

Abstract
The transcription factors hypoxia inducible factors 1 and 2 (HIF-1 and HIF-2) regulate multiple responses to physiological hypoxia such as transcription of the hormone erythropoietin to enhance red blood cell proliferation, vascular endothelial growth factor to promote angiogenesis and glycolytic enzymes to increase glycolysis. Recent studies indicate that HIFs also regulate mitochondrial respiration and mitochondrial oxidative stress. Interestingly, mitochondrial metabolism, respiration and oxidative stress also regulate activation of HIFs. In this review, we examine the evidence that mitochondria and HIFs are intimately connected to regulate each other resulting in appropriate responses to hypoxia.
Keywords: mitochondria, HIF, ROS, respiration
Introduction
Multi-cellular organisms have evolved multiple mechanisms to respond to decreased oxygen levels (hypoxia) [1]. The three important physiological responses to hypoxia are: pulmonary vascular constriction to shunt blood to better oxygenated regions of the lung; neurotransmitter release by the carotid body to increase breathing and production of the hormone erythropoietin (EPO) to enhance red blood cell proliferation to increase the haemoglobin concentration in the blood [2]. Hypoxia is observed in many diseases such as ischemia and cancer [3]. Although physiological responses to hypoxia at the tissue level have been appreciated for decades, the molecular and cellular biology of hypoxia have only been elucidated in the past 15 years. The discovery of the transcription factor hypoxia inducible factor 1 (HIF-1) led to understanding the underlying molecular responses to hypoxia [4]. HIF-1 was discovered as a nuclear factor bound to a cis-acting hypoxia response element (HRE) in the 3′ flanking region of the EPO gene during hypoxia. HIF-1 is a heterodimer of two basic helix loop-helix/PAS proteins, HIF-1α and the aryl hydrocarbon nuclear trans-locator (ARNT or HIF-1β). HIF-1α protein is only detectable under hypoxic conditions, while HIF-1β subunit is constitutively stable [5]. Subsequently, HIF-2α and HIF-3α were discovered, which show similar regulation in response to hypoxia [6, 7]. The regulation and biological consequences of HIF activation have been a major focus of hypoxia research. In the past decade, a major role for the mitochondria in the regulation of HIFs and vice versa has been elucidated. This article reviews how coordinated signalling between HIFs and the mitochondria regulate the cellular response to hypoxia.
Oxidative phosphorylation
Historically, mitochondria have been viewed primarily as consumers of oxygen in order to generate ATP, i.e. oxidative phosphorylation. Mitochondrial oxygen consumption is initiated when reducing equivalents generated primarily as NADH and FADH2 from the TCA cycle provide electrons to the mitochondrial electron transport chain complexes I and II, respectively. Complexes I and II provide two electrons to ubiquinone (Q) resulting ubiquinol (QH2, reduced ubiquinone). Ubiquinol transfers its electrons to complex III (bc1 complex), which donates its electrons to cytochrome c. Reduced cytochrome c can transfer its electrons to complex IV (cytochrome c oxidase). Subsequently, complex IV transfers the electrons to molecular oxygen. The electron transport chain is located in the inner mitochondrial membrane. Cytochrome c is not membrane bound. The movement of electrons through the electron transport chain is coupled to proton translocation from the mitochondrial matrix to the inner mitochondrial membrane space (Fig. 1). The pumping of protons across the inner mitochondrial membrane generates an electrochemical gradient of protons consisting of pH gradient and a membrane potential. These protons return down their gradient either through a proton leak or the ATP synthase (complex V). The ATP synthase couples the transport across the membrane to the synthesis of ATP from ADP to Pi. The phosphate for the phosphorylation is transported into the mitochondria by the phosphate carrier, and the ATP is exported to the cytosol in exchange for ADP by the adenine nucleotide carrier located in the inner mitochondrial membrane.
Fig 1.

Overview of oxidative phosphorylation. Oxidative phosphorylation is the flow of electrons from NADH and FADH2 to O2 through the mitochondrial electron transport chain resulting in pumping of protons across the inner mitochondrial membrane into the intermembrane space. This creates a proton-motive force which is utilized to generate ATP through the ATP synthase.
Hypoxic activation of HIFs
A major breakthrough in our understanding of the regulation of HIFs came from studies demonstrating that complexes containing the tumour suppressor von Hippel-Landau protein (pVHL) serves as an E3 ubiquitin ligase for the degradation of HIF-α protein during normoxia [8]. Subsequently several groups of investigators demonstrated that HIF-α protein is hydroxylated at proline residues by prolyl hydroxylases (PHDs) under normoxia to allow pVHL to interact with HIF-α protein for ubiquitin targeted degradation [9–12]. HIF-α protein is also hydroxylated at an asparagine residue by the asparaginyl hydroxylase factor inhibiting HIF-1 (FIH) under normoxia to prevent interactions with co-activators such as p300 and aberrant transcriptional activation [13, 14]. Hypoxia suppresses the hydroxylation of proline and asparagine residues, thereby allowing full HIF mediated transcriptional activation.
