

Abstract
This paper describes a model system to characterize the rate enhancement that stems from localization of an enzyme with its substrate. The approach is based on a self-assembled monolayer that presents a substrate for the serine esterase cutinase along with a peptide ligand for an SH2 adaptor domain. The monolayer is treated with a fusion protein of cutinase and the SH2 domain, and the rate for the interfacial reaction is monitored using cyclic voltammetry. The rate is approximately 30-fold greater for monolayers that present the ligand for the SH2 domain than for those that omit the ligand. The rate enhancement is due to the interaction of the adaptor domain with the immobilized ligand. Further, the rate enhancement increases with the densities of both the ligand and the substrate. This example provides a well-defined model system for quantitatively assessing the magnitude of rate enhancement that is possible with colocalization of an enzyme with its substrate and may be particularly significant for understanding the signaling events that rely on enzyme localization at the cell membrane.
Introduction
Enzymes are the catalysts that regulate the networks of reactions that occur in the cell. Enzyme activities are themselves regulated in many ways—through the synthesis and degradation of proteins,(1) the post-translational modification of proteins,(2) and the availability of cofactors(3)—which is important in determining which reactions can take place out of an almost unbounded number of possibilities. Eukaryotic cells also make wide use of protein localization to regulate function.4,5 Enzymes frequently contain so-called adaptor domains that bind peptide motifs and can direct enzyme activity to a subset of available substrates and, analogously, sequester enzymes from other pools of substrates.6,7 For example, the Src homology 2 (SH2) domain plays a role in a broad range of signal transduction processes by binding to phosphotyrosine-containing peptide motifs.8,9 Yet, experimental studies of the roles that protein localization plays in regulating and accelerating enzyme-mediated reactions remain challenging. In this paper, we use self-assembled monolayers that present an enzyme substrate and an adaptor ligand as a model system with which to characterize the rate enhancement that is realized when an adaptor domain localizes the enzyme to the surface.
Results and Discussion
Model System
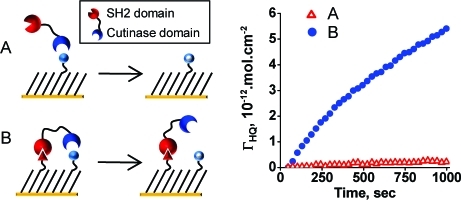
Our model system is illustrated in Figure 1 and is based on the enzyme-mediated conversion of a substrate molecule that is immobilized to the monolayer. We use an enzyme that is fused to an adaptor domain that can interact with a second ligand on the surface. In this way, we can compare the rates of the interfacial reaction for the cases where the surface does and does not present a ligand for the adaptor domain. This system provides a well-controlled case study for assessing the rate enhancement that is possible when adaptor domains are used to localize an enzyme with its substrate. Specifically, the monolayer presents a 4-hydroxyphenyl 2-methylvalerate molecule and a peptide against a background of tri(ethylene glycol) groups (Figure 2). The small molecule is a substrate for the serine esterase, cutinase, and is converted by the enzyme to a hydroquinone product. We showed previously that immobilized substrates of this form are active toward cutinase and that the progress of the reaction can be monitored in real time using cyclic voltammetry to detect the redox-active hydroquinone product.10,11 In the present work, we add to the surface a phosphorylated peptide, Ac−pYEEIEAKKKC−NH2 (pY), that interacts with an SH2 adaptor domain. In this way, by using a fusion protein having cutinase tethered to the SH2 adaptor domain (Cut−SH2), we ask to what extent localization of the cutinase to the surface—by way of the interaction of the adaptor domain and the phosphorylated peptide—can increase the rate of enzyme-mediated deacylation of the small-molecule substrate. As a control, we use monolayers that present the peptide in its nonphosphorylated form, which does not bind to the SH2 domain.
Figure 1.

