the media solids, extracted water from another portion, and determined the stable isotope ratios.
The δ18O and δD values of the media solids were not changed by dissolving and autoclaving in water using typical laboratory methods, even when isotope ratios of the water were very different from the powdered media (Table 1). Thus, there was no evidence of detectable isotopic exchange between the O and H atoms of the media solids and the water during media preparation. Furthermore, autoclaving media in steam made from local deionized water had little to no effect on the isotopic composition of the media water, even when it varied substantially (390‰ in D and 27‰ in 18O) from that of the local water (Table 1). The differences between waters 1–4 and the mixed media, particularly the δD values, can be accounted for by the mixing of ≈24 ml of salt solutions made with deionized lab water per liter of medium.
The δ18O and δD Values of Both Cells and Spores Are a Linear Function of the Culture Water Values.
To determine the relationship between culture water isotope ratios and spore isotope ratios, we grew cultures of B. subtilis in SSM made with waters 1–4. We conducted three independent trials in which cells were harvested in mid-log phase (after ≈3 h of growth) and four trials in which cells were grown until they had sporulated (48 h). The δ18O values of both spores and logarithmically growing cells were linearly correlated with the δ18O values of the culture water (r2 > 0.99, Fig. 1). The slopes and intercepts of the lines describing mid-log-phase and spore-phase growth were statistically different from each other. Although the y intercepts appear close to each other, they fall outside their respective 95% confidence intervals. Likewise, the δD values of both spores and logarithmically growing cells were linear functions of the δD values of the culture waters (r2 > 0.99, Fig. 2). The slopes and intercepts of the lines were again statistically different. We determined the isotopic content of the culture water after the growth of the spore cultures (48 h) and found no substantial change, indicating that the fractionation observed was not due to any changes in culture water isotope ratios.
Figure 1.

δ18O values of spores and logarithmically growing cells correlate with δ18O values of culture water. Spore data are averaged from four independent trials, and cell data are averaged from three independent trials. Error bars represent one standard error of measurement.
Figure 2.

δD values of spores and logarithmically growing cells correlate with δD values of culture water. Spore data are averaged from four independent trials, and cell data are averaged from three independent trials. Error bars represent one standard error of measurement.
Exchange of Spore Hydrogen Atoms with Environmental Water Is Not Significant.
Theoretically, it is possible that hydrogen atoms not bound to carbon undergo exchange with atmospheric water vapor. We tested the extent to which chemical exchange between spore hydrogen atoms and environmental water was occurring in a reciprocal washing experiment. We grew two batches of spores, one in water 1 (δD = −124‰) and the other in water 4 (δD = 271‰). Each of these batches of spores was split into two portions. One portion was washed in water 1 and the other in water 4, which involved soaking the spores in wash water for at least 7 days. After washing, spores were lyophilized and analyzed. No significant difference in spore δD values was observed as a function of wash water, even though waters 1 and 4 have δD values differing by ≈400‰ (t = 0.322, P = 0.769 for spores grown in water 1; t = 0.194, P = 0.859 for spores grown in water 4). In similar experiments with feather keratin, Chamberlain et al. (3) observed limited hydrogen exchange between feathers and environmental water, and most of that exchange occurred in the first 8 days of exposure. We concluded that no significant exchange between spore hydrogen atoms and environmental water took place.
We also tested for a potential effect of hygroscopic adsorption of water on the δD values of our spore samples. The δD values of a number of spore samples grown in each water were determined after storage in a vacuum desiccator and also after equilibration in laboratory air. No significant differences in δD values were observed (t = 0.298, P = 0.786 for spores grown in water 1; t = 1.333, P = 0.314 for spores grown in water 4). Salt Lake City has a dry climate; adsorption might be more significant in a more humid area. It is standard procedure to store spores in a desiccator before analysis to minimize interaction with atmospheric water.
Aggregate Fractionation Factors Between Cellular Organic Matter and Water Are Different Between Log-Phase Cells and Spores.
The O and H isotopes in culture water can be incorporated into cellular macromolecules through the direct incorporation of atoms into macromolecular products or through the exchange of O and H atoms during biosynthetic reactions. Because no isotopic exchange occurred between the media substrates and water before growth of the organisms, all of the isotope fractionation that occurred during cell growth was likely to have been mediated by metabolic activities of the cells.