Mitochondria regulate HIFs
The hydroxylation of HIF-α protein by PHDs is inherently oxygen dependent since the oxygen atom of the hydroxy group is derived from molecular oxygen. In addition, prolyl hydroxylation requires the mitochondrial TCA cycle intermediate 2-oxoglutarate and iron as cofactors. 2-oxoglutarate is required because the hydroxylation reaction is coupled to the decarboxylation of 2-oxoglutarate to succinate, which accepts the other oxygen atom from molecular oxygen. Since the hydroxylation reaction requires oxygen, it has been widely speculated that PHDs act as the direct oxygen sensors for hypoxic activation of HIFs [15]. Clearly, in the absence of oxygen or iron, HIF-α would not undergo proline hydroxylation and subsequent pVHL mediated ubiquitin-targeted degradation. Thus, the PHDs would be sensors under anoxia. However, it is not clear whether the PHDs would intrinsically be inhibited at higher levels of oxygen (1–2% O2), which have also been shown to activate HIFs. These higher levels of oxygen are more likely to be encountered in physiological conditions associated with tissue hypoxia, for example cancer. Recombinant PHDs have a Km close to ambient air in vitro indicating that the PHDs are decreasing their enzymatic activity throughout the physiological range of PO2[16]. Therefore, if the PHDs were in fact the direct oxygen sensors, one would predict a continuous increase in the accumulation of HIF-α protein as oxygen levels fall from 21% O2 to 0% O2. In fact, the HIF-1α protein begins to accumulate around 5% O2 and its concentration increases as the oxygen levels approach anoxia [17]. It is therefore likely that a variety of inputs regulate PHD activity during hypoxia in addition to oxygen concentration [18]. Emerging evidence indicates that the mitochondrial electron transports chain generated reactive oxygen species (ROS) and oxygen consumption regulates hydroxylation of the HIF-α protein during hypoxia [19]. In the next section, we discuss how mitochondrial respiration, ROS and TCA cycle metabolites regulate HIF-α hydroxylation.
Mitochondrial ROS regulates HIFs
During mitochondrial respiration under normal oxygen conditions, O2 is chemically reduced to water by the transfer of four electrons at cytochrome oxidase. The resulting free energy change is conserved in the form of ATP synthesis. It has been estimated that 2–3% of the O2 consumed by mitochondria is incompletely reduced, yielding superoxide [20]. Superoxide can be generated at complexes I, II and III of the mitochondrial electron transport chain (Fig. 2) [21]. Complexes I and II release superoxide into the mitochondrial matrix while complex III can release superoxide into either the mitochondrial intermembrane space or mitochondrial matrix [22]. Complex III generates superoxide during the Q-cycle, which is initiated by the transfer of two electrons to ubiquinone from complex I or complex II resulting in the reduction of ubiquinone to ubiquinol. Subsequently, ubiquinol oxidation requires donation of two electrons: the first electron transfer is to the Rieske iron–sulphur protein (RISP) and cytochrome c1 resulting in the oxidation of ubiquinol to ubisemiquinone at the Qo site of complex III. The second electron is transferred from ubisemiquinone to cytochrome b resulting in the oxidation of ubisemiquinone to ubiquinone. Ubisemiquinone created at the Qo site of complex III is able to donate an electron to oxygen to generate superoxide [23, 24]. The availability of O2, the reduction state of the electron carriers and the mitochondrial membrane potential determine the ability of electron transport chain to generate superoxide [25].
Fig 2.

Mitochondrial electron transport chain generates superoxide. Complexes I, II and III generate superoxide into the mitochondrial matrix. Complex III can also release superoxide into the intermembrane space. Complex IV does not generate superoxide. Complex III generates superoxide through the Ubiquinone (Q) cycle.
