

Abstract
Microbial processes for commodity chemicals have focused on reduced products and anaerobic conditions where substrate loss to cell mass and CO2 are minimal and product yields are high. To facilitate expansion into more oxidized chemicals, Escherichia coli W3110 was genetically engineered for acetate production by using an approach that combines attributes of fermentative and oxidative metabolism (rapid growth, external electron acceptor) into a single biocatalyst. The resulting strain (TC36) converted 333 mM glucose into 572 mM acetate, a product of equivalent oxidation state, in 18 h. With excess glucose, a maximum of 878 mM acetate was produced. Strain TC36 was constructed by sequentially assembling deletions that inactivated oxidative phosphorylation (ΔatpFH), disrupted the cyclic function of the tricarboxylic acid pathway (ΔsucA), and eliminated native fermentation pathways (ΔfocA-pflB ΔfrdBC ΔldhA ΔadhE). These mutations minimized the loss of substrate carbon and the oxygen requirement for redox balance. Although TC36 produces only four ATPs per glucose, this strain grows well in mineral salts medium and has no auxotrophic requirement. Glycolytic flux in TC36 (0.3 μmol⋅min−1⋅mg−1 protein) was twice that of the parent. Higher flux was attributed to a deletion of membrane-coupling subunits in (F1F0)H+-ATP synthase that inactivated ATP synthesis while retaining cytoplasmic F1-ATPase activity. The effectiveness of this deletion in stimulating flux provides further evidence for the importance of ATP supply and demand in the regulation of central metabolism. Derivatives of TC36 may prove useful for the commercial production of a variety of commodity chemicals.
Keywords: metabolic engineering‖glycolytic flux‖acetic acid‖fermentation‖ ATPase
Current dependence on petroleum can be reduced by expanding the production of chemicals, automotive fuels, and plastics from renewable plant biomass using microbial biocatalysts. The commercial production of commodity chemicals by microbial fermentation has been limited to reduced products such as ethanol and lactic acid under anaerobic conditions where substrate loss to cell mass or CO2 is minimal and product yields are high. In aerobic processes, Escherichia coli is widely used as a biocatalyst for higher value products such as recombinant proteins (1–4) and amino acids (5, 6). Many E. coli strains grow well in simple mineral salts medium and readily metabolize all of the hexose and pentose sugar constituents of plant biomass (7, 8). Recent successes have been reported in the engineering of E. coli strains for new commodity chemicals such as 1,3-propanediol (9, 10), 1,2-propanediol (11, 12), adipic acid (13), lactic acid (14–16), succinic acid (17, 18), and ethanol (7, 8). Some are proceeding toward commercialization. In all aerobic and anaerobic processes, acetate production by the native E. coli pathway (phosphotransacetylase and acetate kinase) has been generally regarded as an undesirable consequence of excessive glycolytic flux (1–3, 8, 14, 19). To facilitate expansion into more oxidized commodity chemicals, we developed a genetic approach that combines the attributes of fermentative and oxidative metabolism (rapid growth, external electron acceptor for NADH oxidation) into a single biocatalyst. As an example, this approach was used to engineer E. coli for acetate production.
The biological production of acetic acid is currently limited to food uses. Petrochemical routes have largely displaced biological processes as uses expanded to plastics, solvents, and road de-icers (20–22). World production of acetate for 2001 was estimated at 6.8 million metric tons, half of which was produced in the U.S. (23). In the current biological process, sugars are fermented to ethanol by Saccharomyces. Ethanol in the resulting beer is subsequently oxidized to acetic acid by Acetobacter under aerobic conditions (20–22). This process can be summarized as follows:
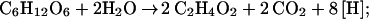 |
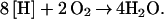 |
In this study, we have genetically modified E. coli W3110 to produce acetic acid as the primary product from glycolysis. Although ethanol was eliminated as an intermediate, the stoichiometry of the reaction remained as shown above. The resulting biocatalyst (TC36) contains multiple chromosomal alterations (Fig. 1) that direct carbon flow to acetate and minimize carbon loss to cell mass, CO2, and alternative products. This strain is devoid of plasmids, foreign genes, and antibiotic resistance markers and grows well in mineral salts medium without complex nutrients.
Figure 1.

Diagram summarizing genetic modifications used to redirect glucose metabolism to acetate. Bold arrows mark principal metabolic routes in TC36. Reactions that have been blocked by gene deletions are marked with filled circles. Gene names are shown in italics. (A) Central carbon metabolism. Acetate is the principal product from sugar metabolism with the net production of four ATP equivalents (∼P) per glucose molecule. (B) Oxidative phosphorylation. The (F1F0)H+-ATP synthase is inactive in TC36, although the electron transport system remains functional as the primary route for NADH oxidation. (C) F1-ATPase. TC36 lacks essential subunits for ATP synthesis but retains a functional cytoplasmic F1-ATPase.
Materials and Methods
Microorganisms and Media.
Strains and plasmids used in this study are listed in Table 1. Working cultures of E. coli W3110 (ATCC 27325) derivatives were maintained on a minimal medium containing mineral salts (per liter: 3.5 g of KH2PO4; 5.0 g of K2HPO4; 3.5 g of (NH4)2HPO4, 0.25 g of MgSO4⋅7 H2O, 15 mg CaCl2⋅2 H2O, 0.5 mg of thiamine, and 1 ml of trace metal stock), glucose (2% in plates; 3% in broth), and 1.5% agar. The trace metal stock was prepared in 0.1 M HCl (per liter: 1.6 g of FeCl3/0.2 g of CoCl2⋅6 H2O/0.1 g of CuCl2/0.2 g of ZnCl2⋅4 H2O/0.2 g of NaMoO4/0.05 g of H3BO3). 4-Morpholinopropanesulfonic acid (0.1 M, pH 7.1) was added to both liquid and solid media (filter-sterilized) when needed for pH control but was not included in medium used for 10-liter fermentations. Minimal medium was also prepared by using succinate (1 g⋅liter−1) as a sole source of carbon (nonfermentable substrate). Succinate (1 g⋅liter−1) was added as a supplement to glucose-minimal medium when needed. During plasmid and strain construction, cultures were grown in LB broth or on LB plates (1.5% agar) (24). Glucose (2%) was added to LB medium for all strains containing mutations in (F1F0)H+-ATP synthase. Antibiotics were included as appropriate (kanamycin, 50 mg⋅liter−1; ampicillin, 50 mg⋅liter−1; and tetracycline, 12.5 or 6.25 mg⋅liter−1). Fusaric acid plates were used to select for loss of Tn10-encoded tetracycline resistance (14).