This study compares the rates for the cutinase-mediated deacylation of an immobilized substrate for a surface that presents the substrate alone (A) and a surface that presents the substrate together with an adaptor ligand, which can bind an SH2 domain that is fused to the cutinase enzyme (B). This model system allows direct measurement of the rate enhancement that is realized when the adaptor domain positions the enzyme for an “intramolecular” reaction with its substrate.
Figure 2.

(A) Monolayers were prepared to present both the enzyme substrate and the adaptor ligand against a background of tri(ethylene glycol) groups. The enzyme converts the substrate to a hydroquinone product, which can be detected with cyclic voltammetry. SAMDI mass spectra of the monolayer confirm the presence of the valerate substrate before treatment with the enzyme (B) and the hydroquinone product following incomplete treatment (C).
Measurement of Rates for Interfacial Reaction
We prepared monolayers presenting both the cutinase substrate and SH2 ligand against a background of tri(ethylene glycol) groups. We started with a monolayer that was assembled from a solution of the following disulfides in 1:1:2 ratio: a disulfide presenting one maleimide group and one tri(ethylene glycol) group, a disulfide presenting one 4-hydroxylphenyl 2-methylvalerate group and one tri(ethylene glycol) group, and a disulfide bearing two tri(ethylene glycol) groups. We then treated the monolayer with the peptide to allow its immobilization to the remaining maleimide groups. Reactions with the enzyme were then performed by immersing the monolayer in phosphate-buffered saline containing Cut−SH2 (5 nM). The formation of the redox-active hydroquinone product was characterized in real time by cyclic voltammetry, where the monolayer served as the working electrode and with a platinum wire counter electrode and a Ag/AgCl reference electrode. Voltammograms were obtained by scanning from −300 to 400 mV at 50 mV/s, and sequential scans showed an increasing amount of the hydroquinone, which reached a limiting, but stable, value when the reaction was complete. We used SAMDI MS (the combination of self-assembled monolayers and matrix-assisted laser desorption/ionization mass spectrometry)12,13 to characterize the presence of the substrate at the start of the reaction and the accumulation of product during the reaction (Figure 2, parts B and C). We determined the density of hydroquinone at each time point by integrating the area under the anodic peak in the voltammograms. The density was plotted against time, and the initial reaction rate was determined from the slope of the linear region of the curves.
Rate Enhancement in the Presence of the Adaptor Domain
We next investigated the rate enhancement of the interfacial reaction for a monolayer that also presented the peptide ligand for the SH2 adaptor domain. We prepared two monolayers that each presented the enzyme substrate at constant density, but that additionally presented the peptide ligand, either in the phosphorylated form (Ac−pYEEIEAKKKC−NH2, pY) or in the nonphosphorylated form (Ac−YEEIEAKKKC−NH2, Y). Only the phosphorylated form binds the SH2 domain, and therefore, the second monolayer serves as a control for the role of the adaptor domain. We treated both monolayers with Cut−SH2 under identical conditions, and we measured the initial rates for each reaction. We followed the reaction with cyclic voltammetry for 35 cycles (∼16 min) (Figure 3, parts A and B), and the initial rates were determined as described above. We found that the initial rate was approximately 30 times faster for the monolayer that presents the pY ligand than for the surface that presents the nonphosphorylated control ligand (Figure 3C). To verify that the difference in rate is due only to the binding of the adaptor domain to the immobilized ligand, and not to the presence of the phosphate group on the peptide, we also compared the rate of reaction of the two surfaces with cutinase alone (that is, cutinase not fused to the adaptor domain). Figure 3D shows that the two reactions proceeded with indistinguishable rates, showing that the enhancement of the enzyme reaction rate owes to the binding between the enzyme-conjugated adaptor domain and its ligand immobilized on surface and not to non-specific effects at the interface. We also performed an experiment that omitted the Cut−SH2 from the electrolyte and observed only the non-faradic current over the same number of scans showing that the ester was not hydrolyzed in the absence of enzyme (data not shown).
Figure 3.