We have proposed a model in which the increased generation of ROS at complex III of the mitochondrial electron transport chain is required for HIF-1α protein stabilization during hypoxia [19]. The earliest evidence in support of this model was the observation hypoxia paradoxically increases ROS generation [26, 27]. Furthermore, cells depleted of their mitochondrial DNA (ρ° cells) were not able to elicit hypoxia induced increase in ROS generation and HIF-1α protein accumulation [27, 28]. The overexpression of catalase abolished the HIF-1α protein accumulation in response to hypoxia [28]. Hydrogen peroxide was able to stabilize HIF-1α protein levels under normoxic conditions in both wild-type and ρ° cells28. These initial observations were corroborated by various investigators [29, 30]. Genetic evidence to support a role for the hypoxic increase in ROS in the activation of HIFs came from the observation that cells lacking cytochrome c fail to stabilize HIF-1α or HIF-2α protein during hypoxia31. Depleting the complex III RISP using shRNAs also prevented hypoxic HIF-1α protein stabilization [32, 33]. The evidence that ROS generated at the Qo site of complex III stabilize HIF-1α protein during hypoxia comes from the observation that cells harbouring a deletion of the cytochrome b gene are able to stabilize HIF-1α during hypoxia34. Cells deficient in the cytochrome b gene are respiratory deficient but are still capable of generating ROS at the Qo site of complex III [34]. Depleting RISP in these cytochrome b-deficient cells with shRNAs abolished ROS generation and the hypoxic stabilization of HIF-1α protein. Collectively these experiments demonstrate that the production of ROS at the Qo site of mitochondrial complex III is crucial for the hypoxic stabilization of HIF-α independent of mitochondrial oxygen consumption. Whether hypoxia increases ROS generation has remained a point of contention. Initial studies relied on using oxidant sensitive dyes such as DCFH to monitor ROS production during hypoxia. These dyes have limitations with respect to sensitivity, specificity and photoactivation [35]. More recently, a mitochondrially localized FRET based redox probe and a mitochondrially localized ratiometric redox sensitive green fluorescent protein (GFP) probe have been used to confirm that hypoxia increases mitochondrial derived ROS production [33, 36]. The levels of ROS generated during hypoxia appear to be substantially lower than those that induce senescence or apoptosis as assessed using a mitochondrially targeted redox sensitive GFP probe [36]. How ROS inhibits the hydroxylation of the HIF-1α protein is currently not known. The hydroxylation reaction requires the reduced form of iron (Fe2+). It is possible that the hydrogen peroxide produced during hypoxia oxidizes Fe2+ to Fe3+ (Fig. 3) [37].
Fig 3.
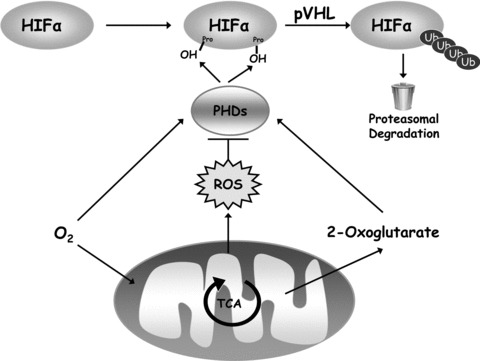
Mitochondria regulate HIFs. HIFs are hetrodimers between the HIFα proteins and HIF1β protein. HIFα proteins are hydroxylated at two distinct proline residues by PHDs under normoxic conditions. The hydroxylation directs the HIFα proteins for pVHL mediated ubiquitin-dependent degradation. The hydroxylation reaction requires oxygen and 2-oxoglutarate as substrates. Mitochondria can regulate hydroxylation by controlling the availability of oxygen and the TCA cycle intermediate 2-oxolgultrate to the PHDs. Furthermore, under hypoxic conditions, the release of ROS from mitochondrial complex III results in prevention of hydroxylation and stabilization of HIFα proteins. Thus, mitochondria regulate HIFα proteins through ROS, oxygen and 2-oxogluatrate availability.
Mitochondrial respiration regulates HIFs
An alternative explanation for the inability of respiratory deficient cells to stabilize HIF-1α protein during hypoxia is the redistribution of oxygen from mitochondria to the PHDs during hypoxia [38]. In this model, normal levels of oxygen consumption in wild-type cells consuming oxygen would result in lower intracellular oxygen levels than those observed in respiratory deficient cells The lower levels of intracellular oxygen would inhibit the PHDs in wild-type cells compared with respiratory deficient cells, stabilizing HIF proteins. This model is not compatible with the observation that certain respiratory deficient mutants such as cells deficient in cytochrome b are able to stabilize HIF-1α protein. These cells show dramatically reduced levels of oxygen consumption but retain their ability to increase the generation of ROS during hypoxia from the Qo site of complex III [34].
Although the redistribution of oxygen from mitochondria to the PHDs during hypoxia is unlikely to be the trigger for hypoxic stabilization of HIF-1α protein, there are examples where high levels of oxygen consumption by the mitochondria would leave the cytosol ‘hypoxic’. The overexpression of PGC1α under normoxia can make the cytosol hypoxic through an increase in mitochondrial biogenesis in which stimulates oxygen consumption [39]. This triggers stabilization of the HIF-1α protein. Also in culture conditions where cells under normoxia are confluent or have limited gas exchange cytosolic hypoxia might trigger stabilization of the HIF-1α protein [40].
TCA cycle intermediates regulate HIFs
PHDs convert the TCA cycle intermediate 2-oxoglutarate to succinate in order to hydroxylate the HIF-α subunit. Thus, a rise in succinate would prevent hydroxylation by mass action product inhibition. Succinate is normally converted into fumarate within the TCA cycle by succinate dehydrogenase (SDH), a membrane-bound enzyme that is also a component (complex II) of the electron transport chain. SDH is a complex of four different polypeptides (SDHA, SDHB, SDHC and SDHD) and several prosthetic groups that include FAD, non-haem iron (iron–sulphur centres), ubiquinone and haemb [41]. The loss of any of the SDH subunits would be predict to trigger HIF activation since they all should increase succinate levels. Indeed, loss of SDHD elevates HIF-1α protein under normoxia [42]. Mutations in SDHB, SDHC or SDHD gene have been associated with paraganglioma [43–45]. By contrast, SDHA mutations have not been linked with paraganglioma. Furthermore, RNAi against SDHA does not elevate HIF-1α protein under normoxia [46]. This indicates that the rise in succinate levels due loss of any of the four subunits of SDH not sufficient to increase HIF activation.