We describe the isotope fractionation occurring during heterotrophic biosynthesis of organic compounds as
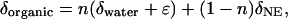 |
1 |
where δorganic, δwater, and δNE are the δD or δ18O values of synthesized organic matter, water, and the nonexchangeable stable isotopes of the substrate, respectively; n is the proportion of oxygen or hydrogen derived from medium water during organic biosynthesis; and ɛ is the isotope fractionation effect for enzyme-mediated exchange or addition (25, 26). Eq. 1 was originally applied to studies of the synthesis of a single product (cellulose) from a single substrate via a pathway of limited, defined steps, assuming that the O and H atoms that were not exchangeable with water were not otherwise fractionated by biosynthetic enzymes.
In considering the aggregate biosynthesis of all cellular products in a microbe from a complex medium, we must assume that even O or H atoms unavailable for exchange with water might undergo enzymatic reactions in which isotope fractionation could occur. We modified Eq. 1 to describe our system by including a potential fractionation factor, ɛNE, for O or H atoms that undergo enzyme-mediated rearrangement but are not available for exchange with water.
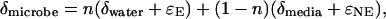 |
2 |
where δmicrobe, δmedia, and δwater are the isotope ratios in the cells or spores, powdered media components, and culture water, respectively; n is the fraction of atoms in the cells or spores derived from culture water; and ɛE is the isotope fractionation that occurs to the atoms derived from water.
In our experiments, the powdered media components and growth conditions were held constant, so that the term (1 − n)(δmedia + ɛNE) in Eq. 2 is constant. Eq. 2 can therefore be rearranged to the linear equation form y = mx + b, in which the slope m is equal to n, the fraction of atoms in the microbial product derived from the media water. The value of the y intercept is equal to
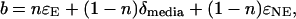 |
3 |
where n and δmedia are known. Rearranging, the aggregate fractionation effects, ɛAG, can be expressed as
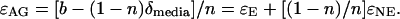 |
4 |
If all of the reactions in which isotopic fractionation occurs make the atoms available for exchange with water, as Sternberg and colleagues observed for heterotrophic cellulose biosynthesis (25, 26), then ɛNE = 0 and ɛAG = ɛE. Table 2 lists ɛAG for each of our experimental systems. By growing cultures in media that are nutritionally identical but have different stable isotope compositions, it should be possible to determine specific values for ɛE and ɛNE.
Table 2.
Aggregate fractionation factors (ɛAG) for cells and spores
| ɛAG δD, ‰ | ɛAG δ18O, ‰ | |
|---|---|---|
| Logarithmically growing cells | 122 | 22.6 |
| Spores | 40 | 16.4 |
For hydrogen in particular, it will be important to determine how changing the culture medium affects spore isotope ratios. The slopes of the line relating water hydrogen isotope ratios to those of spores grown in SSM indicate that only ≈28% of the spore hydrogen atoms were derived from water. Most of the hydrogen atoms in spores, therefore, were derived from the culture medium and not from water. Hydrogen isotope ratio values (δmedia) of culture media would be expected to have a significant effect on δD of spores.
We have ignored any possible contribution of atmospheric oxygen to the final oxygen isotope content of the spores and cells, which seems justified. B. subtilis is aerobic and uses oxygen as its final electron acceptor, creating water molecules. These water molecules might affect the isotopic composition of intracellular water in a way that would be reflected in the isotopic composition of macromolecular products. If so, the effect of atmospheric oxygen should be a constant offset for every culture and, along with the oxygen isotopes found in the media solids, would contribute to the y intercept of the equations in Figs. 1 and 2. It is possible that the amount of oxygen available to a culture during growth (as a function of shaker rpm, for example) could affect the final oxygen isotope ratio in the spores and complicate data interpretation. In the off-campus growth experiment described below, we do not know exactly what shaker rpm or sizes of culture flasks were used to grow the organisms, yet results fell along the predicted line. It would be particularly important to investigate this question if the organism in question were a facultative anaerobe that changed its metabolism with changing oxygen availability, but such potential concerns appear insignificant for B. subtilis.
Logarithmically growing cells and spores have different aggregate fractionation factors and n values. This is not surprising, given that they are distinctly different developmental stages of the organism. Sporulation begins at the end of logarithmic growth in laboratory cultures and requires 6–8 h to complete (27). A suite of sporulation-specific proteins is induced, including extracellular hydrolytic enzymes, internal biosynthetic enzymes, and spore structural proteins. Spores contain novel compounds such as dipicolinic acid and muramic δ-lactam residues, novel structures such as the spore cortex and coat, and storage compounds such as 3-phosphoglyceric acid that comprise 0.1–0.3% of the dry weight of a spore (28). Stable isotope analysis of various cell and spore components should make it possible to identify fractionation factors associated with specific cellular processes.
δ18O and δD Values Correctly Identified the Geographical Source of Spores Produced Across the United States.