Cyclic voltammetry was used to measure the rate of the reaction of a cutinase−SH2 fusion protein with the immobilized substrate. Representative cyclic voltammograms for a surface that presents substrate together with phosphorylated peptide ligand (pY) for the SH2 domain (A) and for a surface that presents the substrate with an inactive form of the ligand (Y) (B). (C) The density of hydroquinone product on the monolayer was measured during the reaction for pY (●) and Y (△) presenting surfaces, showing an approximately 30-fold greater initial rate for the former. (D) An analogous experiment using cutinase—but without the SH2 domain—reveals similar rates for the interfacial reaction on the two monolayers, demonstrating that the rate enhancement is only due to binding between the SH2 domain and the pY ligand.
Dependence of Rate Enhancement on Density of Substrate and Adaptor Ligand
We reasoned that the rate enhancement should depend on the density of the cutinase substrate as well as the density of the adaptor ligand. At low densities of substrate, a bound enzyme does not have substrate molecules available within reach and therefore should show no rate enhancement. Above a threshold density of substrate, the rate enhancement might be expected to increase with the density of substrate because the bound enzyme has an increasing number of substrates that it can access. Similarly, the rate enhancement will increase with the density of the adaptor ligand until the density is sufficiently high that all substrates are within reach of a bound enzyme. We characterized the dependence of the rate enhancement on density by first preparing monolayers that had a constant density of adaptor ligand, but with densities of cutinase substrate varying from 1 to 12% (relative to total alkanethiolate). We also compared the rate enhancement for a series of monolayers that had a constant density of substrate and with densities of adaptor ligand pY varying from 0% to 12% (relative to total alkanethiolate). In both cases monolayers were treated with Cut−SH2 under identical conditions to determine the initial rates for the reactions (Figure 4). We found that the magnitude of initial rate enhancement (vpY/vY) increased approximately linearly with substrate density and that this trend held even at densities of substrate of 12%. We also found that the rate enhancement increased rapidly with the density of the pY peptide but reached a maximum value at a density of 1.5%. Hence, the rate increase is more sensitive to changes in the density of the ligand than it is to the density of the substrate. This trend is consistent with a simple geometric model. Using a structural model of the protein,14,15 we estimate that the mean separation of the cutinase active site and the SH2 binding site is 10 nm and we further assume that flexibility of the protein allows this distance to vary by ±0.5 nm. With these assumptions, the cutinase can sweep a ring area on the monolayer of about 63 nm2, which would correspond to approximately 290 alkanethiolates. At a density of substrate of 12%, there are approximately 35 substrates accessible to each bound cutinase. Thus, for each bound cutinase, this number may represent a maximum rate enhancement for conversion of substrates to product, which is consistent with the rate enhancement of about 30-fold that we observe. Accordingly, the rate enhancement can then be expected to decrease with lower densities of substrate.
Figure 4.

Initial rate enhancement (vpY/vY) for the SH2-mediated reaction depends on both the density of substrate (A) and the pY ligand (B). (A) A series of monolayers having a fixed density of the pY peptide ligand of 12% and densities of substrate varying from 1% to 12% was treated with the Cut−SH2 fusion protein. The plot shows the initial rate enhancement for each substrate density. (B) The second experiment used a series of monolayers having a constant density of substrate of 12% and densities of the pY ligand ranging from 0% to 12%. Again, the plot shows the initial rate enhancement for each monolayer. Error bars represent the average of three measurements ± one standard deviation.
We also note that, for low densities of the peptide ligand, the maximal rate enhancement cannot be achieved (Figure 4B), because not all substrates are in the vicinity of an occupied adaptor ligand—that is, not all substrates can be reached by a fusion protein that is bound to a ligand on the monolayer. We offer the following simple geometrical model that is consistent with these trends. We assume each bound Cut−SH2 covers a circular region of 10 nm diameter, whose area is 314 nm2, or 1446 alkanethiolates. For a density of pY ligand of 1.5%, there are approximately 20 pY ligands in this region. The equilibrium dissociation constant, Kd, for the SH2 domain and the phosphorylated peptide ligand is approximately 100 nM.(16) Therefore, at the concentration of 5 nM Cut−SH2 that we use, approximately 5% of the pY ligands are bound to a Cut−SH2, and there is approximately one bound pY ligand localized to each circular region for monolayers presenting the ligand at a density of 1.5%, which is consistent with our observation of a maximal rate enhancement at this density (Figure 4B). For lower densities of the pY ligand, not all substrate molecules will be accessible by a bound enzyme, leading to lower rate enhancements. For higher densities of the pY ligand, the surface is completely covered with surface-bound enzymes, leading to the limiting rate enhancement.