Table 1.
Sources and characteristics of strains and plasmids
| Strains/plasmids | Relevant characteristics | Ref. |
|---|---|---|
| Strains | ||
| W3110 | Wild type | ATCC 27325 |
| TOP10F | lacIq (episome) | Invitrogen |
| SE2279 | MG1655, pflB ldhA∷Tn10 | This study |
| SZ33 | W3110, ldhA∷Tn10 | This study |
| SZ40 | W3110, Δ(focA-pflB)∷FRT ΔfrdBC | 14 |
| SZ46 | W3110, Δ(focA-pflB)∷FRT ΔfrdBC ldhA∷Tn10 | This study |
| SZ47 | W3110, Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA | This study |
| TC20 | W3110, ΔadhE∷FRT-tet-FRT | 8 |
| TC21 | W3110, ΔatpFH∷FRT-tet-FRT | This study |
| TC23 | W3110, Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT-tet-FRT | This study |
| TC24 | W3110, Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT | This study |
| TC25 | W3110, ΔsucA∷FRT-tet-FRT | This study |
| TC30 | W3110, Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT ΔadhE∷FRT-tet-FRT | This study |
| TC31 | W3110, Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT ΔadhE∷FRT | This study |
| TC32 | W3110, (Succ−), Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT ΔadhE∷FRT ΔsucA∷FRT-tet-FRT | This study |
| TC35 | W3110, (Succ+, Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT ΔadhE∷FRT ΔsucA∷FRT-tet-FRT | This study |
| TC36 | W3110, (Succ+, Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT ΔadhE∷FRT ΔsucA∷FRT | This study |
| Plasmids | ||
| pCR2.1-TOPO | bla kan, TOPO™ TA cloning vector | Invitrogen |
| pFT-A | bla flp low-copy vector containing recombinase and temperature-conditional pSC101 replicon | 28 |
| pKD46 | bla γ β exo low-copy vector containing red recombinase and temperature-conditional pSC101 replicon | 26 |
| pLOI2065 | bla, SmaI fragment with FRT flanked tet gene | 8 |
| pLOI2800 | bla kan sucA | This study |
| pLOI2801 | bla kan sucA∷FRT-tet-FRT | This study |
| pLOI2802 | bla kan adhE | 8 |
| pLOI2803 | bla kan adhE∷FRT-tet-FRT | This study |
| pLOI2805 | bla kan atpEFH | This study |
| pLOI2807 | bla kan atpFH∷FRT-tet-FRT | This study |
Genetic Methods.
Standard methods were used for plasmid construction, phage P1 transduction, electroporation, and PCR (24, 25). Chromosomal DNA from E. coli W3110 (and derivatives) served as a template to amplify genes using primers complementary to coding regions (ORFmers) purchased from the Sigma–Genosys (The Woodlands, TX). PCR products were initially cloned into plasmid vector pCR2.1-TOPO. During plasmid constructions, restriction products were converted to blunt ends by using either the Klenow fragment of DNA polymerase I (5′ overhang) or T4 DNA polymerase (3′ overhang) as needed. Integration of linear DNA was facilitated by using pKD46 (temperature conditional) containing an arabinose-inducible Red recombinase (26). Integrants were selected for tetracycline resistance (6.25 mg⋅liter−1) and screened for appropriate antibiotic resistance markers and phenotypic traits. At each step, mutations were verified by analyses of PCR products and fermentation products. FRT-flanked antibiotic resistance genes used for selection were deleted by using a temperature-conditional plasmid (pFT-A) expressing FLP recombinase from a chlortetracycline-inducible promoter (27, 28).
Fermentation.
Acetate production was examined in glucose-minimal medium containing 167 mM glucose by using a New Brunswick Bioflow 3000 fermentor (New Brunswick Scientific) with a 10-liter working volume (37°C, dual Rushton impellers, 450 rpm). Dissolved oxygen was maintained at 5% of air saturation (unless otherwise stated) by altering the proportion of N2 and O2. Broth was maintained at pH 7.0 by the automatic addition of 11.4 M KOH. During fed batch experiments, additional glucose was added from a sterile 60% stock. Three fed batch regimes were investigated: (i) 3% glucose initially with the addition of 3% after 12 h (6% total); (ii) 6% glucose initially with the addition of 4% glucose after 16 h (10% total); (iii) 3% glucose initially with multiple additions to maintain glucose levels above 100 mM.
Seed cultures were prepared by inoculating colonies from a fresh plate (48 h) into 3 ml of glucose-minimal medium (13 × 100-mm tube) containing 0.1 M 4-morpholinopropanesulfonic acid. After incubation for 14 h (120-rpm rotator), cultures were diluted 400-fold into 1-liter baffled flasks containing 200 ml of mineral salts medium (37°C, 280 rpm). When cells reached 1.5–2.2 OD550 nm, sufficient culture volume was harvested (5,000 × g, 25°C) to provide an inoculum of 33 mg of dry cell weight liter−1 in the 10-liter working volume.
Broth samples were removed to measure organic acids, residual glucose, and cell mass. Volumetric and specific rates were estimated from measured values for glucose and acetate by using graphpad prism (GraphPad, San Diego). A smooth curve was generated with 10 points per min (Lowess method) to fit measured results. The first derivative (acetate or glucose versus time) of each curve served as an estimate of volumetric rate. Specific rates (mmols liter−1⋅h−1⋅mg−1 dry cell weight) were calculated by dividing volumetric rates by respective values for cell mass.
ATPase.
