

Abstract
The method of voltage clamp fluorometry combined with site-directed fluorescence labeling was used to detect local protein motions of the fully active Na+/K+-ATPase in real time under physiological conditions. Because helix M5 extends from the cytoplasmic site of ATP hydrolysis into the cation binding region, we chose the extracellular M5–M6 loop of the sheep α1-subunit for the insertion of cysteine residues to identify reporter positions for conformational rearrangements during the catalytic cycle. After expression of the single cysteine mutants in Xenopus oocytes and covalent attachment of tetramethylrhodamine-6-maleimide, only mutant N790C reported molecular rearrangements of the M5–M6 loop by showing large, ouabain-sensitive fluorescence changes (≈5%) on addition of extracellular K+. When the enzyme was subjected to voltage jumps under Na+/Na+-exchange conditions, we observed fluorescence changes that directly correlated to transient charge movements originating from the E1P–E2P transition of the transport cycle. The voltage jump-induced fluorescence changes and transient currents were abolished after replacement of Na+ by tetraethylammonium or on addition of ouabain, showing that conformational flexibility is impaired under these conditions. Voltage-dependent fluorescence changes could also be observed in the presence of subsaturating K+ concentrations. This allowed to monitor the time course of voltage-dependent relaxations into a new stationary distribution of states under turnover conditions, showing the acceleration of relaxation kinetics with increasing K+ concentrations. As a result, the stationary distribution between E1 and E2 states and voltage-dependent relaxation times can be determined at any time and membrane potential under Na+/Na+ exchange as well as Na+/K+ turnover conditions.
P-type ATPases form a major class of primary active membrane transport proteins, so called because they become transiently phosphorylated on ATP hydrolysis. The most prominent member is the ubiquitously occurring Na+/K+-ATPase, which exports three Na+ ions and imports two K+ ions in each transport cycle and thereby maintains the electrochemical gradients of Na+ and K+ across the plasma membrane of most animal cells.
The reaction cycle of the Na+/K+-ATPase is described in terms of the Albers–Post scheme (see Fig. 1A) (1, 2). The transduction of primary energy from ATP hydrolysis to active ion transport is brought about by conformational changes that occur for both the α and β subunit of the Na+/K+-ATPase (3–6). Several approaches were undertaken to reveal Na+/K+-ATPase conformational changes by using fluorescence labeling of the native enzyme with fluorescein-5′-isothiocyanate, N-(p-(2-benzimidazolyl)phenyl)-maleimide and styrylpyridinium dyes like RH421 (7–12), which were limited for several reasons. Purely biochemical assays to investigate conformation-dependent proteolysis patterns cannot provide time-resolved data, in the case of fluorescence labeling with styryl dyes the site of interaction with the enzyme is not defined, and site-directed labeling (e.g., by fluorescein) severely affects enzyme function. In addition, none of these methods allows to study the enzyme under physiological conditions, e.g., defined ion gradients or membrane potentials.
Figure 1.

(A) Albers–Post scheme for the Na+/K+-ATPase reaction cycle. The enzyme can assume two distinct conformations: E1 with ion binding sites facing the cytoplasm, and E2 with ion binding sites open to the extracellular space. The main electrogenic event was assigned to Na+ transport steps that are kinetically coupled to the E1P–E2P transition (underlaid in gray). (B) Na+/K+-ATPase α subunit modeled into the 1EUL structure of the SERCA Ca2+-ATPase (25) by using SWISSMODEL (courtesy of Jan B. Koenderink). Helix M5 and residue N790 (circle) are marked in red. Mutant N790C allowed for site-specific labeling by TMRM and yielded strong fluorescence changes in response to extracellular K+ or voltage pulses. Amino acids contributing to cation binding are colored as follows: E327, blue (helix M4); D776 and E779, red (helix M5); D804 and D808, green (helix M6). Two Ca2+ ions (yellow) from the 1EUL structure are also shown.
The intention of this study was to detect site-specific conformational changes in real time on the fully active Na+/K+-ATPase, and assign them to partial reactions of the Albers–Post cycle. Therefore, we expressed the Na+/K+-ATPase heterologously in Xenopus oocytes and used the method of site-directed fluorescence labeling combined with voltage clamp fluorometry (VCF), which was pioneered in the laboratories of E. Y. Isacoff (13) and F. Bezanilla (14, 15) on the Shaker potassium channel.
The Na+/K+-ATPase can assume two principal conformations, E1 with ion binding sites facing the cytoplasm, and E2 with ion binding sites open to the extracellular space. Phosphorylation of E1 (after intracellular Na+ binding) by ATP forms E1P and occludes three Na+ ions that are released to the extracellular medium after the conformational change to E2P. Subsequently, two K+ ions bind from the extracellular medium leading to dephosphorylation and K+ occlusion. After ATP binding the E1 conformation is reached again and the K+ ions are released to the intracellular solution. Because of this 3Na+/2K+ stoichiometry the Na+/K+-ATPase mediates net transport of charge. There is general agreement, that Na+-dependent transport steps are electrogenic (Fig. 1A, underlaid in gray) and kinetically coupled to the E1P–E2P conformational change (16–23).