Discussion
This study offers an early model system that provides a quantitative assessment of the rate enhancement that derives from localization of an enzyme with its substrate. Here, we show that the rate of an enzyme acting on an immobilized substrate is increased by up to 30-fold when the enzyme is associated with the surface. Our previous study has derived the rate of the interfacial enzyme reaction as v = (kcat/Km)[E0]ΓS.(11) Colocalization of the enzyme with its substrate would not influence the value of kcat but, rather, decreases the value of Km, resulting in an apparent increase in the enzyme reaction rate. The use of monolayers was critical to this study for several reasons. First, the well-defined structure and synthetic flexibility allow excellent control in presenting multiple ligands at the surface, with control over the densities and microenvironments of each. Second, the compatibility of the monolayers with electrochemistry permits the use of cyclic voltammetry to monitor the progress of the reaction progress in real time and therefore to obtain high-quality kinetic data. Finally, the availability of surface chemistries that are effective at preventing the non-specific adsorption of proteins keeps the surface free from fouling and is important to realizing well-behaved kinetics throughout the reaction. We believe these characteristics will make this approach also important for looking at effects of non-equilibrium systems, including the role of spatial patterning of enzymes and their substrates.
Other recent studies have also characterized rate enhancements that stem from colocalization of an enzyme and substrate. Gureasko et al. studied the activation of Ras (a GTP-binding protein) by a guanine nucleotide exchange factor Son of Sevenless (SOS).(17) SOS has two binding sites for Ras, one of which is an allosteric site and binds to activated Ras and the other of which is a catalytic site and promotes activation of Ras. The activity of SOS is dramatically increased (up to 500-fold) when Ras is bound to the membrane. Localization of SOS to the membrane by the interaction of its allosteric site with activated Ras increases the local concentration of its catalytic domain as well as enhances the activity of the catalytic domain by inducing favorable conformational change, thus accelerating the activation of more membrane-bound Ras in its vicinity. Similarly, in our system, localization of cutinase to the surface by way of an adaptor domain increases the concentration of cutinase near the surface, which then accelerates conversion of nearby cutinase substrates to product. In another example, Dueber et al. prepared fusion proteins that localized three enzymes that sequentially modify a substrate.(18) By localizing the enzymes—and by adjusting the stoichiometry of the enzymes—the product released by the first enzyme is then present at a high effective concentration for the second, where it is more efficiently converted to the second product. In this way, these authors could engineer a 77-fold increase in the concentration of product in the reaction mixture relative to a system where the metabolic enzymes were present in monomeric form.