Based on the structure and mechanism of complex II it is predicted that mutations in SDH B, C or D would increase ROS generation while mutations in SDHA would not [41]. Indeed, RNAi against SDHA protein does not increase ROS, HIF activation or tumorigenicity [46]. In contrast, RNAi against SDHB protein increases ROS production, HIF activation and tumorigenicity [46]. It is likely that the increase in ROS coupled with an increase in succinate levels cooperate to activate HIFs under normoxia. Another TCA metabolite that regulates HIF activation under normoxia is fumarate [47]. Mutations in the TCA cycle enzyme fumarate hydratase (FH) are associated hereditary leiomyomatosis and renal cell cancer (HLRCC) [48]. Fumarate can inhibit the forward hydroxylation reaction similar to succinate [49]. Recent evidence suggests that loss of FH results in ROS-dependent activation of HIF [50]. Thus, the FH mutation activates HIF by increases both in ROS and fumarate levels.
Hypoxia decreases cellular ATP utilization to diminish mitochondrial respiration
Cells exposed to hypoxia acutely (seconds) do not display a decrease in oxygen consumption [51–53]. However, as cells are exposed to hypoxia for longer periods (minutes to hours) they display a reversible suppression of oxygen consumption. Cells display this decrease in metabolism at levels of hypoxia (1–3% O2) well above those associated with an inhibition of mitochondrial respiration and ATP production secondary to oxygen limitation (typically <0.5%) [54]. The persistence of mitochondrial respiration in the face of severe hypoxia is explained by the low Km of the cytochrome c oxidase, which is less than 1 μM O2[53]. The decrease in oxygen consumption under moderate hypoxia is likely to be an adaptive mechanism to avoid the development of anoxia. During hypoxia, cells that fail to decrease their oxygen consumption are likely to become anoxic faster than cells that can suppress their rate of oxygen consumption [55].
What controls mitochondrial respiration during hypoxia? In their seminal work Chance and Williams proposed that mitochondrial respiration in cells is controlled by cellular ATP utilization. Their model suggested that increased cytoplasmic ATP utilization decreases cytosolic ATP levels and increases cytosolic ADP and Pi levels [56]. The rise in cytosolic ADP levels leads to a rise in mitochondrial ADP via the increased activity of the adenine nucleotide carrier. The increased mitochondrial ADP concentration stimulates the ATP synthase to augment the rate of ATP synthesis. The increased ATP synthesis results in a decrease in the mitochondrial membrane potential, which stimulates the respiratory chain to consume oxygen. Aside from cellular ATP utilization, the other factors that control mitochondrial respiration are the NADH supply, the respiratory chain and the degree of proton leak [57]. Metabolic control analysis combines experimental data with mathematical models to estimate the contribution of a given enzyme or pathway to the overall rate of metabolism. Brand and colleagues performed metabolic control analysis on liver cells under normoxia and observed that 15–30% of respiration is controlled by the NADH supply (these include pyruvate supply to the mitochondria, the TCA cycle and any other NADH-supplying reaction); 20% is controlled by the proton leak; and 50% is controlled by ATP utilization by cellular ATPases [58]. Interestingly, the respiratory chain contributes only 0–15% to respiratory control suggesting that the maximal rate of electron transport contributes little to the overall rate of cellular respiration. We did a similar analysis of liver cells under hypoxic conditions and found that although hypoxia decreased oxygen consumption by 50%, the control of respiration had not changed from normoxic conditions [53]. In this study, cellular ATP utilization remained the major factor controlling respiration under hypoxia. Thus, the major reason for the decrease in respiration during hypoxia is due to a decrease in cellular ATP utilization.
A major ATP consumer that hypoxia inhibits is the function of Na/K-ATPase. The activity of the Na/K-ATPase alone can account for 20–70% of the oxygen expenditure of mammalian cells [59]. Na/K-ATPase is a transmembrane protein found in higher eukaryotes that transports Na+ and K+ across the plasma membrane to maintain ionic gradients [60]. The Na/K-ATPase is a heterodimer composed of α and β subunits [61]. The α subunit is a transmembrane protein that cleaves high-energy phosphate bonds and exchanges intracellular Na+ for extracellular K+ coupled to the hydrolysis of ATP. The smaller β subunit is a glycosylated transmembrane molecule that controls the heterodimer assembly and insertion into the plasma membrane. Multiple investigators have reported that hypoxia reversibly suppresses Na/K-ATPase activity [62–66]. The hypoxia-induced decrease of the Na/K-ATPase activity is due to endocytosis of the α subunit from the plasma membrane by PKC ζ. Hypoxia stimulates AMPK α1 isoform, which directly phosphorylates PKC ζ at Thr410 to promote Na,K-ATPase endocytosis [67]. The endocytosis of the Na,K-ATPase during hypoxia triggers pVHL-mediated degradation of plasma membrane Na-K-ATPase in an HIF-independent manner [68]. Interestingly, the endocytosis and degradation of the plasma membrane Na-K-ATPase during hypoxia is dependent on mitochondrial generated ROS (Fig. 4) [63, 69].