Cells were grown for enzyme assays as described above for seed cultures. On reaching 0.75–1.0 OD550 nm, cultures were chilled on ice and harvested by centrifugation (8,000 × g, 5 min at 4°C). Cell pellets were washed five times with 0.1 M Tris⋅HCl (pH 7.55), resuspended in 1 ml of this buffer, and broken by using a model W220F ultrasonic cell disruptor (Heat Systems/Ultrasonics). Total ATPase activity in disrupted cell preparations was assayed at pH 7.55 essentially as described by Evans (29). Inorganic phosphate was measured by the method of Rathbun and Betlach (30). Results represent an average of three cultures for each strain. Specific activity is expressed as μmol Pi released min−1⋅mg−1 protein.
Analyses.
Organic acids and glucose concentrations were determined by using a Hewlett–Packard HPLC (HP 1090 series II) equipped with a UV monitor (210 nm) and refractive index detector. Products were separated by using a Bio-Rad HPX-87H column (10 μl injection) with 4 mM H2SO4 as the mobile phase (0.4 ml⋅min−1, 45°C). Cell mass was estimated by measuring OD550 nm (1.0 OD550 nm is equivalent to 0.33 g⋅liter−1 dry cell weight) by using a Bausch & Lomb Spectronic 70 spectrophotometer with 10 × 75-mm culture tubes as cuvettes. Protein concentration was determined by using the BCA Protein Assay Kit from Pierce.
Results
Elimination of Fermentation Products (Strain SZ47).
Inspection of native pathways in E. coli (Fig. 1) indicated that the production of acetate and CO2 as sole metabolic products from glucose would require an external electron acceptor such as oxygen. Due to low oxygen solubility, however, it is difficult to satisfy the oxygen demand from active E. coli metabolism. Typically a portion of substrate is converted into fermentation products (31, 32). This problem was eliminated by combining deletions in genes encoding lactate dehydrogenase, pyruvate formatelyase, and fumarate reductase as follows. The ldhA∷Tn10 mutation in E. coli SE2279 was transduced into E. coli W3110 by using phage P1 to produce strain SZ33. P1 phage grown on SZ33 was used to transfer this mutation into SZ40(Δ(focA-pflB)∷FRT ΔfrdBC) to produce SZ46. Tetracycline-sensitive derivatives of SZ46 were selected by using fusaric acid medium (14). One clone was designated SZ47 (Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA). The ΔldhA mutation in SZ47 was confirmed by the absence of lactate in fermentation broth, inability to grow anaerobically in glucose-minimal medium, and PCR analysis by using ldhA ORFmers (1.0 kbp for the wild-type ldhA compared with 1.1 kbp for SZ47). The slightly larger size of the amplified product from SZ47 is attributed to remnants of Tn10.
Disruption of Oxidative Phosphorylation (Strain TC24).
Growth under oxidative conditions is characterized by conversion of up to 50% of substrate carbon to cell mass (33). To reduce the potential drain of substrate into cell mass, a mutation was introduced into SZ47 that deleted portions of two subunits in (F1F0)H+-ATP synthase concerned with assembly to the plasma membrane (34), disrupting oxidative phosphorylation while preserving the hydrolytic activity of F1-ATPase in the cytoplasm. To construct this deletion, the atpEFH coding region of the atpIBEFHAGDC operon was amplified by PCR by using primers (ORFmers) complementary to the 5′-end of the atpE gene and the 3′-end of the atpH. The amplified fragment (1.3 kbp) was cloned into pCR2.1-TOPO, and one clone was selected in which the atpEFH genes were oriented to permit expression from the lac promoter (pLOI2805; Fig. 2). The atpF gene and 117 nucleotides at the 5′-end of atpH gene were removed from pLOI2805 by digestion with HpaI and BstEII (Klenow-treated). This region was replaced with a 1.7-kbp SmaI fragment from pLOI2065 containing the FRT-tet-FRT cassette to produce pLOI2807 (Fig. 2). After digestion with ScaI, pLOI2807 served as a template for amplification of the atpEΔ(FH)∷FRT-tet-FRT region (2.4 kbp) by using the 5′ atpE and 3′ atpH primers. Amplified DNA was precipitated, digested again with ScaI to disrupt any residual plasmid, and purified by phenol extraction. This DNA was introduced into E. coli W3110(pKD46) by electroporation while expressing Red recombinase. Plasmid pKD46 was eliminated by growth at 42°C. Recombinants (double crossover) were identified by using antibiotic markers (tetracycline resistant; sensitive to ampicillin and kanamycin) and by the inability to grow on succinate-minimal plates in the absence of glucose (fermentable carbon source). Integration was further confirmed by PCR analysis by using the 5′ atpE and 3′ atpH primers (1.3-kbp fragment for W3110; 2.3-kbp fragment for mutants). One clone was selected and designated TC21(Δatp(FH)∷FRT-tet-FRT).
Figure 2.

Diagram summarizing plasmid constructions.
Phage P1 was used to transduce the Δatp(FH)∷FRT-tet-FRT mutation in TC21 to SZ47 and produce TC23. The tet gene was removed from TC23 by the FLP recombinase (pFT-A). After elimination of pFT-A by growth at 42°C, the Δatp(FH)∷FRT mutation was further confirmed by PCR analysis by using the 5′ atpE and 3′ atpH primers (0.8 kbp for deletion and 1.3 kbp for SZ47). The resulting strain was designated TC24(Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA ΔatpFH∷FRT). TC24 was unable to grow in minimal medium without a fermentable carbon source (substrate level phosphorylation) but retained the ability to oxidize NADH by the electron transport system.
Inactivation of Alcohol Dehydrogenase E (Strain TC31).
The native bifunctional alcohol/aldehyde dehydrogenase (adhE) in E. coli catalyzes the reduction of acetyl∼CoA to ethanol, a reaction that directly competes with the conversion of acetyl∼CoA to acetate by phosphotransacetylase and acetate kinase (Fig. 1). To eliminate this activity, phage P1 was used to transduce the ΔadhE∷FRT-tet-FRT mutation in TC20 to TC24 and produce TC30. Chromosomal integration was confirmed by PCR analysis by using adhE primers (2.7 kbp for TC24 and 3.2 kbp for the ΔadhE∷FRT-tet-FRT mutant). The tet gene was deleted from TC30 by FLP recombinase by using pFT-A. After elimination of pFT-A by growth at 42°C, a clone was selected and designated TC31(Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT ΔadhE∷FRT).
Interruption of the Tricarboxylic Acid Cycle (Strain TC32).