Several reports have described the analogous use of colocalization to improve the rates and yields of reactions in synthesis as well. For example, Wang et al. used a hydrogel matrix that was functionalized with palladium catalysts to promote reactions of encapsulated reactants. Suzuki and Heck reactions within the hydrogel were accelerated by deswelling the hydrogel and therefore concentrating both reactants.(19) Keilitz and Haag reported a 5-fold increase in the rate for asymmetric epoxide ring-opening reaction when a dendritic polymer was used to organize the reactants. Here, the reaction proceeded through an intermediate having the reactants coordinated to the immobilized catalysts.(20) Finally, Kusukawa et al. used molecular capsules to sequester isoprene and naphthoquinone and observed a 113-fold increase in the rate for the Diels−Alder reaction.(21)
Our example has an analogy to the role of exosites in promoting the activity of a substrate. An exosite is a region of the substrate that does not contact the active site of the enzyme but, rather, interacts with a separate region of the enzyme in order to increase the binding affinity of the substrate for the enzyme. For example, Pellicena et al. compared the rate of phosphorylation of peptide substrates by the Hck kinase. They compared the phosphorylation rates for two peptides that either include or omit a phosphotyrosine motif that is a high-affinity ligand for the SH2 domain of the kinase and find that the peptide that contains the adaptor ligand has 10-fold higher activity.(22) In our example, the pY peptide on the monolayer effectively serves as an exosite in increasing the affinity of the enzyme for the substrate, but with two significant distinctions. First, the distance between the adaptor ligand and the valerate substrate is not fixed, as it would be in a protein substrate that has a defined structure. With the monolayer, there is a distribution of distances that separate the adaptor ligand and cutinase substrate and therefore a distribution in rate enhancements that are realized. Our measurement of the rate enhancement reflects an average of this distribution. Our finding that the rate enhancement falls with lower densities of substrate and adaptor ligand agrees with this interpretation. Second, the localization of the substrate molecules to an interface means that a single adaptor ligand (or, as we discuss here, “exosite”) can direct the cutinase enzyme to multiple valerate substrates, giving an improved efficiency of the reaction.
We also found that the magnitude of the rate enhancement depends on the densities of both the substrate and the ligand. The rate increased approximately linearly with the density of the substrate up to a density of 12%. The rate also increased with the density of ligand, but reached a limiting value at a 10-fold lower density. These observations are consistent with the stoichiometry of the reaction, wherein one ligand can direct the cutinase enzyme to many substrate molecules. Therefore, the threshold density of ligand that would allow access of a bound enzyme to all the substrate molecules is substantially lower than the density of substrate molecules that can lead to the maximum rate enhancement. Hence, this study shows that the rate enhancement is more sensitive to changes in the density of the adaptor ligand than it is to changes in the density of the substrate.
In the present work, the density of the adaptor ligand is constant throughout the course of the reaction, while the density of the substrate decreases as the reaction proceeds; therefore, it gives the maximum rate enhancement at the very beginning of the reaction. We recently described another model system wherein the enzyme-mediated reaction results in a product that is itself an adaptor ligand for the enzyme.(23) This reaction therefore displays a rate enhancement that increases in magnitude as it proceeds—that is, the reaction is autocatalytic. That example used the Abl kinase and a monolayer that presented a single peptide substrate. Once phosphorylated, the peptide serves as a ligand for the SH2 domain of Abl and then recruits the enzyme to the surface where it can catalyze additional reactions. Unlike the work described in the present paper, the density of the adaptor ligand was not constant throughout the reaction, but increased with progression of the reaction. This distinction gives rise not only to the autocatalytic profile but also to unique spatial characteristics of the reaction. For example, peptide substrates are more active only if they are within a short distance of a phosphopeptide product. Therefore, the autocatalytic reaction is expected to proceed to give zones of product that increase in size, whereas the rate-enhanced reaction described here is expected to proceed uniformly across the substrate. The comparison of these two systems highlights the lesson that systems in which the substrate and adaptor ligand are unconnected give a diminishing rate as the reaction proceeds and systems in which the enzyme-mediated product is itself the adaptor ligand give an autocatalytic profile.
Conclusions
This paper provides a quantitative study of the rate enhancement that can be realized when an enzyme and its substrate are brought together through a separate binding interaction of an adaptor domain. The work is significant because it provides quantitative information on the rate enhancement and its dependence on the density of both the adaptor ligand and the substrate. These studies may be important for better understanding and perhaps predicting the impact of adaptor domains in controlling signaling pathways in cells. We believe that the use of self-assembled monolayers provides an effective route for modeling a broader range of reactions that are spatially and temporally structured.
Experimental Section
Materials
All reagents were used as received without further purification. All amino acids and peptide synthesis reagents were purchased from Anaspec. E. coli (BL21) and plasmid pET-22b(+) were purchased from Novagen. Restriction enzymes for digestion were purchased from New England Biolabs. Phosphate-buffered saline (PBS) used in electrochemical experiments was purchased from Gibco. Glass coverslips for gold deposition were obtained from Fisher Scientific.