From Figs. 1 and 2, one can predict δD and δ18O of spores grown in SSM in water of known isotopic content. Likewise, given δD and δ18O values of spores grown in SSM, one should be able to predict the δD and δ18O values of the water in which they were grown. To test this hypothesis, we asked colleagues in North Carolina, Ohio, Louisiana, and New Mexico to grow spores for us in SSM and supplied them with the dry media components. We grew spore cultures in our local deionized water to make a fifth test batch of spores. Our colleagues provided us with samples of the water they used to make the media. Using the δD and δ18O values of the water samples, we predicted the δD and δ18O values of the spores.
The measured δ18O of the resulting spores matched the predicted values within the 95% confidence limits of the regression line in Fig. 1 in every case (Table 3, Fig. 3), as did the δD values. In each case, except those of the samples from North Carolina and Ohio, which were grown in waters of identical isotope ratios, we could unambiguously assign the correct origin to the spores from our list of potential sources, based on their isotopic content and the predictions we made from the water isotope values.
Table 3.
Predicted and measured δD and δ18O values of spores grown at remote sites
| Site | Water
|
Spores
|
||||
|---|---|---|---|---|---|---|
| δD, ‰ | δ18O, ‰ | Predicted δD, ‰ | Measured δD, ‰ | Predicted δ18O, ‰ | Measured δ18O, ‰ | |
| Baton Rouge, LA | −27.3 | −4.7 | −64.9 | −59.5 | 13.3 | 13.0 |
| Durham, NC | −46.2 | −7.4 | −70.2 | −70.3 | 11.3 | 11.2 |
| Columbus, OH | −46.0 | −7.5 | −70.1 | −72.7 | 11.2 | 11.3 |
| Los Alamos, NM | −77.8 | −10.7 | −79.0 | −76.3 | 8.8 | 9.3 |
| Salt Lake City, UT | −120.4 | −14.9 | −91.0 | −95.9 | 5.7 | 6.0 |
Figure 3.

(A) δ18O values of test spore samples versus their local water δ18O plotted with standard curve from Fig. 1. (B) δD values of test spore samples versus their local water δD plotted with standard curve from Fig. 2. In both, the points representing the samples from Ohio and North Carolina are superimposed.
Stable Isotope Ratios Provide a Potential Forensic Tool for Tracing Geographic Origins of Microbial Cultures.
The δ18O and δD values of precipitation are predictable based on topography, distance from an ocean, and climate, and maps based on measured values have been assembled (18). Although not unique for every geographic region, the δ18O and δD values nevertheless constitute a geographic signature for a given region that can often be used to distinguish waters from two geographically different regions. Purification techniques such as distillation or ion-exchange chromatography, typically used to prepare water for microbiological uses, do not change stable isotope ratios. Our results have delineated the relationship between water isotopes and spore isotopes when the spores are grown in SSM. Given a batch of spores grown in this medium, it is now possible to identify those geographic regions from which the culture water could have originated. Further work is needed to extend these results to other strains and growth media.
With bioterrorism a threat, stable isotope analyses may help constrain the range of regions from which microbiological materials might have originated. Although DNA analysis can reveal specific genetic information about the strain of an organism, it does not provide direct evidence of the environment in which a particular batch of organisms was produced (29, 30). Stable isotope analyses are a potential forensic tool to complement genetics-based studies.
Acknowledgments
We thank Dan Zeigler of Ohio State University, Mark Batzer of Louisiana State University, Ken Wilson of Duke University, and Paul Jackson of Los Alamos National Laboratories for growing spore cultures for us, and L. Chesson, W. Ike, T. Martin, and E. Stange for technical assistance. This study was supported by a research contract from the U.S. Central Intelligence Agency.
Abbreviation
- SSM
Schaeffer's sporulation medium
References
- 1.Hobson K, Wassenaar L I, Taylor O R. Oecologia. 1999;120:397–404. doi: 10.1007/s004420050872. [DOI] [PubMed] [Google Scholar]
- 2.Hobson K, Wassenaar L I. Oecologia. 1997;109:142–148. doi: 10.1007/s004420050068. [DOI] [PubMed] [Google Scholar]
- 3.Chamberlain C P, Blum J D, Holmes R T, Feng X, Sherry T W, Graves G R. Oecologia. 1997;109:132–141. doi: 10.1007/s004420050067. [DOI] [PubMed] [Google Scholar]
- 4.Vogel J C, Eglinton B, Auret J M. Nature. 1990;346:747–749. [Google Scholar]
- 5.van der Merwe N J, Lee-Thorp J A, Thackeray J F, Hall-Martin A, Kruger F J, Coetzee H, Bell R H V, Lindeque M. Nature. 1990;346:744–746. [Google Scholar]
- 6.Hobson K A. Oecologia. 1999;120:314–326. doi: 10.1007/s004420050865. [DOI] [PubMed] [Google Scholar]
- 7.Conte M H, Weber J C. Nature. 2002;417:639–641. doi: 10.1038/nature00777. [DOI] [PubMed] [Google Scholar]
- 8.Ometto J P, Flanagan L B, Martinelli L A, Marcelo Z, Moreira-Niro H, Ehleringer J R. Global Biogeochem Cycles. 2002;16:1109. [Google Scholar]
- 9. Pataki, D., Ehleringer, J., Flanagan, L. B., Yakir, D., Bowling, D. R., Still, C., Buchman, N. & Berry, J. (2002) Global Biogeochem. Cycles, in press.