The importance of helix M5 in functional rearrangements of the Na+/K+-ATPase was suggested by Lutsenko et al. (5). After trypsination of the enzyme the M5–M6 hairpin is released into the supernatant in a K+-dependent manner. The same phenomenon was also observed in the closely related H+/K+-ATPase (24). Modeling of the Na+/K+-ATPase primary sequence into the 3D structure of the sarcoplasmic reticulum Ca2+-ATPase (SERCA pump) (25) shows that the M5 helix assumes a piston-like structure extending from the site of ATP binding and protein phosphorylation into the cation binding region (Fig. 1B). The recently published E2 structure of the SERCA pump (26) revealed a movement of ≈5 Å of helix M5 relative to the membrane plane, which would be sufficient to bring about hydrophobicity changes at an extracellular reporter fluorophore (27, 28). Helix M5 and the subsequent extracellular M5–M6 loop are highly conserved among all P-type ATPases (29) and may play an important role for energy transduction and coupling ATP hydrolysis to ion transport.
It will be shown, that the application of the site-directed fluorescence labeling/VCF technique allows to study specific conformational changes during the transport cycle of the Na+/K+-ATPase. The observed fluorescence changes of a reporter fluorophore attached to a spatially defined position are Na+/K+-ATPase-specific. It is demonstrated that the highly conserved extracellular M5–M6 loop is involved in voltage- and Na+/K+-dependent conformational changes. Insights on the reaction mechanism of the Na+/K+-ATPase were obtained: in the Na+/Na+ exchange mode, the distribution of the E1P and the E2P conformational states can be determined both under stationary and time-resolved conditions; under turnover conditions, the occupancy of the E1 and E2 states as well as the time course of the transition in between can be quantified depending on the transmembrane voltage and Na+/K+ concentrations. The results will be discussed regarding the recently published structural changes of the Ca2+-ATPase, a homolog of the α subunit of the Na+/K+-ATPase.
Materials and Methods
Molecular Biology.
The cDNAs of sheep Na+/K+-ATPase α1-subunit, a modified construct with no extracellularly exposed cysteine residues (carrying mutations C911S and C964A) (30), and rat β1-subunit were subcloned into vector pTLN (31). The mutations Q111R and N122D were introduced into α1-subunit cDNAs to obtain reduced ouabain sensitivity (32) allowing for selective inhibition of the endogenous oocyte Na+/K+-ATPase. The Na+/K+-ATPase construct with reduced ouabain sensitivity is termed NaKWT, the extracellularly Cys-less and ouabain-insensitive construct NaKØCys. Single cysteine mutations in M5–M6 were introduced into NaKØCys by PCR and verified by sequencing.
Oocyte Preparation and cRNA Injection.
Oocytes were obtained by collagenase treatment after partial ovarectomy from Xenopus laevis females and injected with 15–25 ng of α-subunit and 1.5–2.5 ng of β-subunit cRNA, which was prepared using the SP6 mMessage mMachine kit (Ambion, Austin, TX). After injection, oocytes were kept in ORI buffer (90 mM NaCl/2 mM KCl/2 mM CaCl2/5 mM Mops, pH 7.4) containing 1 mg/ml penicillin/streptomycin at 18°C for three to four days.
Oocyte Pretreatment and Fluorescence Labeling.
Before measurements, oocytes were kept for 30 min in LS buffer (110 mM NaCl/2.5 mM Na-citrate/10 mM Mops/Tris, pH 7.4) and 30 min in PS buffer [100 mM NaCl/1 mM CaCl2/20 mM tetraethylammonium chloride (TEACl)/5 mM BaCl2/5 mM NiCl2/5 mM Mops/Tris, pH 7.4] to elevate intracellular Na+ concentration (22). Cys-specific fluorescence labeling was achieved by incubating Na+-loaded oocytes in PS buffer containing 5 μM tetramethylrhodamine-6-maleimide (TMRM, Molecular Probes) for 2–5 min at room temperature in the dark, followed by extensive washes in dye-free buffer. This period was optimal to achieve robust labeling and to avoid unspecific dye incorporation. Measurements under Na+/Na+-exchange conditions were peformed in Na+ test solution (100 mM NaCl/20 mM TEACl/5 mM BaCl2/5 mM NiCl2/5 mM Mops/Tris/10 μM ouabain, pH 7.4). For K+ titration experiments and stationary pump current recordings, 0.3–30 mM K+ were replaced for equimolar amounts of Na+. Heterologously expressed Na+/K+-ATPase was inhibited by 5 mM ouabain.
Two-Electrode Voltage Clamp Epifluorescence Measurements.