During oxidative growth, up to 50% of substrate carbon can be lost as CO2 (33). This loss of carbon can be attributed in large measure to the high efficiency of the tricarboxylic acid cycle and the electron transport system (NADH oxidation). During fermentative metabolism, the production of CO2 and NADH is reduced primarily by strong repression of sucAB encoding 2-ketoglutarate dehydrogenase (35, 36), disrupting the cyclic function of the tricarboxylic acid cycle. We have genetically imposed a similar restriction in carbon flow during oxidative metabolism by deleting part of the sucA gene.
The sucA coding region was amplified by using ORFmers. The resulting 2.8-kbp PCR product was cloned into pCR2.1-TOPO to produce pLOI2800 (Fig. 2) in which the sucA coding region was oriented to permit expression from the lac promoter. A 1.1-kbp fragment was removed from the central region of sucA by digestion of pLOI2800 with SnaBI and AccI (Klenow-treated). This region was replaced with a 1.7-kbp SmaI fragment containing the FRT-tet-FRT cassette from pLOI2065 to produce pLOI2801 (Fig. 2). Plasmid pLOI2801 was digested with PvuI and ScaI and used as a template to amplify the 3.3-kbp region containing sucA::FRT-tet-FRT by using sucA ORFmers. Amplified DNA was precipitated, digested with PvuI and ScaI to disrupt any residual circular plasmid, and extracted with phenol. Purified DNA was electroporated into E. coli W3110(pKD46) while expressing Red recombinase. Plasmid pKD46 was eliminated by growth at 42°C. Disruption of sucA was confirmed by PCR analysis by using sucA ORFmers (2.8-kbp fragment for wild type and 3.3 kbp for sucA∷FRT-tet-FRT mutants) and designated TC25.
Phage P1 was used to transduce the sucA∷FRT-tet-FRT mutation from TC25 into TC31. Transfer of this mutation was verified by PCR analysis (2.8 kbp for wild-type sucA and 3.3 kbp for sucA∷FRT-tet-FRT mutants) and phenotype (Succ−). Inactivation of 2-ketoglutarate dehydrogenase (ΔsucA) in this ΔfrdBC background resulted in an undesirable auxotrophic requirement for succinate (Succ–) during growth on glucose-minimal medium. The resulting strain was designated TC32(Succ–, Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT ΔadhE∷FRT ΔsucA∷FRT-tet-FRT).
Elimination of Succinate Requirement (Strain TC36).
Spontaneous Succ+ mutants of TC32 were readily obtained after serial transfers in glucose-minimal broth containing decreasing amounts of succinate (4 to 0.4 mM) followed by selection on glucose-minimal plates without succinate. Over 170 clones were recovered per milliliter of culture after enrichment (≈3% of viable cells). Ten clones were tested, and all grew well in glucose minimal broth without succinate and produced acetate as the dominant product. One was selected (TC35) for deletion of the tet gene by using the FLP recombinase. This deletion was confirmed by analysis of PCR products by using sucA primers (3.3 kbp for TC35 and 1.8 kbp after tet deletion). The resulting strain was designated TC36 (Succ+; Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT ΔadhE∷FRT ΔsucA∷FRT). Although the genetic change that eliminated the succinate requirement was not identified, likely candidates include mutations that provide a low constitutive expression of aceAB (glyoxylate shunt) or citC (citrate lyase).
Total ATPase activity was examined in disrupted cell extracts of TC36 and W3110 (wild type). The activity in TC36 (0.355 units⋅mg−1 protein) was equivalent to 71% of the unmodified parent (0.502 units⋅mg−1 protein), confirming that F1-ATPase was not inactivated by the ΔatpFH∷FRT mutation. This is similar to the levels of ATPase reported for an atpH mutant of E. coli, which blocked membrane assembly and coupling to oxidative phosphorylation (34).
Effects of Gene Disruptions on Growth and Glycolytic Flux.
TC36 was genetically engineered for the production of acetate from carbohydrates such as glucose (Fig. 1). Batch fermentations with pH control were used to compare the performance of this strain with W3110 (wild type) and two intermediate strains used for construction, SZ47(ΔfocA-pflB ΔfrdBC ΔldhA) and TC24(ΔfocA-pflB ΔfrdBC ΔldhA ΔatpFH). Based on preliminary experiments, 5% oxygen saturation and 3% glucose (37°C) were selected as test conditions. Broth pH was maintained at neutrality to minimize toxicity from undissociated acids.
Disruption of oxidative phosphorylation and the cyclic function of the tricarboxylic acid cycle, elimination of the primary fermentation pathways, and the production of acetate as the primary end-product from glycolysis had relatively little effect on the growth of E. coli. The maximum growth rates for strains W3110 (wild type) and SZ47 (lacking the three native fermentation pathways) were similar although the cell yield for SZ47 was higher (Fig. 3A; Tables 2 and 3). Inactivation of oxidative phosphorylation (ΔatpFH) resulted in a small reduction in growth rate and cell yield (TC24). Cell yield and growth rate were lowest for strain TC36 containing additional mutations in 2-ketoglutarate dehydrogenase (ΔsucA) and alcohol dehydrogenase (ΔadhE), ≈80% of the unmodified parent W3110.
Figure 3.

Effects of selected mutations on growth (A), glucose utilization (B), and base consumption (C). ■, W3110 (wild type); □, SZ47(Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA); ○, TC24(Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT); ●, TC36 (Succ+; Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT ΔadhE∷FRT ΔsucA∷FRT).
Table 2.
Comparison of metabolic rates
| Strain | Specific growth rate, μ | Max vol* glucose utilization, mmol⋅liter−1⋅h−1 | Max spec glucose† utilization, mmol⋅h−1⋅g−1 | Max vol* acetate production, mmol⋅liter−1⋅h−1 | Max spec† acetate production, mmol⋅h−1⋅g−1 |
|---|---|---|---|---|---|
| W3110 | 0.87 | 18 | 9 | 9.5 | 10 |
| SZ47 | 0.87 | 22 | 11 | 9 | 10 |
| TC24 | 0.78 | 28 | 20 | 26 | 16 |
| TC36 | 0.69 | 33 | 18 | 23 | 16 |
Maximum volumetric rates for glucose utilization and acetate production.