Fig 4.
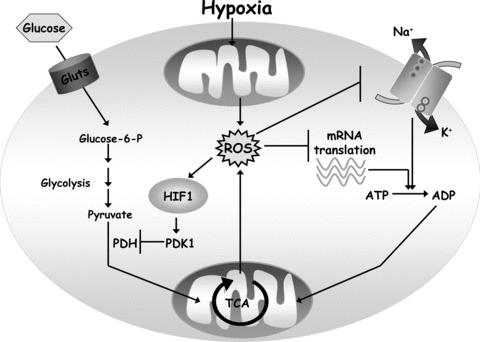
Hypoxia regulates mitochondrial respiration. Mitochondrial respiration is regulated by the oxygen, ADP and reducing equivalents (NADH, FADH2 from TCA cycle) availability. Oxygen is limiting for respiration under severe hypoxic conditions (<0.5% O2). Thus under physiological hypoxia (1–3% O2) oxygen is not limiting to conduct maximal respiration. The major controller of mitochondrial respiration is ADP availability from the cellular ATP utilization. Hypoxia through mitochondrial ROS also diminishes the activity of Na/K ATPase and mRNA translation. This results in a decrease in cellular ATP utilization and a decrease in ADP availability to mitochondria. Hypoxia also stimulates the release of mitochondrial ROS from complex III to activate HIF-1, which induces the transcription of PDK1. PDK1 negatively regulates pyruvate dehydrogenase, an enzyme that converts pyruvate to acetyl-CoA. Thus, an increase in HIF-1-dependent PDK1 expression results in diminished availability of acetyl-CoA. This also contributes to diminished respiration during hypoxia by decreasing TCA cycle activity.
The other major ATP consumer that hypoxia inhibits is mRNA translation [70]. The initiation of mRNA translation is regulated by the active eukaryotic initiation factors eIF4F and eIF2 [71]. The mammalian target of rapamycin (mTOR) and pancreatic eIF2α kinase (PERK) are the key regulators of translation during hypoxia [72, 73]. In growth-promoting conditions, mTOR sustains translation by phosphorylating the eIF4E-binding proteins (4E-BPs) and ribosomal protein S6 kinases (S6Ks) [74]. The PI3K/Akt signalling up-regulates mTOR activity by alleviating repression of mTOR by the TSC2 complex [75]. Hypoxia (1.5% O2) causes rapid (within 15 min.) and reversible hypophosphorylation of mTOR and its effectors 4E-BP1 and S6K [72]. The rapid inhibition of mTOR is HIF independent and occurs through the activation of AMPK76. AMPK phosphorylates TSC2 causing repression of mTOR [76]. Thus, loss of TSC2 effectively suppresses AMPK-induced mTOR inhibition during hypoxia [77].
We recently demonstrated that rapid activation of AMPK during hypoxia is dependent on mitochondrial ROS [78]. The sustained inhibition of mTOR over hours involves the HIF-dependent transcription of REDD1, which suppress mTOR-dependent mRNA translation [79]. The other major contributor to the decrease in mRNA translation during hypoxia is the activation of PERK73. PERK activation results in eIF2α phosphorylation, which inhibits mRNA translation initiation. Mitochondrial ROS have been implicated in the activation of PERK [80].
HIF-1 regulates mitochondrial respiration
The regulation of NADH supply by HIF-1 is another regulator of respiration during hypoxia (Fig. 4). As noted above, the supply of NADH to the respiratory chain accounts for approximately 15–30% of respiratory control. HIF-1 regulates pyruvate supply to mitochondria through activation of pyruvate dehydrogenase kinase 1 (PDK1) [81, 82]. PDK1 inactivates pyruvate dehydrogenase, the enzyme responsible for converting pyruvate into acetyl CoA. The HIF-1-induced increase in PDK1 during hypoxia reduces pyruvate conversion to acetyl-CoA resulting in diminished stimulation of the TCA cycle. This decreases the supply of NADH to the respiratory chain. As consequence the HIF-1-dependent decrease in respiration during hypoxia results in an increase in intracellular oxygen tension [81]. In addition, the decrease in HIF-1-dependent respiration during hypoxia might limit the generation of ROS [82]. Thus, the expression of HIF target genes, for example PDK1 might provide a negative feedback loop limiting the supply of NADH and inhibiting further mitochondrial ROS production.