An oocyte perfusion chamber was mounted on the stage of a fluorescence microscope (Axioskop 2FS, Carl Zeiss), equipped with a ×40 water immersion objective (numerical aperture = 0.8). Currents were measured using a two-electrode voltage clamp amplifier CA-1B (Dagan Instruments, Minneapolis). Fluorescence was excited by a 100 W tungsten lamp using 535DF50 excitation filter, 565EFLP emission filter, and a 570DRLP dichroic mirror (Omega Optical, Brattleboro, VT). Fluorescence was measured with a PIN-020A photodiode (United Detector Technologies) mounted to the microscope camera port. Photodiode signals were amplified by a patch clamp amplifier EPC-5 (HEKA Electronics, Lamprecht, Germany). Fluorescence and current signals were simultaneously recorded with pClamp 8 software (Axon Instruments, Foster City, CA). Data analysis and presentation were carried out with ORIGIN 5.0 (Microcal Software, Northampton, MA).
Measurement and Analysis of Transient Currents.
Transient Na+/K+-ATPase currents under Na+/Na+ exchange conditions were obtained as the difference between current responses to a specific voltage step first in Na+ test solution, then in Na+ test solution containing 5 mM ouabain. Time constants were obtained from monoexponential fits to the data. The first few milliseconds after the voltage step were excluded to avoid artifacts arising from capacitive charging of the oocyte membrane. The transported charge Q is the time integral of the fitted currents, extrapolated to onset of voltage pulses. The resulting Q–V curves were fitted according to a Boltzmann function:
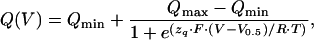 |
1 |
where Qmax and Qmin are the saturation values of translocated charge, V0.5 is the voltage of half-maximal activation, zq is the equivalent charge, F is the Faraday constant, R is the molar gas constant, T is temperature in K, and V is transmembrane potential. All experiments were carried out at room temperature (20–22°C).
Results and Discussion
The aim of this study was real-time and site-specific detection of conformational changes of the fully active Na+/K+-ATPase α subunit under physiological conditions, based on fluorescence responses of a reporter fluorophore. The technique of site-directed fluorescence labeling in combination with VCF has extensively been used for the detection of structural rearrangements underlying voltage sensor movements in voltage-gated cation channels (13–15). VCF implies electrophysiological measurements on integral membrane proteins in which single cysteines are introduced in putative reporter sites. On expression of these constructs in Xenopus oocytes, a fluorescent dye with a reactive sulfhydryl-specific moiety (in this case, TMRM) is covalently coupled to this cysteine. The fluorescence emission of the dye responds to environmental changes, which are brought about by the conformational rearrangements of the protein.
In the following, we will describe results obtained by application of this method to the Na+/K+-ATPase, link these findings to published data, and outline novel information that could not be obtained so far. Finally, we will discuss our findings in the frame of recently published structural information for P-type ATPases.
Functional Expression and Labeling by TMRM.
Single cysteine mutations within the extracellular M5–M6 loop (Fig. 2A) were introduced into a sheep Na+/K+-ATPase α1 subunit devoid of other extracellular cysteines (termed NaKØCys, see Materials and Methods) (30). Functional expression of this NaKØCys construct and of the single cysteine mutants was assessed by measuring stationary pump currents on addition of 10 mM K+ in two-electrode voltage clamp experiments. All solutions contained 10 μM ouabain to inhibit the endogenous oocyte Na+/K+-ATPase. It should be noted that all constructs had a reduced ouabain sensitivity in the millimolar range (32), as known from the rat Na+/K+-ATPase α1 subunit (see Materials and Methods). All 11 cysteine mutants we generated were functional and apart from mutant F786C produced currents similar to NaKWT (Fig. 2 A and B). However, only the N790C mutant showed high fluorescence changes after TMRM labeling and responded to changes in conditions that alter the occupancy of the E1P and E2P conformational states, as described below. Oocytes expressing the NaKØCys(N790C) construct exhibited a much higher fluorescence on TMRM labeling than uninjected controls (Fig. 2C), showing that labeling of endogenous membrane proteins produces only minor background fluorescence.
Figure 2.

(A) Amino acids of the extracellular M5–M6 loop of the Na+/K+-ATPase. Amino acids of helices M5 and M6 are shown in black boxes. (B) Stationary pump currents of Na+/K+-ATPase single cysteine mutants at 0 mV in response to 10 mM K+. The dotted line indicates the stationary current level of the NaKWT and NaKØCys constructs. Data originated from three to five oocytes; values are means ± SE. (C) Fluorescence images of TMRM-labeled oocytes. (Upper) Uninjected control. (Lower) Oocyte injected with NaKØCys(N790C) α and β subunit cRNA. Labeling and illumination conditions were identical. (D) Parallel recording of pump current (Lower) and fluorescence change (Upper) from an oocyte expressing NaKØCys(N790C) in response to 10 mM K+ and 5 mM ouabain (see perfusion protocol) at 0 mV.
Stationary Current and Fluorescence Measurements.