Maximum specific rates (dry cell weight basis) for glucose utilization and acetate production. Values for glucose represent a measure of maximal glycolytic flux.
Table 3.
Summary of fermentation products
| Strain | Conditions | Cell yield, g/liter | Fermentation products,* mM
|
Yield, %† | Carbon recovery, % substrate C‡ | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Acetate | 2-ketoglutarate | Fumarate | Lactate | Pyruvate | Succinate | |||||
| W3110 | 3% glucose 5% DO | 4.5 | 30 | 39 | 0.8 | 33 | <1 | 5 | 9 | 60 |
| SZ47 | 3% glucose 5% DO | 5.3 | 6 | 11 | 0.9 | <1 | 1 | 3 | 2 | 20 |
| TC24 | 3% glucose 5% DO | 4.4 | 156 | 1 | 1.0 | <1 | <1 | 2 | 47 | 66 |
| TC36 | 3% glucose 5% DO | 3.5 ± 0.2 | 224 ± 14 | 16 ± 6 | 0.4 ± 0.1 | <1 | 0 ± 0.5 | 4 ± 1 | 68 | 89 |
| TC36 | 3% glucose 15% DO | 3.2 | 190 | 24 | <1 | <1 | <1 | 3 | 57 | 88 |
| TC36 | 3% Glucose 5% DO N-limited | 2.5 | 220 | 31 | <1 | <1 | <1 | 10 | 66 | 95 |
| TC36 | 3 + 3% glucose 5% DO | 3.8 | 523 | 21 | <1 | 3 | 14 | 2 | 78 | 95 |
| TC36 | 3 + 3% glucose 5% DO N-limited | 3.0 | 572 | 33 | <1 | <1 | <1 | 6 | 86 | 102 |
| TC36 | 6% glucose 5% DO | 4.18 | 415 | 47 | 0.3 | <1 | 46 | 7 | 62 | 92 |
| TC36 | 6 + 4% glucose 5% DO§ | 4.5 | 767 | 37 | 0.5 | <1 | 72 | 5 | 72 | 97 |
| TC36 | Fed batch 5% DO¶ | 4.1 | 878 | 33 | 3.4 | <1 | <1 | 25 | 75 | 88 |
DO, dissolved oxygen.
Concentrations in broth after all glucose had been depleted, except as noted.
Yield expressed as a percentage of the maximal theoretical yield (0.67 g of acetate per gram of glucose).
Carbon recovery represents the percentage of substrate carbon recovered. Recovered carbon was calculated as the sum of carbon in cell mass, fermentation products, and CO2.
In the final sample, 44 mM glucose was present.
Excess glucose (9.5%) was added to maintain levels above 100 mM; 107 mM glucose was present in the final sample.
Maximal rates for glucose utilization (specific and volumetric) were higher for TC36 and TC24 than for W3110 and SZ47 (Table 2). This increase in metabolic activity can be primarily attributed to the ΔatpFH mutation. ATP levels serve as an allosteric regulator of several key glycolytic enzymes (33) and acetate kinase (37). In strains containing the ΔatpFH mutation, increased glycolytic flux and substrate level phosphorylation could partially compensate for the loss of ATP production from respiration-coupled phosphorylation. Differences between strains were particularly evident when comparing incubation times required to complete sugar metabolism (Fig. 3B). With TC36 and TC24, glucose was exhausted in 16–18 h compared with 26 h for SZ47 and 30 h for W3110. The maximum specific rate of glucose utilization (glycolytic flux) was 9 mmol⋅h−1⋅g−1 dry cell weight in the unmodified parent (W3110), 20 mmol⋅h−1⋅g−1 dry cell weight in TC24, and 18 mmol⋅h−1⋅g−1 dry cell weight in TC36. The slightly lower glycolytic flux in TC36 as compared with TC24 may be related to the increase in ATP yield resulting from improvements in acetate yield (1 ATP per acetate). Assuming protein represents 55% of dry cell weight, maximal glycolytic flux in TC36 is ≈0.55 μmols glucose min−1⋅mg−1 protein.
Production of Acetate and Other Organic Acids.
A substantial portion of glucose carbon was not recovered in the carbon balance (Table 3) for W3110 (40%) and SZ47 (80%). This loss is attributed to production of volatile products by high flux through the tricarboxylic acid cycle (CO2) but may also include the reduction of acetyl∼CoA to acetaldehyde and ethanol (Fig. 1). Although ethanol was absent in broth samples from all pH-controlled fermentations (sparged at 1 liter⋅min−1), a small amount of ethanol (6 mM) was found in seed cultures of W3110 (shaken flasks). No ethanol was present in seed cultures of TC36, which contains a mutation in alcohol dehydrogenase E. In W3110, the electron transport system (5% dissolved oxygen) and native fermentation pathways (Table 3) serve as complementary routes for NADH oxidation. Eliminating the fermentation pathways (SZ47) doubled the loss of carbon as volatile products (Table 3) and increased cell yield but decreased the rate of acetate production in comparison to W3110 (Tables 2 and 3).
Inactivation of oxidative phosphorylation (ΔatpFH) in SZ47 to produce TC24 resulted in a 26-fold increase in acetate yield and a 3-fold improvement in carbon recovery (Table 3, Fig. 4A). Acetate yield and carbon recovery increased by another 30% with the introduction of the sucA and adhE mutations (TC36). TC36 produced an average of 224 mM acetate in 16 h with small amounts of other products. This represents 68% of the maximum theoretical yield using native pathways (two acetates per glucose), remaining carbon being divided between cell mass, dicarboxylic acids, and CO2. Maximal rates of acetate production (specific and volumetric) were ≈2-fold higher for TC24 and TC36 than for SZ47 and W3110 (Table 2), a difference that can be attributed solely to the mutation in the (F1F0)H+-ATP synthase. This mutation eliminated ATP production by oxidative phosphorylation while retaining cytoplasmic F1-ATPase for the gratuitous consumption of ATP.
Figure 4.