Synthesis of Substrate and Peptides
4-Hydroxy-3-(3-mercaptopropyl)phenyl 2-methylvalerate was synthesized in seven steps using a previously reported method, but with 2-methylvaleryl chloride in place of the valeric anhydride used in the original report.(24) Peptides were synthesized using standard protocols on Fmoc-Rink amide 4-methylbenzhydrylamine resin. All peptides were purified by reversed-phase HPLC on a C18 column.
Preparation of Monolayers
Gold substrates were prepared by electron beam vacuum deposition of 10 nm titanium followed by 90 nm gold onto glass coverslips. A disulfide presenting one maleimide group and one tri(ethylene glycol) group(25) was treated with 4-hydroxy-3-(3-mercaptopropyl)phenyl 2-methylvalerate by mixing these two in 2:1 ratio in Tris−HCl buffer (pH = 8.0) for 30 min and was then combined with an equal amount of a disulfide bearing two tri(ethylene glycol) groups and diluted with ethanol to 0.1 mM total disulfide concentration. A gold-coated slide was immersed in this solution for 24 h at room temperature. The slide was then taken out, rinsed with water and ethanol, and dried under a stream of nitrogen. The monolayer was then treated with either the phosphorylated or nonphosphorylated peptide (0.02 mM total concentration) in Tris−HCl buffer (pH = 8.0) for 1 h to allow its immobilization to the remaining maleimide groups. To vary the substrate density, 2-mercaptoethanol was mixed with substrate at molar ratios ranging from 9:1 to 1:3 to dilute the fraction of alkanethiol that presents the active substrate. In each case, cyclic voltammetry was used to verify the density of the substrate after it had been treated with cutinase. The density of the pY peptide was adjusted by forming mixtures with the Y peptide at molar ratios ranging from 3:1 to 1:99, and the mixture was immobilized on to monolayers with maleimide groups. The monolayers presenting both substrates and peptides were then treated with 0.05 mM 4-bromobenzyl mercaptan in 20% EtOH in PBS for 30 min to cap any trace amount of unreacted maleimide, after which the monolayers were treated with 2 mg/mL bovine serum albumin (BSA) in PBS buffer for 30 min, rinsed with copious water, and dried with N2.
Electrochemical Measurements
Cyclic voltammetry was performed with a Bioanalytical Systems potentiostat using 0.5 mg/mL BSA, 0.2 mg/mL NaN3 in PBS as the electrolyte. The electrochemical cell was configured with the monolayer as the working electrode, Ag/AgCl as the reference electrode, and a platinum wire as the counter electrode.(11) The potential was scanned from −300 to 400 mV at 50 mV/s. The density of hydroquinone on the surface was determined by integrating the area under the anodic peak in the voltammograms.
Plasmid Construction
Polymerase chain reaction (PCR) was performed to amplify cutinase from pCut22b.(26) The following primers were used for PCR amplification of cutinase gene: 5′-TTTCATATGTCCATGGGCCTGCCTACTTCTAACC-3′ (primer 1) and 5′-AGAGCCACCGCCACCAGAGCCACCGCCACCGGATCCTCCTCCAGCAGAACCACGGACAGCCCGA-3′ (primer 2). The v-Src SH2 gene was a gift from Professor Piers Nash (the Ben May Department for Cancer Research, the University of Chicago). The SH2 gene was amplified by using the following primers: 5′-GGCTCTGGTGGCGGTGGCTCTTGGTACTTTGGGAAGATCACTCGTC-3′ (primer 3) and 5′-TTTTAAGCTTACGACCTTCGATACCACCGTCAGTCACGATGAATTCCTGGGG-3′ (primer 4). The two amplified products were joined by another PCR amplification using primer 1 and primer 4. The cutinase−SH2 (pCut−SH2) gene was purified by agarose gel (2%) electrophoresis. The DNA was then digested with NcoI and HindIII and then cloned in pET-22b(+). The pCut−SH2 gene insertion was confirmed by bidirectional DNA sequencing. The resulting plasmid encodes a gene for cutinase−SH2 fusion protein with a 14 residue Gly-Ser linker between the C-terminal of cutinase and N-terminal of SH2. The N-terminal part of the gene had a pelB leader sequence, which allows for periplasmic localization of the expressed protein.