Fig. 2D shows a parallel recording of pump current and fluorescence of TMRM-labeled mutant NaKØCys(N790C). Addition of K+ induces a stationary current that is accompanied by a fluorescence increase. Both stationary current and fluorescence increase could be inhibited by 5 mM ouabain, showing that the observed fluorescence change is Na+/K+-ATPase-specific. The similarity between pump currents and the fluorescence responses on K+ concentration changes provided initial evidence that the fluorescence changes correlate with pump conformational transitions. This behavior is explained in terms of the Albers–Post reaction scheme for the Na+/K+-ATPase (1, 2). In the absence of K+, dephosphorylation is slow and the E2P conformation highly accumulates, as described (19, 20, 22). Addition of K+ stimulates dephosphorylation, turnover is increased, and the E2P conformation is depleted in favor of E1 states. Therefore, the low fluorescence level measured under K+-free conditions can be assigned mainly to the E2P conformation, whereas high fluorescence measured in presence of K+ is indicative of E1 states (which is corroborated by the data in the next paragraph). Note that a fluorescence increase hints at a more hydrophobic environment of the dye, whereas an aqueous environment leads to fluorescence quenching.
Voltage Pulse Experiments and the E1P–E2P Transition.
To allow for a more specific attribution of fluorescence signals to partial reactions of the Albers–Post cycle, we tested whether the fluorescence changes reflect the E1P–E2P transition under certain conditions, and performed voltage pulse experiments in the absence of K+. Under these conditions, the Na+/K+-ATPase is restricted to Na+/Na+ exchange (21). Because dephosphorylation in the absence of K+ is slow, the enzyme shuttles in a voltage-dependent manner almost exclusively between E1P and E2P, which allows the isolated investigation of the E1P–E2P conformational change. Na+ transport steps are intimately linked to the E1P–E2P transition (Fig. 1A, underlaid in gray) and involve charge movement across the membrane. Changes in membrane potential determine the kinetics of electrogenic reactions and shift the distribution between E1P and E2P, giving rise to transient currents. The main electrogenic events, namely sodium binding steps, occur on a microsecond time scale, but the detected transient charge movement is rate-limited by the conformational change E2P(Na) ↔ E1P(Na) (19). Relaxation into a new distribution of states occurs with the sum of forward and backward reaction rate constants, and transient currents decay with first order kinetics (18–23).
In Fig. 3A, transient currents in response to voltage pulses are shown. Jumps to positive potentials favor the E2P conformation and release of positive charges to the extracellular side, resulting in a positive transient current. The simultaneously recorded fluorescence signal decays with a very similar time course (Fig. 3B). Negative potentials drive the pump into the E1P conformation, which enables electrogenic Na+ reuptake resulting in a negative transient current accompanied by fluorescence increases. These observations agree with the interpretation of the stationary current and fluorescence data shown in Fig. 2D.
Figure 3.

(A) Fluorescence change signals in the absence of K+ on voltage pulses from 0 mV to values as stated. (B) Voltage jump-induced transient currents, obtained as ouabain-sensitive difference currents (see Materials and Methods), recorded in parallel to traces in A. (C) Rate constants (reciprocal of time constants) for fluorescence changes (□) and transient currents (●) as depicted in A and B. Data are means ± SE from five oocytes. (D) Voltage dependence of fluorescence saturation values (□) and translocated charge (●) from experiments as shown in A and B. Data are means ± SE from five oocytes. Fits of a Boltzmann function are superimposed, yielding the following parameters: ΔF-V curve (dashed): V0.5 = −117 mV ± 1.4mV, zq = 0.76 ± 0.2; Q–V curve (solid): V0.5 = −110 mV ± 9 mV, zq = 0.85 ± 0.12.
Comparison of the signals from Fig. 3 A and B shows that, for all potentials tested, the kinetics of fluorescence changes and transient currents are almost identical. Current and fluorescence signals were analyzed by fitting with a monoexponential function to obtain the voltage dependence of the apparent rate constants (Fig. 3C). The resulting curves are very similar, indicating that the process detected by fluorescence changes under Na+/Na+ exchange conditions parallels the charge translocation coupled to the E1P–E2P transition.
The transient currents of the NaKWT construct measured under Na+/Na+ exchange conditions decayed with ≈80 s−1 at 21°C and 0 mV (data not shown). This value agrees well with published data (≈50–200 s−1, refs. 16–21) especially when compared with data obtained from Xenopus oocytes (22, 23). For the NaKØCys(N790C) construct, this rate constant is reduced to 20 s−1. Note that the observed slow relaxation time constant of the E1P–E2P transition of the NaKØCys(N790C) construct is brought about by the introduced mutation. Therefore, it does not reflect similarly slow rate constants, which had initially been reported for the E1P–E2P conformational change of the native Na+/K+-ATPase (33).