Effects of selected mutations on the production of acetate (A), dicarboxylic acids (B), and pyruvate (C). ■, W3110 (wild type); □, SZ47(Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA); ○, TC24(Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT); ●, TC36 (Succ+; Δ(focA-pflB)∷FRT ΔfrdBC ΔldhA Δatp(FH)∷FRT ΔadhE∷FRT ΔsucA∷FRT).
Strain W3110 accumulated the highest levels of dicarboxylic acids (primarily succinate and 2-ketoglutarate) during glucose metabolism, ≈3-fold that of the engineered strains (Fig. 4B). The order of appearance of dicarboxylic acids in the broth correlated with growth rate and the order of entry into stationary phase for the four strains examined. Dicarboxylic acids were partially consumed as glucose levels declined and may represent spillover products from excessive glycolysis during the transition from exponential to stationary phase. It is interesting to note that pyruvate levels in the broth of TC36 also increased (16 mM at 12 h) during this transition (Fig. 4C). Although this pyruvate was subsequently metabolized, the excretion of pyruvate indicates that glucose uptake and glycolysis per se may not be limiting for acetate production. No significant accumulation of pyruvate was observed for the three other strains (W3110, SZ47 or TC24).
The consumption of base to maintain pH 7.0 provides an overall measure of total organic acid production (Fig. 3C). Higher rates and maxima for TC24 and TC36 are consistent with more rapid glucose metabolism. In general, variations in glucose utilization were accompanied by corresponding changes in base utilization. The exponential nature of the early time points reflects growth of the biocatalysts.
Improving Acetate Yields.
Dicarboxylic acids and cell mass were the dominant competing co-products from glucose. A limited set of experiments was conducted to evaluate the potential for process changes to improve acetate yield (Table 3). Acetate yield was not improved by increasing the dissolved oxygen level from 5% to 15%, reducing ammonia nitrogen (2 g⋅liter−1 ammonium phosphate) by 40% to limit growth or increasing the initial concentration of glucose from 3% to 6% (Table 3). However, a simple two-step batch feeding strategy was beneficial. A second addition of 3% glucose at the end of the growth phase (12 h) was metabolized to completion and produced 523 mM acetate with minimal loss to cell mass (Fig. 5). Acetate yield for this one-step addition (6% total glucose) was 78% of the theoretical maximum as compared with 68% for 3% glucose. The highest acetate yield, 86% of the theoretical maximum, was obtained by combining the one-step addition of 3% glucose with the nitrogen limitation to further limit loss of carbon into cell mass (Table 3). Additional fed-batch experiments were conducted in which multiple additions were made to glucose levels >100 mM. With this approach, 878 mM acetate was produced representing 75% of the maximum theoretical yield (Table 3).
Figure 5.

Fermentation of 6% glucose to acetate by TC36 in mineral salts medium. Fermentation was begun with 3% glucose followed by a second addition of 3% glucose after 12 h. □, cell mass; ○, glucose; ●, acetate.
Discussion
Strain TC36 was genetically engineered to combine the attributes of fermentative metabolism (low cell mass, high product yields) and oxidative metabolism (external electron acceptor) into a single biocatalyst for the production of oxidized chemicals such as acetate (Fig. 1). Chromosomal deletions were used instead of point mutations to maximize stability. All antibiotic resistance genes and auxotrophic requirements were eliminated to permit growth in simple mineral salts medium. During oxidative metabolism, up to half of the substrate carbon can be converted to roughly equal amounts of cell mass and CO2 (2, 32, 33) with minimal carbon flow into alternative products. To reduce the opportunity for excessive growth during oxidative metabolism, ATP production from NADH oxidation (oxidative phosphorylation) was eliminated by deleting the portion of (F1F0)H+-ATP synthase involved in membrane assembly while preserving a functional cytoplasmic F1-ATPase to provide gratuitous hydrolysis of ATP. With this mutation, a maximum of four ATP molecules (net) can be produced per glucose (assumes all pyruvate is metabolized to acetyl∼CoA and acetate) compared with a theoretical maximum of 36 ATP molecules for wild-type strains of E. coli. Excessive oxidation of substrate to CO2 and production of NADH were eliminated by disrupting the cyclic function of the tricarboxylic acid cycle (ΔsucA) with the added benefit of reducing oxygen demand for NADH oxidation. Additional mutations were introduced to eliminate all major fermentation pathways as alternative routes for NADH oxidation, minimizing the formation of alternative products. The resulting strain, TC36, has absolute requirements for a fermentable carbon source (substrate level phosphorylation) and for an external electron acceptor that can couple to the electron transport system during growth in mineral salts medium to maintain redox balance. With genetic blocks in all major fermentation pathways and oxidative phosphorylation, this strain is relatively insensitive to variations in dissolved oxygen.
The (F1F0)H+-ATP synthase and 2-ketoglutarate dehydrogenase mutations introduced into TC36 to minimize the levels of ATP and NAD(P)H produced from glucose under oxidative conditions would also be expected to promote glycolysis through native allosteric controls (8, 33, 38), providing a mechanism for the observed 2-fold increase in glycolytic flux compared with W3110 (wild type). With additional mutations in fermentation pathways, further metabolism of pyruvate was limited primarily to small biosynthetic needs and conversion to acetyl∼CoA by the pyruvate dehydrogenase complex. Although pyruvate dehydrogenase is activated by low NADH, acetyl∼CoA production may be limited by the availability of free CoA (note pyruvate accumulation in TC36 broth between 9 and 15 h; Fig. 4C). Resulting rises in pyruvate pools would serve as an allosteric activator of phosphotransferase (37), the first committed step for acetate production from acetyl∼CoA. Gratuitous ATP hydrolysis by F1-ATPase should ensure the availability of ADP for the final step in acetate production catalyzed by acetate kinase (Fig. 1). Excess pyruvate can also be directly oxidized to acetate by pyruvate oxidase (poxB), an enzyme that is induced during the latter stages of growth, and by environmental stress (39). This enzyme may also contribute to acetate production by TC36.