- 10.Flanagan L B, Wever L A, Carlson P J. Global Change Biol. 2002;8:599–615. [Google Scholar]
- 11.Carter J F, Titterton E L, Murray M, Sleeman R. Analyst. 2002;127:830–833. doi: 10.1039/b201496n. [DOI] [PubMed] [Google Scholar]
- 12.Ehleringer J R, Casale J F, Lott M J, Ford V L. Nature. 2000;408:311–312. doi: 10.1038/35042680. [DOI] [PubMed] [Google Scholar]
- 13.Brooks J, Buchman N, Phillips S, Ehleringer J, Evans R, Lott M J, Martinelli L A, Pockman W, Sandquist D, Sparks J, et al. J Agric Food Chem. 2002;50:6413–6418. doi: 10.1021/jf020594k. [DOI] [PubMed] [Google Scholar]
- 14.Pissinato L, Martinelli L, Victoria R, Camargo P. Food Res Int. 1999;32:665–668. [Google Scholar]
- 15.Parker I G, Kelly S D, Sharman M, Dennis M, Howie D. Food Chem. 1998;63:423–428. [Google Scholar]
- 16.Craig H. Science. 1961;133:1702–1703. doi: 10.1126/science.133.3465.1702. [DOI] [PubMed] [Google Scholar]
- 17.Gat J R. In: The Terrestrial Environment, Handbook of Environmental Isotope Geochemistry. Fritz P, Fontes J C, editors. Vol. 1. Amsterdam: Elsevier Science; 1980. pp. 21–47. [Google Scholar]
- 18.Kendall C, Coplen T B. Hydrolog Processes. 2001;15:1363–1393. [Google Scholar]
- 19.Roden J S, Ehleringer J R. Oecologia. 1999;121:467–477. doi: 10.1007/s004420050953. [DOI] [PubMed] [Google Scholar]
- 20.Yapp C J, Epstein S. Nature. 1982;297:636–639. [Google Scholar]
- 21.Longinelli A. Geochim Cosmochim Acta. 1984;48:385–390. [Google Scholar]
- 22.Harwood C, Cutting S, editors. Molecular Biological Methods for Bacillus. Chichester, U.K.: Wiley; 1990. [Google Scholar]
- 23.Nicholson W, Setlow P. In: Molecular Biological Methods for Bacillus. Harwood C, Cutting S, editors. Chichester, U.K.: Wiley; 1990. pp. 391–450. [Google Scholar]
- 24.Fessenden J, Cook C, Lott M J, Ehleringer J R. Rapid Commun Mass Spectrom. 2002;16:1257–1260. doi: 10.1002/rcm.711. [DOI] [PubMed] [Google Scholar]
- 25.Luo Y, Sternberg L. J Exp Bot. 1992;43:47–50. [Google Scholar]
- 26.Sternberg L, DeNiro M, Savidge R. Plant Physiol. 1986;82:423–427. doi: 10.1104/pp.82.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doi R H. In: Bacillus. Harwood C, editor. New York: Plenum; 1989. pp. 169–216. [Google Scholar]
- 28.Paidhungat M, Setlow P. In: Bacillus subtilis and Its Closest Relatives: From Genes to Cells. Sonenshein A L, Hoch J A, Losick R, editors. Washington, DC: Am. Soc. Microbiol.; 2002. [Google Scholar]
- 29.Read T, Salzberg S L, Pop M, Shumway M, Umayam L, Jiang L, Holtzapple E, Busch J, Smith K, Schupp J M, et al. Science. 2002;296:2028–2033. doi: 10.1126/science.1071837. [DOI] [PubMed] [Google Scholar]
- 30.Enserink M. Science. 2002;296:1002–1003. doi: 10.1126/science.296.5570.1002. [DOI] [PubMed] [Google Scholar]