A slight increase in relaxation time of the NaKØCys(N790C) construct was observed due to TMRM binding (data not shown). The same observations, i.e., slowed kinetics on cysteine substitution and further reduction of the rate constant after TMRM attachment, were previously reported in Shaker K+ channel studies (14). However, the slower kinetics observed for NaKØCys(N790C) does not impair the assignment of the observed fluorescence signals to the E1P–E2P conformational change. Residual kinetic differences between fluorescence and transient current responses at negative potentials may arise from unlabeled pump molecules, which contribute to the currents with faster kinetics than that of the fluorescence response (see Materials and Methods).
Voltage-Dependent Charge Movement and Fluorescence Change.
Further information about the charge translocation process caused by the E1P–E2P conformational transition can be derived from analysis of the translocated charge (integral of the transient currents). When plotted against the membrane potential, the amount of charge moved during a transient current follows a Boltzmann distribution (21), which is shown in Fig. 3D (Q–V curve, filled circles). When the voltage dependence of the saturation values of the corresponding fluorescence responses is plotted in the same way, the resulting ΔF-V curve (Fig. 3E, open squares) superimposes with the Q–V curve. Because the amount of charge in response to a voltage jump is limited in either direction, positive voltage pulses lead to maximal accumulation of E2P, which is indeed observed as saturating fluorescence. Negative voltage pulses shift the pump into E1P in a saturating fashion and fluorescence saturation is expected at strongly hyperpolarizing potentials, which was indeed observed at high Na+ concentrations (200 mM) at extremely hyperpolarizing potentials (−160 mV, data not shown). Because the charge movement occurs during the E1P–E2P conformational change, the Q–V curve allows a direct measure of the amounts of E1P and E2P at any voltage. From the equivalence of the Q–V curve and the ΔF-V curve, we conclude that, under Na+/Na+ exchange conditions, the fluorescence level is an absolute measure of the concentrations of E1P and E2P. The structural rearrangement during the E1P–E2P transition is directly correlated to the main electrogenic step in the transport cycle, i.e., extracellular Na+ release/reuptake. The fluorescence label at amino acid 790 demonstrates the involvement of the M5–M6 loop and most probably also of helix M5 in this crucial conformational change.
Compared with NaKWT or NaKØCys the Q–V-curve of NaKØCys(N790C) is shifted to hyperpolarizing potentials.NaKWT or NaKØCys exhibited a value for the half-maximal voltage (V0.5) of ≈−65 mV (data not shown), which agrees well with data reported by others (−30, … , −90 mV, refs. 18–21 and 34). For NaKØCys(N790C), the V0.5 value was ≈−110 mV (Fig. 3D). This shift in voltage dependence is obviously caused by the introduced mutation and can be interpreted as a reduced cation affinity (especially for Na+). This finding is surprising because residue 790 is not part of the cation binding pocket itself.
TEA+ Replacement and Ouabain Inhibition.
Voltage pulse experiments were carried out to test for effects of TEA+ and ouabain on transient currents and fluorescence changes (see Fig. 6, and Supporting Text which are published as supporting information on the PNAS web site, www.pnas.org). Replacement of Na+ for TEA+ did not change the stationary fluorescence at holding potential and abolished fluorescence changes. Addition of 5 mM ouabain to the 100 mM Na+ solution also did not shift stationary fluorescence at holding potential and inhibited voltage-dependent fluorescence changes as well as transient currents, no matter whether K+ was present or not (see Figs. 4G and 6). Ouabain binds to the Na+/K+-ATPase in the E2P conformation (35, 36), in agreement with the observed low fluorescence level (indicative of E2P). Transient currents in the Na+/Na+ exchange mode are often defined as ouabain-sensitive currents in the literature (19, 21–23). The fluorescence data show that ouabain blocks the E2P–E1P transition, which also precludes transient charge translocation.
Figure 4.

(A–E) Voltage pulse-induced fluorescence responses from a continuous recording of an oocyte expressing NaKØCys(N790C) at different K+ concentrations. Fluorescence increase is indicated by the black arrow. (F) Control measurement after removal of extracellular K+. (G) Inhibition by 5 mM ouabain in the presence of 30 mM K+. (Inset) The applied voltage protocol.
[K+] Dependence of the Transient Fluorescence Signals.
VCF enables investigation of the Na+/K+-ATPase under physiological turnover conditions, as voltage-dependent fluorescence responses can also be measured in the simultaneous presence of Na+ and K+ (Fig. 4). Under these conditions, observation of transient currents is impossible for kinetic reasons, because of the redistribution of reaction cycle intermediates. To interpret the fluorescence signals under turnover conditions we assume, that the fluorophore discriminates between the two principal conformations of the enzyme. E1 states are then characterized by high fluorescence and E2 states by low fluorescence. Two observations justify this assumption. First, the voltage dependence of the ΔF-V curve (Fig. 3E) covers the complete range from maximal accumulation of E2P at depolarizing potentials to maximal accumulation of E1P at hyperpolarizing potentials. Second, the saturating stationary fluorescence levels at high [K+] (highest fluorescence and maximal accumulation of E1 states) or in presence of ouabain (lowest fluorescence and maximal accumulation of E2P) do not significantly exceed the saturation values of the ΔF-V curve under Na+/Na+ exchange conditions (see description of Figs. 4 and 5A below). Therefore, the ΔF-V curve under Na+/Na+ exchange conditions serves as a calibration for the determination of the distribution between E1 and E2 states under turnover conditions, which cannot be addressed by purely electrophysiological approaches.
