

Abstract
This study aimed at contributing to the elucidation of the genetic basis of age-related hearing impairment (ARHI), a common multifactorial disease with an important genetic contribution as demonstrated by heritability studies. We conducted a genome-wide association study (GWAS) in the Finnish Saami, a small, ancient, genetically isolated population without evidence of demographic expansion. The choice of this study population was motivated by its anticipated higher extent of LD, potentially offering a substantial power advantage for association mapping. DNA samples and audiometric measurements were collected from 352 Finnish Saami individuals, aged between 50 and 75 years. To reduce the burden of multiple testing, we applied principal component (PC) analysis to the multivariate audiometric phenotype. The first three PCs captured 80% of the variation in hearing thresholds, while maintaining biologically important audiometric features. All subjects were genotyped with the Affymetrix 100 K chip. To account for multiple levels of relatedness among subjects, as well as for population stratification, association testing was performed using a mixed model. We summarised the top-ranking association signals for the three traits under study. The top-ranked SNP, rs457717 (P-value 3.55 × 10−7), was associated with PC3 and was localised in an intron of the IQ motif-containing GTPase-activating-like protein (IQGAP2). Intriguingly, the SNP rs161927 (P-value 0.000149), seventh-ranked for PC1, was positioned immediately downstream from the metabotropic glutamate receptor-7 gene (GRM7). As a previous GWAS of a European and Finnish sample set already suggested a role for GRM7 in ARHI, this study provides further evidence for the involvement of this gene.
Keywords: Saami, isolated population, mixed model, genome-wide association study, age-related hearing impairment, presbycusis
Introduction
Age-related hearing impairment (ARHI), or presbycusis, is the most frequent sensory disability among the elderly.1, 2 The involvement of genetic factors in the development of this multifactorial disorder was demonstrated by several heritability studies.3, 4, 5 Only recently have the first studies aiming at the identification of these genetic factors been conducted. Hitherto, three linkage studies have been performed,6, 7, 8 but only one region on chromosome 8q24.13–q24.22 reached genome-wide significance.8 In addition, several association studies based on a candidate gene approach and two genome-wide association studies (GWAS)8, 9 have been conducted. Early candidate gene-based association studies are reviewed by Van Eyken et al.10 Recently, based on a candidate gene approach, we reported associations between ARHI and variants in GRHL2,11 encoding a transcription factor that is also involved in monogenic hearing loss. In addition, based on a pooling GWAS, we reported association with variants in the glutamate receptor-7 gene (GRM7),9 encoding a metabotropic glutamate receptor assumed to modulate hair cell excitability and synaptic efficacy. Acquired mitochondrial mutations have also been proposed as a cause of ARHI in humans. These include both the ‘common' 4977-bp mitochondrial DNA deletion associated with ageing12 and accumulation of many different mitochondrial mutations.13
Despite these first series of studies, the genetic aetiology of ARHI remains elusive. In this paper, we present the results of a GWAS for ARHI in the Finnish Saami population. The Saami, previously known as Lapps, live across Northern Scandinavia and the Kola Peninsula. Traditionally, Saami have been fishers, hunters and reindeer herders. A minority of the Saami continue to be engaged in these traditional economic activities, whereas the majority has adopted other occupations and a more westernised lifestyle. Among European populations, the Saami are considered a population genetic ‘outlier'. Despite the fact that both the Saami and Finnish languages belong to the Uralic language group, there is strong genetic differentiation between the Saami and Finns.14 Several genetic studies have been conducted to shed light on the genetic origin of the Saami.15, 16, 17, 18 These studies suggested that the Saami are descendants of Europeans who, several thousand years ago, reached northern Fennoscandia from both a western and an eastern route and subsequently got admixed. They also found a small East Asian contribution to the gene pool.18, 19 Several genetic studies have further suggested that the extent of linkage disequilibrium (LD) in the Saami is dramatically higher compared to that of outbred populations.20, 21, 22, 23 Higher levels of LD are observed in other population isolates as well.24, 25 Most of the genetic studies of genetically isolated populations were conducted involving young founder populations, in which the high extent of LD can be attributed to a founder effect. However, in ancient, small, genetically isolated populations without evidence of demographic expansion, high levels of LD are believed to have been generated by genetic drift.26, 27 The Saami represent such a population. Therefore, the choice of this study population was based on its anticipated statistical power advantage for association mapping, offering better genomic coverage for a given set of markers, as compared to outbred populations.
Owing to the expected higher levels of LD in the Saami population and based on what was known about the structure of the human genome at the time this study was designed, we anticipated that the Affymetrix 100 K array pair would entail sufficient genomic coverage for identification of putative ARHI-susceptibility genes.
Materials and methods
Subjects
Saami subjects, aged between 50 and 75 years, were enrolled through the public population register from four municipalities in Northern Finland (Ivalo, Utsjoki, Vuotso and Enontekiö) in three stages. In the first stage, geographical criteria were applied; only subjects from an area with a high probability of finding Saami individuals were selected. In the second stage, putative study subjects were invited based on an expert evaluation of Saami communities. Finally, the Saami identity of the subject in question was confirmed through a discussion involving the subject and the researcher. Except for the age range that was used, the ARHI selection criteria were as described previously.28 Briefly, all volunteers completed an extended questionnaire detailing medical history and exposure to environmental factors putatively influencing hearing. Subjects suffering from one or more medical pathologies from a standardised list of conditions that could potentially affect hearing ability were excluded. All subjects who passed this initial screening underwent an otoscopic investigation before pure-tone audiometry. Air conduction thresholds were measured at 0.125, 0.25, 0.5, 1, 2, 3, 4, 6 and 8 kHz, and bone conduction at 0.5, 1, 2 and 4 kHz for all participating volunteers. Audiological exclusion criteria were an air-bone gap of more than 15 dB averaged over 0.5, 1 and 2 kHz in at least one ear or asymmetrical hearing impairment with a difference in air conduction thresholds exceeding 20 dB in at least two frequencies between 0.5, 1 and 2 kHz. Written informed consent was obtained from all volunteers. The study was approved by the Finnish National Advisory Board on Health Care Ethics and by the ethical committees or the appropriate local institutional review boards at all participating institutions.