HIF-1 also controls mitochondrial respiration during hypoxia by exchanging subunit 4–1 of cytochrome c oxidase (COX4–1) for the more efficient COX4–2 isoform [83]. Most mammalian cells express COX4–1 isoform under normoxia [84]. Hypoxia through HIF-1 induces COX4–2 mRNA and protein expression [83]. By contrast, COX4–1 mRNA levels do not change in response to alterations in the cellular O2 concentration; however, hypoxia increases the degradation of COX4–1 protein levels through HIF-1-dependent expression of LON, a mitochondrial protease. A consequence of COX4–1 switching to COX4–2 enhances the efficiency of the electron flux through the respiratory chain during hypoxia. This enhances ATP generation and decreases hypoxia induced ROS production. The HIF-1-dependent transcriptional effects on COX4 subunits swapping is likely to occur over prolonged hypoxia. To date, most studies indicate that COX activity has minimal effect on respiration. For example, in young adult mice a 95% reduction in skeletal muscle COX activity is not associated with reduced maximal muscle force generation, enhanced fatigue or signs of oxidative damage or apoptosis [85]. This is compatible with the observation in cells that the maximal level of electron transport contributes at most 15% to the control of cellular respiration [58].
HIF-2 regulates mitochondrial oxidative stress
Although, HIF-1 and HIF-2 are activated by similar mechanism by hypoxia, they have both overlapping and distinct gene targets. HIF-1 regulates metabolic genes while HIF-2 regulates EPO and the mitochondrial matrix protein superoxide dismutase 2 (SOD2) [86–88]. Depending on the genetic background, loss of HIF-2 in adult mice results in profound anaemia [87] or multiple organ pathology due to increase in oxidative stress [88]. The livers of HIF2 null mice display increased oxidative stress due to reduced expression of SOD2 as well as frataxin, a chaperone for the oxidant sensitive protein aconitase [89]. The role of HIF-2 regulation of oxidative stress is further supported by the observation that SOD2 levels are markedly increased in VHL null renal cell carcinoma cells [90]. Furthermore, reduction in HIF-2α makes mice more susceptible to oxidative stress induced ischemia-reperfusion injury [91]. It will be interesting whether activation of HIF-2 prevents oxidative stress induced injury in other organs such as brain, lung and heart.
Conclusions
The discovery of the HIFs has provided a molecular explanation for the physiological responses to hypoxia. Furthermore, in appropriate activation of HIFs has been implicated in a variety of pathologies such as cancer. In the past decade, studying the regulation of HIFs has led to an appreciation that mitochondrial metabolism and ROS are essential to regulators of HIFs. Conversely, HIFs have also been shown to regulate mitochondrial metabolism and ROS levels. Since mitochondria are the major consumers of oxygen, it is not surprising that HIFs and mitochondria are inter-connected. The hypoxic response is essential for survival of metazoans thus would have to be linked to mitochondrial metabolism. Understanding the details of mitochondrial regulation of HIFs could pave the way for new therapies to modulate HIF function for HIF associated pathologies.
Acknowledgments
This work was supported by a NIH Grant R01CA123067–04 as well as the LUNGevity Foundation and a Consortium of Independent Lung Health Organizations convened by ‘Respiratory Health Association of Metropolitan Chicago’ to NSC. K.V.T is supported by a NIH training grant T32CA009560–22.
References
- 1.Semenza GL. Life with oxygen. Science. 2007;318:62–4. doi: 10.1126/science.1147949. [DOI] [PubMed] [Google Scholar]
- 2.Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 1996;76:839–85. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- 3.Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–54. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–7. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 6.Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- 7.Gu YZ, Moran SM, Hogenesch JB, et al. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998;7:205–13. [PMC free article] [PubMed] [Google Scholar]
- 8.Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 9.Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 10.Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 11.Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 12.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–40. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 13.Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–86. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lando D, Peet DJ, Gorman JJ, et al. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–71. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Hirsila M, Koivunen P, Gunzler V, et al. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–80. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- 17.Jiang BH, Semenza GL, Bauer C, et al. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol. 1996;271:C1172–80. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- 18.Pan Y, Mansfield KD, Bertozzi CC, et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol. 2007;27:912–25. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15:660–6. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- 20.Chance B, Williams GR. The respiratory chain and oxidative phosphorylation. Adv Enzymol Relat Subj Biochem. 1956;17:65–134. doi: 10.1002/9780470122624.ch2. [DOI] [PubMed] [Google Scholar]
- 21.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–73. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 23.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–14. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 24.Sun J, Trumpower BL. Superoxide anion generation by the cytochrome bc1 complex. Arch Biochem Biophys. 2003;419:198–206. doi: 10.1016/j.abb.2003.08.028. [DOI] [PubMed] [Google Scholar]