Figure 5.

(A) Voltage dependence of fluorescence saturation values at different K+ concentrations: 0 mM (□), 0.3 mM (○),1 mM (▵), 3 mM (▿),10 mM (⋄). (B) [K+] dependence of apparent rate constants of fluorescence signals on off voltage jumps to −80 mV. Data are means ± SE from three oocytes.
Fig. 4 shows voltage jump-induced fluorescence changes in the presence of different K+ concentrations. All data traces were obtained from continuous recording on the same oocyte expressing NaKØCys(N790C) to allow for comparison of the absolute fluorescence levels under the varying conditions of the experiment. After solution exchanges to the stated K+ or inhibitor concentrations at −80 mV holding potential the cell was subjected to 200-ms voltage pulses between +60 and −160 mV (on pulse) in 20 mV steps, followed by a step back to −80 mV (off pulse). The stationary fluorescence level at −80 mV, visible at the beginning of each set of fluorescence signals, rises with increasing K+ concentration corresponding to the increase in stationary fluorescence in Fig. 2D on addition of 30 mM K+. Fig. 4A shows fluorescence responses to on and off voltage pulses at zero K+, the same conditions as for Fig. 3 A and B. Voltage jumps to positive potentials elicited only small fluorescence decreases, which did not change with voltage. In contrast, negative potentials induced strongly voltage-dependent fluorescence increases. With 1 mM extracellular K+ (Fig. 4B) fluorescence signals started from an already increased stationary value. Positive voltage pulses induced fluorescence decreases, and jumps to negative potentials led to fluorescence increases with the onset of saturation becoming apparent at extremely positive or negative voltages. With further increasing extracellular [K+] (Fig. 4 C and D) stationary fluorescence at −80 mV successively increases. Negative voltage pulses induced only small fluorescence increases, whereas positive potentials led to comparatively large fluorescence decreases. At 30 mM extracellular K+ stationary fluorescence at −80 mV reaches a maximum, and voltage pulses in either direction no longer induce any fluorescence change (Fig. 4E). To demonstrate reversibility, the voltage protocol was applied on removal of K+ (Fig. 4F) and signals are equivalent to those from Fig. 4A. Addition of ouabain at 30 mM K+ reduces fluorescence to a minimum (Fig. 4G), even slightly lower than obtained in the absence of K+ at depolarizing voltages. Voltage pulses do not induce fluorescence changes, showing that the pump is arrested in E2P.
[K+] Dependence of ΔF-V Curves.
The distribution of voltage-dependent saturation values for fluorescence changes (ΔF-V curves) at different K+ concentrations is depicted in Fig. 5A. The change in the characteristics of the ΔF-V curves with increasing [K+] is composed of two effects, a positive shift of the V0.5 value and a decrease in slope (Fig. 5A). The latter can be interpreted as a decreased voltage sensitivity. The value for the equivalent charge, zq, from Boltzmann fits of the ΔF-V curves decreases from ≈0.8 at zero K+ to ≈0.45 at 1 mM K+. The value zq represents the amount of charges translocated or, equivalently, the fraction of the transmembrane field which an unitary charge passes during a charge-translocating event (which may be a composite of several charge-translocating steps under these conditions). Therefore, the easiest interpretation is that activation of the K+ branch of the Albers–Post cycle leads to an inward transport of ≈0.35 unitary positive charges, in accordance with the reported lower electrogenicity of K+ transport steps (34, 37, 38). This value rather is a lower limit as K+ concentrations >3 mM induce such a large shift of the ΔF-V curve that Boltzmann parameters cannot be determined (Fig. 6A). At [K+] > 10 mM the absence of detectable fluorescence changes indicates that the stationary distribution between E1 and E2 states can no longer be shifted by voltage pulses within the accessible voltage range (−160 mV to +60 mV). In contrast to inhibitor experiments, however, the pump is fully active under these conditions.
[K+] Dependence of Relaxation Kinetics.
The transient fluorescence responses on voltage jumps at subsaturating K+ concentrations directly yield kinetic information about the electrogenic event(s) or preceding electroneutral reaction steps, which are rate-limiting for these charge-moving events. However, in contrast to K+-free conditions in which the major electrogenic event occurs during extracellular Na+ release/reuptake, the full Albers–Post cycle probably also contains charge translocating steps within the K+ branch (34, 36–38). The apparent rate constant of the voltage-dependent relaxation into a new stationary distribution of states at 0 mV increases from 20 s−1 to 70 s−1 at saturating K+ concentrations with an apparent Km of ≈1 mM (Fig. 5B), which is close to the value which can be inferred from the K+ dependence of the stationary current (data not shown).