Expression and Purification of Cut−SH2 Protein
Cut−SH2 was expressed in E. coli (BL21) harboring the pCut−SH2 gene with a T7 expression system. The cells were grown at 37 °C to A600 = 0.4 before induction with 0.1 mM of isopropyl β-d-thiogalactoside (IPTG) at 28 °C for 5 h. Cells were then harvested by centrifugation at 5000g for 30 min. The cells were sonicated to release the protein and then centrifuged at 10 000g to separate the protein from the cellular debris. The supernatant was collected for further purification of the protein. The protein was purified using a Ni−NTA resin, where 10 mL of the resin was added to the supernatant and then allowed to equilibrate for 2 h. The solution was then poured in a column and washed with 20 mM imidazole solution. The protein was eluted using 100 mM imidazole solution. All fractions were combined and then passed through a 10 kDa centrifuge filter which resulted in concentrating the protein solution. SDS−PAGE showed that the protein was ∼90% pure with the major band at ∼40 kDa which is the expected size (38.29 kDa) of the fusion protein. The protein was further purified by ion exchange chromatography using a Resource Q column. Enzyme activity was confirmed by monitoring the hydrolysis of p-nitrophenyl butyrate as described.(27)
Acknowledgments
This work was supported by the NSF (CCF-0829552).
References
- Asano K.; Chee C. B. E.; Gaston B.; Lilly C. M.; Gerard C.; Drazen J. M.; Stamler J. S. Constitutive and inducible nitric-oxide synthase gene-expression, regulation, and activity in human lung epithelial-cells. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 10089–10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens V.; Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H.; Pollock J. S.; Nakane M.; Gorsky L. D.; Forstermann U.; Murad F. Purification of a soluble isoform of guanylyl cyclase-activating-factor synthase. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T.; Scott J. D. Signaling through scaffold, anchoring, and adaptor proteins. Science 1997, 278, 2075–2080. [DOI] [PubMed] [Google Scholar]
- Kuriyan J.; Eisenberg D. The origin of protein interactions and allostery in colocalization. Nature 2007, 450, 983–990. [DOI] [PubMed] [Google Scholar]
- Pawson T. Protein modules and signaling networks. Nature 1995, 373, 573–580. [DOI] [PubMed] [Google Scholar]
- Pawson T.; Gish G. D.; Nash P. SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 2001, 11, 504–511. [DOI] [PubMed] [Google Scholar]
- Anderson D.; Koch C. A.; Grey L.; Ellis C.; Moran M. F.; Pawson T. Binding of Sh2 domains of phospholipase-C-gamma-1, Gap, and Src to activated growth-factor receptors. Science 1990, 250, 979–982. [DOI] [PubMed] [Google Scholar]
- Moran M. F.; Koch C. A.; Anderson D.; Ellis C.; England L.; Martin G. S.; Pawson T. Src homology region-2 domains direct protein−protein interactions in signal transduction. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 8622–8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo W. S.; Mrksich M. Self-assembled monolayers that transduce enzymatic activities to electrical signals. Angew. Chem., Int. Ed. 2003, 42, 3121–3124. [DOI] [PubMed] [Google Scholar]
- Nayak S.; Yeo W. S.; Mrksich M. Determination of kinetic parameters for interfacial enzymatic reactions on self-assembled monolayers. Langmuir 2007, 23, 5578–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J.; Mrksich M. Using MALDI-TOF mass spectrometry to characterize interfacial reactions on self-assembled monolayers. Langmuir 2003, 19, 4867–4870. [Google Scholar]