Eliminating oxidative phosphorylation while preserving (F1) H+ATPase resulted in a 2-fold increase in glycolytic flux (TC24 and TC36). Previously, Chao and Liao (40) and Patnaik et al. (41) demonstrated a similar 2-fold stimulation of glycolytic flux in E. coli using plasmids to express genes that created futile cycles to consume ATP. Recently, Koebmann et al. (42) independently concluded that glycolytic flux is limited by ATP utilization during the oxidative metabolism of glucose. In their studies, flux increased in a dose-dependent manner with controlled expression of F1-ATPase genes from a plasmid. Our results with TC24 and TC36 containing a chromosomal deletion (ΔatpFH) provide further support for this hypothesis. Thus glycolytic flux appears to be regulated by the economy of supply and demand as proposed by Hofmeyer and Cornish-Bowden (43). During the oxidative metabolism of glucose, glycolytic flux is limited by the metabolic ability to use ATP (availability of ADP) rather than by glucose transport or the catalytic capacities of central glycolytic enzymes. With this in mind, similar strategies that delete subunits concerned with the membrane assembly of the (F1F0)H+ ATP synthase, create futile cycles for ATP consumption, or increase cytoplasmic levels of the ATPase activities may prove useful to decrease cell yield, increase metabolic flux, and increase product yield in many other bioconversion processes.
Strain TC36 can be used as a biocatalyst platform for the efficient production of oxidized products. Under conditions of excess glucose, strain TC36 produced a maximum of 878 mM acetate, 75% of the maximum theoretical yield (Table 3) or 0.50 g of acetate per gram of glucose. Only cell mass and small amounts of organic acids were produced as coproducts with acetate. It is likely that 878 mM acetate approaches the upper limit of tolerance for metabolism in TC36. Concentrations as low as 50 mM acetate have been shown to induce a stress response in E. coli (44). The minimal inhibitory concentration for growth has been previously reported as 300–400 mM acetate at neutral pH (45, 46). Oxygen transfer often becomes limiting during aerobic bioconversion processes, promoting the accumulation of reduced products (31, 32). Synthesis of reduced products was eliminated by mutations in genes (ΔfocA-pflB ΔfrdBC ΔldhA ΔadhE) encoding the four major fermentation pathways. Excessive oxygen demand and NADH production were also reduced by a deletion in 2-ketoglutarate dehydrogenase (ΔsucA). The resulting strain, TC36(ΔfocA-pflB ΔfrdBC ΔldhA ΔatpFH ΔsucA ΔadhE), metabolizes sugars to acetate with the efficiency of fermentative metabolism, diverting a minimum of carbon to cell mass (biocatalyst) and CO2. By replacing the acetate pathway, a variety of alternative oxidized products can be produced with the mutational strategies used for the construction of TC36.
Commercial production of acetate with biocatalysts involves a two-step process in which sugar is first fermented to ethanol by yeasts followed by aerobic oxidation to acetate by Acetobacter (20, 21). Although titers of ≈650 mM are typically produced, higher titers can be readily achieved by the addition of complex nutrients in fed-batch processes requiring 60–120 h. Overall yields for commercial processes have been estimated to be 76% of the theoretical maximum (two acetate per glucose; 0.67 g of acetic acid per gram of glucose). Genetically engineered E. coli TC36 can produce acetate in a simpler single-step process using glucose with titers and yields equivalent to or higher than current batch processes. Although maximum titers with TC36 were lower than can be achieved by ethanol oxidation using Acetobacter (20), acetate production rates from glucose by TC36 were almost 2-fold higher than ethanol oxidation and required only mineral salts as nutrients. E. coli TC36 offers a unique set of advantages over currently used biocatalysts for the commercial production of acetate: a single-step process using sugars as substrates, high rates of acetate production, high acetate yields, simple nutrition (mineral salts), and a robust metabolism permitting the bioconversion of hexoses, pentoses, and many dissacharides. Derivatives of this strain may prove useful as biocatalysts for other products that are equivalent or more oxidized than sugars.
Acknowledgments
This research was supported by the Florida Agricultural Experiment Station (No. R-09127) and grants from the U.S. Department of Agriculture (01-35504-10669 and 00-52104-9704) and the U.S. Department of Energy (FG02-96ER20222).
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on May 1, 2001.
References
- 1.Akesson M, Hagander P, Axelsson J P. Biotechnol Bioeng. 2001;73:223–230. doi: 10.1002/bit.1054. [DOI] [PubMed] [Google Scholar]
- 2.Contiero J, Beatty C, Kumar S, DeSanti C L, Strohl W R, Wolfe A. J Ind Microbiol. 2000;24:421–430. [Google Scholar]
- 3.Aristidou A A, San K, Bennet G N. Biotechnol Prog. 1995;11:475–478. doi: 10.1021/bp00034a019. [DOI] [PubMed] [Google Scholar]
- 4.Luli G W, Strohl W R. Appl Environ Microbiol. 1990;56:1004–1011. doi: 10.1128/aem.56.4.1004-1011.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chotani G, Dodge T, Hsu A, Kumar M, LaDuca R, Trimbur D, Weyler W, Sanford K. Biochim Biophys Acta. 2000;1543:434–455. doi: 10.1016/s0167-4838(00)00234-x. [DOI] [PubMed] [Google Scholar]
- 6.Eggeling L, Pfefferle W, Sahm H. In: Basic Biotechnology. Ratledge C, Kristiansen B, editors. Vol. 2. Cambridge, U.K.: Cambridge Univ. Press; 2001. pp. 281–304. [Google Scholar]
- 7.Ingram L O, Aldrich H C, Borges A C, Saleh A, Causey T B, Martinez A, Morales F, Saleh A, Underwood S A, Yomano L P, York S W, Zaldivar J, Zhou S. Biotechnol Prog. 1999;15:855–866. doi: 10.1021/bp9901062. [DOI] [PubMed] [Google Scholar]
- 8.Underwood S A, Zhou S, Causey T B, Yomano L P, Shanmugam K T, Ingram L O. Appl Environ Microbiol. 2002;68:6263–6272. doi: 10.1128/AEM.68.12.6263-6272.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakamura, C. E., Gatenby, A. A., Hsu, A. K.-H., La Reau, R. D., Haynie, S. L., Diaz-Torres, M., Trimbur, D. E., Whited, G. M., Nagarajan, V., Payne, M. S., et al. (2000) U.S. Patent 6,013,494.