Link of Conformational Changes to Structural Information.
The fluorescence quantum yield of the rhodamine dye is sensitive to environmental hydrophobicity changes (13, 14). The assumption that the fluorophor can discriminate between E1 (high fluorescence) and E2 (low fluorescence) states implies that it resides in a sheltered, hydrophobic environment in E1 states, whereas it encounters a more exposed, quenching environment in E2 states. This can be interpreted either as a movement of the extracellular end of helix M5 (which is in close vicinity to residue 790) relative to the other transmembrane helices and/or opening of an extracellular crevice, which allows for release or binding of cations. This notion is supported by the recently published E2 structure of the SERCA pump (26), which is a homolog of the Na+/K+-ATPase α subunit. The reported movement of the extracellular end of helix M5 of the SERCA pump transversal to the membrane plane is ≈5 Å (see figure 4a in ref. 26), which is small compared with the vast rearrangements of intracellular domains. However, minute positional changes are sufficient to influence the properties of the TMRM label as demonstrated for the Shaker K+ channel (27, 28). The movement of helix M5 relative to the other transmembrane helices is much more pronounced (26). In addition, a rotation of helix M5 was observed which could also cause TMRM fluorescence changes as inferred from Shaker K+ channel studies (28). The fact that only one residue within the extracellular M5–M6 loop can serve as a reporter site for conformational changes votes for tight spacial constraints within the region through which cations leave or reach their binding sites. However, we would like to be cautious in using the published SERCA pump structures for the interpretation of our results. Because the SERCA E2 structure represents a dephosphorylated enzyme stabilized by binding of the inhibitor thapsigargin in the transmembrane region, a true E2P structure with respect to the membrane helices might look different. In addition, the Na+/K+-ATPase contains a β subunit with unpredictable impact on structural details at the extracellular face.
Concluding Remarks.
VCF allows the determination of spatially defined conformational changes of the fully functional Na+/K+-ATPase in real time and under physiological conditions. The reporter fluorophore attached to amino acid 790 close to the extracellular end of helix M5 is involved in a conformational change, which under Na+/Na+ exchange conditions can be attributed to a single reaction step of the catalytic cycle, the E1P–E2P transition. The ΔF-V curve obtained under Na+/Na+ exchange conditions can be used as a calibration to determine the stationary distribution between E1 and E2 states under Na+/K+ turnover conditions. The fluorophore encounters a sheltered, hydrophobic environment in the E1P state and a more exposed quenching, aqueous medium in the E2P state. An obvious explanation for such a change in the fluorophore environment would be a movement of helix M5 relative to the other helices accompanied by extracellular opening of cation binding sites. Given the high homology within helix M5 and the M5–M6 loop among all P-type ATPases, this method will allow detailed studies on conformational transitions of other P-type ATPases, most notably electroneutral pumps such as the H+/K+-ATPase.
Supplementary Material
Acknowledgments
We thank A. Becker for technical assistance, Y.-K. Hu for the extracellularly Cys-less construct, J. B. Koenderink and K. Fendler for discussions, and K. Hartung for help with the experimental setup. We are grateful to Francisco Bezanilla for providing the Shaker H4(Δ6-46,W434F,M356C) cDNA as a valuable test system. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 472), the Max-Planck-Gesellschaft zur Förderung der Wissenschaften, the Johann Wolfgang Goethe-Universität Frankfurt, and National Institutes of Health Grant GM 39500 (to J.H.K.).