- 25.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duranteau J, Chandel NS, Kulisz A, et al. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem. 1998;273:11619–24. doi: 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- 27.Chandel NS, Maltepe E, Goldwasser E, et al. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–20. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chandel NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–8. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 29.Agani FH, Pichiule P, Chavez JC, et al. The role of mitochondria in the regulation of hypoxia-inducible factor 1 expression during hypoxia. J Biol Chem. 2000;275:35863–7. doi: 10.1074/jbc.M005643200. [DOI] [PubMed] [Google Scholar]
- 30.Killilea DW, Hester R, Balczon R, et al. Free radical production in hypoxic pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L408–12. doi: 10.1152/ajplung.2000.279.2.L408. [DOI] [PubMed] [Google Scholar]
- 31.Mansfield KD, Guzy RD, Pan Y, et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1:393–9. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brunelle JK, Bell EL, Quesada NM, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–14. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–8. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 34.Bell EL, Klimova TA, Eisenbart J, et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol. 2007;177:1029–36. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tampo Y, Kotamraju S, Chitambar CR, et al. Oxidative stress-induced iron signaling is responsible for peroxide-dependent oxidation of dichlorodihydrofluorescein in endothelial cells: role of transferrin receptor-dependent iron uptake in apoptosis. Circ Res. 2003;92:56–63. doi: 10.1161/01.res.0000048195.15637.ac. [DOI] [PubMed] [Google Scholar]
- 36.Bell EL, Klimova TA, Eisenbart J, et al. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol Cell Biol. 2007;27:5737–45. doi: 10.1128/MCB.02265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerald D, Berra E, Frapart YM, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–94. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 38.Hagen T, Taylor CT, Lam F, et al. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–8. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 39.O’Hagan KA, Cocchiglia S, Zhdanov AV, et al. PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc Natl Acad Sci USA. 2009;106:2188–93. doi: 10.1073/pnas.0808801106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doege K, Heine S, Jensen I, et al. Inhibition of mitochondrial respiration elevates oxygen concentration but leaves regulation of hypoxia-inducible factor (HIF) intact. Blood. 2005;106:2311–7. doi: 10.1182/blood-2005-03-1138. [DOI] [PubMed] [Google Scholar]
- 41.Yankovskaya V, Horsefield R, Tornroth S, et al. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003;299:700–4. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 42.Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 43.Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–51. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 45.Baysal BE, Willett-Brozick JE, Lawrence EC, et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet. 2002;39:178–83. doi: 10.1136/jmg.39.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guzy RD, Sharma B, Bell E, et al. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–31. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pollard PJ, Briere JJ, Alam NA, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–9. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 48.Alam NA, Rowan AJ, Wortham NC, et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet. 2003;12:1241–52. doi: 10.1093/hmg/ddg148. [DOI] [PubMed] [Google Scholar]
- 49.Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–53. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 50.Sudarshan S, Sourbier C, Kong HS, et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and HIF-1{alpha} stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;29:4080–90. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schumacker PT, Chandel N, Agusti AG. Oxygen conformance of cellular respiration in hepatocytes. Am J Physiol. 1993;265:L395–402. doi: 10.1152/ajplung.1993.265.4.L395. [DOI] [PubMed] [Google Scholar]
- 52.Budinger GR, Chandel N, Shao ZH, et al. Cellular energy utilization and supply during hypoxia in embryonic cardiac myocytes. Am J Physiol. 1996;270:L44–53. doi: 10.1152/ajplung.1996.270.1.L44. [DOI] [PubMed] [Google Scholar]
- 53.Chandel NS, Budinger GR, Choe SH, et al. Cellular respiration during hypoxia. Role of cytochrome oxidase as the oxygen sensor in hepatocytes. J Biol Chem. 1997;272:18808–16. doi: 10.1074/jbc.272.30.18808. [DOI] [PubMed] [Google Scholar]
- 54.Jones DP. Intracellular diffusion gradients of O2 and ATP. Am J Physiol. 1986;250:C663–75. doi: 10.1152/ajpcell.1986.250.5.C663. [DOI] [PubMed] [Google Scholar]
- 55.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–13. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 56.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem. 1955;217:383–93. [PubMed] [Google Scholar]
- 57.Jones DP, Shan X, Park Y. Coordinated multisite regulation of cellular energy metabolism. Annu Rev Nutr. 1992;12:327–43. doi: 10.1146/annurev.nu.12.070192.001551. [DOI] [PubMed] [Google Scholar]
- 58.Brown GC, Hafner RP, Brand MD. A ‘top-down’ approach to the determination of control coefficients in metabolic control theory. Eur J Biochem. 1990;188:321–5. doi: 10.1111/j.1432-1033.1990.tb15406.x. [DOI] [PubMed] [Google Scholar]