- Mrksich M. Mass spectrometry of self-assembled monolayers: A new tool for molecular surface science. ACS Nano 2008, 2, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez C.; Degeus P.; Lauwereys M.; Matthyssens G.; Cambillau C. Fusarium-solani cutinase is a lipolytic enzyme with a catalytic serine accessible to solvent. Nature 1992, 356, 615–618. [DOI] [PubMed] [Google Scholar]
- Waksman G.; Kominos D.; Robertson S. C.; Pant N.; Baltimore D.; Birge R. B.; Cowburn D.; Hanafusa H.; Mayer B. J.; Overduin M.; Resh M. D.; Rios C. B.; Silverman L.; Kuriyan J. Crystal-structure of the phosphotyrosine recognition domain Sh2 of V-Src complexed with tyrosine-phosphorylated peptides. Nature 1992, 358, 646–653. [DOI] [PubMed] [Google Scholar]
- Gilmer T.; Rodriquez M.; Jordan S.; Crosby R.; Alligood K.; Green M.; Kimery M.; Wagner C.; Kinder D.; Charifson P.; Hassell A. M.; Willard D.; Luther M.; Rusnak D.; Sternbach D. D.; Mehrotra M.; Peel M.; Shampine L.; Davis R.; Robbins J.; Patel I. R.; Kassel D.; Burkhart W.; Moyer M.; Bradshaw T.; Berman J. Peptide inhibitors of Src Sh3−Sh2-phosphoprotein interactions. J. Biol. Chem. 1994, 269, 31711–31719. [PubMed] [Google Scholar]
- Gureasko J.; Galush W. J.; Boykevisch S.; Sondermann H.; Bar-Sagi D.; Groves J. T.; Kuriyan J. Membrane-dependent signal integration by the Ras activator Son of Sevenless. Nat. Struct. Mol. Biol. 2008, 15, 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueber J. E.; Wu G. C.; Malmirchegini G. R.; Moon T. S.; Petzold C. J.; Ullal A. V.; Prather K. L. J.; Keasling J. D. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 2009, 27, 753–U107. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang J. Z.; Zhang W. Q.; Zhang M. C. Pd-catalyzed C−C cross-coupling reactions within a thermoresponsive and pH-responsive and chelating polymeric hydrogel. J. Org. Chem. 2009, 74, 1923–1931. [DOI] [PubMed] [Google Scholar]
- Keilitz J.; Haag R. Intramolecular acceleration of asymmetric epoxide ring-opening by dendritic polyglycerol salen−Cr-III complexes. Eur. J. Org. Chem. 2009, 3272–3278. [Google Scholar]
- Kusukawa T.; Nakai T.; Okano T.; Fujita M. Remarkable acceleration of Diels−Alder reactions in a self-assembled coordination cage. Chem. Lett. 2003, 32, 284–285. [Google Scholar]
- Pellicena P.; Stowell K. R.; Miller W. T. Enhanced phosphorylation of Src family kinase substrates containing SH2 domain binding sites. J. Biol. Chem. 1998, 273, 15325–15328. [DOI] [PubMed] [Google Scholar]
- Liao X. L.; Su J.; Mrksich M. An adaptor domain-mediated autocatalytic interfacial kinase reaction. Chem.—Eur. J. 2009, 15, 12303–12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier J. H.; Mrksich M. Engineering a biospecific communication pathway between cells and electrodes. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 2021–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman B. T.; Gawalt E. S.; Mrksich M. Maleimide-functionalized self-assembled monolayers for the preparation of peptide and carbohydrate biochips. Langmuir 2003, 19, 1522–1531. [Google Scholar]
- Hodneland C. D.; Lee Y. S.; Min D. H.; Mrksich M. Selective immobilization of proteins to self-assembled monolayers presenting active site-directed capture ligands. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 5048–5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolattukudy P. E.; Purdy R. E.; Maiti I. B. Cutinases from fungi and pollen. Methods Enzymol. 1981, 71, 652–664. [Google Scholar]