Abbreviations
- VCF
voltage clamp fluorometry
- TEA
tetraethylammonium
- SERCA
sarcoplasmic reticulum Ca2+-ATPase
- TMRM
tetramethylrhodamine-6-maleimide
References
- 1.Post R L, Hegyvary C, Kume S. J Biol Chem. 1972;247:6530–6540. [PubMed] [Google Scholar]
- 2.Albers R W. Annu Rev Biochem. 1967;36:727–756. doi: 10.1146/annurev.bi.36.070167.003455. [DOI] [PubMed] [Google Scholar]
- 3.Jorgensen P L. Biochim Biophys Acta. 1975;401:399–415. doi: 10.1016/0005-2736(75)90239-4. [DOI] [PubMed] [Google Scholar]
- 4.Lutsenko S, Kaplan J H. J Biol Chem. 1994;269:4555–4564. [PubMed] [Google Scholar]
- 5.Lutsenko S, Anderko R, Kaplan J H. Proc Natl Acad Sci USA. 1995;92:7936–7940. doi: 10.1073/pnas.92.17.7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mikhailova L, Mandal A K, Arguello J M. Biochemistry. 2002;41:8195–8202. doi: 10.1021/bi025721k. [DOI] [PubMed] [Google Scholar]
- 7.Karlish S J. J Bioenerg Biomembr. 1980;12:111–136. doi: 10.1007/BF00744678. [DOI] [PubMed] [Google Scholar]
- 8.Smirnova I N, Faller L D. J Biol Chem. 1993;268:16120–16123. [PubMed] [Google Scholar]
- 9.Glynn I M, Hara Y, Richards D E, Steinberg M. J Physiol (London) 1987;383:477–485. doi: 10.1113/jphysiol.1987.sp016422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kane D J, Fendler K, Grell E, Bamberg E, Taniguchi K, Froehlich J P, Clarke R J. Biochemistry. 1997;36:13406–13420. doi: 10.1021/bi970598w. [DOI] [PubMed] [Google Scholar]
- 11.Taniguchi K, Mardh S. J Biol Chem. 1993;268:15588–15594. [PubMed] [Google Scholar]
- 12.Klodos I. In: The Sodium Pump. Bamberg E, Schoner W, editors. Darmstadt, Germany: Steinkopff; 1994. pp. 517–528. [Google Scholar]
- 13.Mannuzzu L M, Moronne M M, Isacoff E Y. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- 14.Cha A, Bezanilla F. Neuron. 1997;19:1127–1140. doi: 10.1016/s0896-6273(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 15.Cha A, Bezanilla F. J Gen Physiol. 1998;112:391–408. doi: 10.1085/jgp.112.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fendler K, Grell E, Haubs M, Bamberg E. EMBO J. 1985;4:3079–3085. doi: 10.1002/j.1460-2075.1985.tb04048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fendler K, Jaruschewski S, Hobbs A, Albers W, Froehlich J P. J Gen Physiol. 1993;102:631–666. doi: 10.1085/jgp.102.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hilgemann D W. Science. 1994;263:1429–1432. doi: 10.1126/science.8128223. [DOI] [PubMed] [Google Scholar]
- 19.Holmgren M, Wagg J, Bezanilla F, Rakowski R F, De Weer P, Gadsby D C. Nature. 2000;403:898–901. doi: 10.1038/35002599. [DOI] [PubMed] [Google Scholar]
- 20.Friedrich T, Nagel G. Biophys J. 1997;73:186–194. doi: 10.1016/S0006-3495(97)78059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakao M, Gadsby D C. Nature. 1986;323:628–630. doi: 10.1038/323628a0. [DOI] [PubMed] [Google Scholar]
- 22.Rakowski R F. J Gen Physiol. 1993;101:117–144. doi: 10.1085/jgp.101.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmgren M, Rakowski R F. Biophys J. 1994;66:912–922. doi: 10.1016/s0006-3495(94)80867-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gatto C, Lutsenko S, Shin J M, Sachs G, Kaplan J H. J Biol Chem. 1999;274:13737–13740. doi: 10.1074/jbc.274.20.13737. [DOI] [PubMed] [Google Scholar]
- 25.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Nature. 2000;405:647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 26.Toyoshima C, Nomura H. Nature. 2002;418:605–611. doi: 10.1038/nature00944. [DOI] [PubMed] [Google Scholar]
- 27.Glauner K S, Mannuzzu L M, Gandhi C S, Isacoff E Y. Nature. 1999;402:813–817. doi: 10.1038/45561. [DOI] [PubMed] [Google Scholar]
- 28.Cha A, Snyder G E, Selvin P R, Bezanilla F. Nature. 1999;402:809–813. doi: 10.1038/45552. [DOI] [PubMed] [Google Scholar]
- 29.Sweadner K J, Donnet C. Biochem J. 2001;356:685–704. doi: 10.1042/0264-6021:3560685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Y K, Kaplan J H. J Biol Chem. 2000;275:19185–19191. doi: 10.1074/jbc.M000641200. [DOI] [PubMed] [Google Scholar]
- 31.Lorenz C, Pusch M, Jentsch T J. Proc Natl Acad Sci USA. 1996;93:13362–13366. doi: 10.1073/pnas.93.23.13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price E M, Lingrel J B. Biochemistry. 1988;27:8400–8408. doi: 10.1021/bi00422a016. [DOI] [PubMed] [Google Scholar]
- 33.Borlinghaus R, Apell H J, Läuger P. J Membr Biol. 1987;97:161–178. doi: 10.1007/BF01869220. [DOI] [PubMed] [Google Scholar]
- 34.Rakowski R F, Vasilets L A, LaTona J, Schwarz W. J Membr Biol. 1991;121:177–187. doi: 10.1007/BF01870531. [DOI] [PubMed] [Google Scholar]
- 35.Glynn I M. The Na+, K+-Transporting Adenosine Triphosphatase. New York: Plenum; 1985. [Google Scholar]
- 36.Läuger P. Electrogenic Ion Pumps. Sunderland, MA: Sinauer Associates; 1991. [Google Scholar]
- 37.Peluffo R D, Berlin J R. J Physiol (London) 1997;501:33–40. doi: 10.1111/j.1469-7793.1997.033bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lafaire A V, Schwarz W. J Membr Biol. 1986;91:43–51. doi: 10.1007/BF01870213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.