- 59.Milligan LP, McBride BW. Energy costs of ion pumping by animal tissues. J Nutr. 1985;115:1374–82. doi: 10.1093/jn/115.10.1374. [DOI] [PubMed] [Google Scholar]
- 60.Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta. 1957;23:394–401. doi: 10.1016/0006-3002(57)90343-8. [DOI] [PubMed] [Google Scholar]
- 61.Kaplan JH. Biochemistry of Na,K-ATPase. Annu Rev Biochem. 2002;71:511–35. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- 62.Carpenter TC, Schomberg S, Nichols C, et al. Hypoxia reversibly inhibits epithelial sodium transport but does not inhibit lung ENaC or Na-K-ATPase expression. Am J Physiol Lung Cell Mol Physiol. 2003;284:L77–83. doi: 10.1152/ajplung.00181.2002. [DOI] [PubMed] [Google Scholar]
- 63.Dada LA, Chandel NS, Ridge KM, et al. Hypoxia-induced endocytosis of Na,K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J Clin Invest. 2003;111:1057–64. doi: 10.1172/JCI16826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mairbaurl H, Wodopia R, Eckes S, et al. Impairment of cation transport in A549 cells and rat alveolar epithelial cells by hypoxia. Am J Physiol. 1997;273:L797–806. doi: 10.1152/ajplung.1997.273.4.L797. [DOI] [PubMed] [Google Scholar]
- 65.Planes C, Friedlander G, Loiseau A, et al. Inhibition of Na-K-ATPase activity after prolonged hypoxia in an alveolar epithelial cell line. Am J Physiol. 1996;271:L70–8. doi: 10.1152/ajplung.1996.271.1.L70. [DOI] [PubMed] [Google Scholar]
- 66.Wodopia R, Ko HS, Billian J, et al. Hypoxia decreases proteins involved in epithelial electrolyte transport in A549 cells and rat lung. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1110–9. doi: 10.1152/ajplung.2000.279.6.L1110. [DOI] [PubMed] [Google Scholar]
- 67.Gusarova GA, Dada LA, Kelly AM, et al. Alpha1-AMP-activated protein kinase regulates hypoxia-induced Na,K-ATPase endocytosis via direct phosphorylation of protein kinase C zeta. Mol Cell Biol. 2009;29:3455–64. doi: 10.1128/MCB.00054-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou G, Dada LA, Chandel NS, et al. Hypoxia-mediated Na-K-ATPase degradation requires von Hippel Lindau protein. FASEB J. 2008;22:1335–42. doi: 10.1096/fj.07-8369com. [DOI] [PubMed] [Google Scholar]
- 69.Comellas AP, Dada LA, Lecuona E, et al. Hypoxia-mediated degradation of Na,K-ATPase via mitochondrial reactive oxygen species and the ubiquitin-conjugating system. Circ Res. 2006;98:1314–22. doi: 10.1161/01.RES.0000222418.99976.1d. [DOI] [PubMed] [Google Scholar]
- 70.Wouters BG, Van Den Beucken T, Magagnin MG, et al. Control of the hypoxic response through regulation of mRNA translation. Semin Cell Dev Biol. 2005;16:487–501. doi: 10.1016/j.semcdb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 71.Dever TE. Gene-specific regulation by general translation factors. Cell. 2002;108:545–56. doi: 10.1016/s0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- 72.Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278:29655–60. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- 73.Koumenis C, Naczki C, Koritzinsky M, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22:7405–16. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gingras AC, Raught B, Gygi SP, et al. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15:2852–64. doi: 10.1101/gad.912401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 76.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 77.Liu L, Cash TP, Jones RG, et al. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–31. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Emerling BM, Weinberg F, Snyder C, et al. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic Biol Med. 2009;46:1386–91. doi: 10.1016/j.freeradbiomed.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu L, Wise DR, Diehl JA, et al. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J Biol Chem. 2008;283:31153–62. doi: 10.1074/jbc.M805056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Papandreou I, Cairns RA, Fontana L, et al. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–97. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 82.Kim JW, Tchernyshyov I, Semenza GL, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 83.Fukuda R, Zhang H, Kim JW, et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–22. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 84.Huttemann M, Kadenbach B, Grossman LI. Mammalian subunit IV isoforms of cytochrome c oxidase. Gene. 2001;267:111–23. doi: 10.1016/s0378-1119(01)00385-7. [DOI] [PubMed] [Google Scholar]
- 85.Diaz F, Thomas CK, Garcia S, et al. Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum Mol Genet. 2005;14:2737–48. doi: 10.1093/hmg/ddi307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hu CJ, Wang LY, Chodosh LA, et al. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–74. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gruber M, Hu CJ, Johnson RS, et al. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci USA. 2007;104:2301–6. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Scortegagna M, Ding K, Oktay Y, et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat Genet. 2003;35:331–40. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- 89.Oktay Y, Dioum E, Matsuzaki S, et al. Hypoxia-inducible factor 2alpha regulates expression of the mitochondrial aconitase chaperone protein frataxin. J Biol Chem. 2007;282:11750–6. doi: 10.1074/jbc.M611133200. [DOI] [PubMed] [Google Scholar]
- 90.Hervouet E, Cizkova A, Demont J, et al. HIF and reactive oxygen species regulate oxidative phosphorylation in cancer. Carcinogenesis. 2008;29:1528–37. doi: 10.1093/carcin/bgn125. [DOI] [PubMed] [Google Scholar]
- 91.Kojima I, Tanaka T, Inagi R, et al. Protective role of hypoxia-inducible factor-2alpha against ischemic damage and oxidative stress in the kidney. J Am Soc Nephrol. 2007;18:1218–26. doi: 10.1681/ASN.2006060639. [DOI] [PubMed] [Google Scholar]