- 10.Tong I, Liao H H, Cameron D C. Appl Environ Microbiol. 1991;57:3541–3546. doi: 10.1128/aem.57.12.3541-3546.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cameron D C, Altaras N E, Hoffman M L, Shaw A J. Biotechnol Prog. 1998;14:116–125. doi: 10.1021/bp9701325. [DOI] [PubMed] [Google Scholar]
- 12.Altaras N E, Cameron D C. Appl Environ Microbiol. 1999;65:1180–1185. doi: 10.1128/aem.65.3.1180-1185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niu W, Draths K M, Frost J W. Biotechnol Prog. 2002;18:201–211. doi: 10.1021/bp010179x. [DOI] [PubMed] [Google Scholar]
- 14.Zhou S, Causey T B, Hasona A, Shanmugam K T, Ingram L O. Appl Environ Microbiol. 2003;69:399–407. doi: 10.1128/AEM.69.1.399-407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang D, Shin S, Rhee J, Pan J. Appl Environ Microbiol. 1999;65:1384–1389. doi: 10.1128/aem.65.4.1384-1389.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dien B S, Nichols N N, Bothast R J. J Ind Microbiol Biotechnol. 2001;27:259–264. doi: 10.1038/sj.jim.7000195. [DOI] [PubMed] [Google Scholar]
- 17. Donnelly, M. I., Sanville-Millard, C. & Chatterjee, R. (1998) U.S. Patent 6,159,738.
- 18.Vemuri G N, Altman M A, Altman E. J Bacteriol. 2002;68:1715–1727. doi: 10.1128/AEM.68.4.1715-1727.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farmer W R, Liao J C. Appl Environ Microbiol. 1997;63:3205–3210. doi: 10.1128/aem.63.8.3205-3210.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berraud C. Biotechnol Lett. 2000;22:451–454. [Google Scholar]
- 21.Cheryan M, Parekh S, Shah M, Witjitra K. Adv Appl Microbiol. 1997;43:1–33. doi: 10.1016/s0065-2164(08)70221-1. [DOI] [PubMed] [Google Scholar]
- 22.Freer S N. World J Microbiol Biotechnol. 2002;18:271–275. [Google Scholar]
- 23.Anonymous. Chem Week. 2002;164:33. [Google Scholar]
- 24.Miller J H. A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Plainview, NY: Cold Spring Harbor Lab. Press; 1992. [Google Scholar]
- 25.Sambrook J, Russell D W. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 2001. [Google Scholar]
- 26.Datsenko K A, Wanner B L. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Morales F, Borges A C, Martinez A, Shanmugam K T, Ingram L O. J Bacteriol. 1999;181:7143–7148. doi: 10.1128/jb.181.22.7143-7148.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Posfai G, Koob M D, Kirkpatrick H A, Blattner F C. J Bacteriol. 1997;179:4426–4428. doi: 10.1128/jb.179.13.4426-4428.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evans D J., Jr J Bacteriol. 1969;100:914–922. doi: 10.1128/jb.100.2.914-922.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rathbun W B, Betlach M V. Anal Biochem. 1969;20:436–445. doi: 10.1016/0003-2697(69)90198-5. [DOI] [PubMed] [Google Scholar]
- 31.Tsai P S, Nageli M, Bailey J E. Biotechnol Bioeng. 2002;79:558–567. doi: 10.1002/bit.10440. [DOI] [PubMed] [Google Scholar]
- 32.Varma A, Boesch B W, Palsson B O. Appl Environ Microbiol. 1993;59:2465–2473. doi: 10.1128/aem.59.8.2465-2473.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neidhardt F C, Ingraham J L, Schaechter M. Physiology of the Bacterial Cell: A Molecular Approach. Sunderland, MA: Sinauer; 1990. [Google Scholar]
- 34.Sorgen P L, Caviston T L, Perry R C, Cain B D. J Biol Chem. 1998;273:27873–27878. doi: 10.1074/jbc.273.43.27873. [DOI] [PubMed] [Google Scholar]
- 35.Cunningham L, Guest J R. Microbiology. 1998;144:2113–2123. doi: 10.1099/00221287-144-8-2113. [DOI] [PubMed] [Google Scholar]
- 36.Park S-J, Chao G, Gunsalus R P. J Bacteriol. 1997;179:4138–4142. doi: 10.1128/jb.179.13.4138-4142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki T. Biochim Biophys Acta. 1969;191:559–569. doi: 10.1016/0005-2744(69)90349-0. [DOI] [PubMed] [Google Scholar]
- 38.Underwood S A, Buszko M L, Shanmugam K T, Ingram L O. Appl Environ Microbiol. 2002;68:1071–1081. doi: 10.1128/AEM.68.3.1071-1081.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang Y-Y, Wang A-Y, Cronan J E., Jr Mol Microbiol. 1994;11:1019–1028. doi: 10.1111/j.1365-2958.1994.tb00380.x. [DOI] [PubMed] [Google Scholar]
- 40.Chao Y, Liao J C. J Biol Chem. 1994;269:5122–5126. [PubMed] [Google Scholar]
- 41.Patnaik R, Roof W D, Young R F, Liao J C. J Bacteriol. 1992;174:7527–7532. doi: 10.1128/jb.174.23.7527-7532.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koebmann B J, Westerhoff H V, Snoep J L, Nilsson D, Jensen P R. J Bacteriol. 2002;184:3909–3916. doi: 10.1128/JB.184.14.3909-3916.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hofmeyer J-H S, Cornish-Bowden A. FEBS Lett. 2000;476:47–51. doi: 10.1016/s0014-5793(00)01668-9. [DOI] [PubMed] [Google Scholar]
- 44.Kirkpatrick C, Maurer L M, Oyelakin N E, Yoncheva Y N, Maurer R, Slonczewski J L. J Bacteriol. 2001;183:6466–6477. doi: 10.1128/JB.183.21.6466-6477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lasko D R, Zamboni N, Sauer U. Appl Microbiol Biotechnol. 2000;54:243–247. doi: 10.1007/s002530000339. [DOI] [PubMed] [Google Scholar]
- 46.Zaldivar J, Ingram L O. Biotechnol Bioeng. 1999;66:203–210. doi: 10.1002/(sici)1097-0290(1999)66:4<203::aid-bit1>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]