Description of the phenotype
For each subject, only hearing thresholds from the best ear, defined as the ear with the lowest average threshold over all measured frequencies, were used in the analyses. Supplementary Figure 1 shows that hearing thresholds increase with age and that, for the higher frequencies, thresholds for men are higher and display greater variability (i.e., there is heteroscedasticity) than those for females. We therefore adjusted hearing thresholds for age and sex. The effect of age was removed by regressing log-transformed threshold values (log10(20+threshold)) for each of the measured frequencies on age, age squared and age cubed. To resolve the heteroscedasticity problem and to correct for sex, this adjustment for age was carried out for males and females separately, after which the residuals of each regression were scaled and subsequently combined. Note that adding the age and sex covariates in the model for association testing does not deal with this problem satisfactorily.
To deal with the multivariate audiometric phenotype, we applied principal component (PC) analysis as a data-reduction technique. Performing association tests for each frequency separately would have resulted in a large number of tests, worsening the multiple-testing problem. In addition, such analysis would have completely ignored the correlation structure of the audiometric data, that is, the shape of the audiogram. The latter would also apply when simple pure-tone averages (PTAs), which are more widely used in hearing research, would have been analysed. Our first PC (PC1) was highly correlated with the PTA over all frequencies, adjusted for sex, age, age square and age cubed (Spearman correlation=0.96). Spearman correlations between PC1 and sex- and age-adjusted PTAs for the high (2–8 kHz) and low frequencies (0.125–1 kHz) were 0.88 and 0.61, respectively. The association testing results for PC1, therefore, would not differ much from those that would be obtained when PTAs would have been analysed.
Table 1 lists the eigenvalues and eigenvectors for the first three PCs, PC1 to PC3. The results of separate calculations for males and females yielded similar values, justifying data pooling (results not shown). Each of the PCs provides independent bits of information. The first three PCs were retained for the analysis as they captured approximately 80% of the total variation. The coefficients of the eigenvectors (loadings) are directly proportional to the correlations between the transformed hearing thresholds corrected for age and gender, and each of the PCs (see, for example, Johnson and Wichern29). PC1 is clearly a ‘size' variable, providing an overall measure of a subject's hearing ability. PC2 and PC3 are ‘shape' variables. PC2 contrasts low with high frequencies and is a measure of the slope of the audiogram. PC3 contrasts the middle frequencies with the lower and higher frequencies and can be considered a measure of the concavity of an audiogram (Table 1 and Figure 1). These phenotypes are biologically meaningful30, 31 and we have previously shown that they have significant heritabilities.8
Table 1. Eigenvectors and eigenvalues for the first three PCs calculated from age- and sex-corrected hearing thresholds.
| Principal component (PC) | PC1 | PC2 | PC3 |
|---|---|---|---|
| Eigenvalue | 4.421 | 1.790 | 0.904 |
| % of variation explaineda | 0.491 | 0.199 | 0.100 |
| Coefficients of eigenvectors (loadings)b | |||
| res125 | 0.208 | 0.460 | 0.527 |
| res250 | 0.288 | 0.470 | 0.287 |
| res500 | 0.333 | 0.382 | −0.186 |
| res1000 | 0.348 | 0.186 | −0.481 |
| res2000 | 0.373 | 0.020 | −0.408 |
| res3000 | 0.393 | −0.237 | −0.109 |
| res4000 | 0.358 | −0.324 | 0.088 |
| res6000 | 0.356 | −0.328 | 0.276 |
| res8000 | 0.305 | −0.345 | 0.335 |
Eigenvalue for the PC divided by the sum of all eigenvalues, which corresponds to the total number of variables in the original dataset (9).
The correlations between the PC and the age- and sex-corrected, transformed hearing thresholds for each frequency can be obtained by multiplying each coefficient by the square root of its corresponding eigenvalue.
Figure 1.

Comparison of typical audiograms between subjects with high and low values for PC1, PC2 and PC3. For each of the three traits, three subjects from the lower and upper 10% extremes of the distribution of the trait are shown. The audiograms for both ears are given, to illustrate the extent of intra-subject variability. The left and right ears correspond to the filled and open circles, respectively. For PC1, on average, hearing loss is much more pronounced in individuals from the higher extremes (top row) as compared with those from the lower extremes (second row from the top). Subjects with low values for PC2 (fourth row from the top) tend to have a much more sloping audiogram, compared with those with high values for PC2 (third row from the top). Subjects with high values for PC3 (second row from the bottom) tend to have a more concave audiogram, compared with those in the lower extreme for PC3 (bottom row). Note that the displayed audiometric data have not been adjusted for sex and age.
Genotyping and quality control of data
Genomic DNA was extracted using standard protocols and diluted to 50 ng/μl. The quality of the genomic DNA was assessed by spectrophotometric analysis and gel electrophoresis. Each sample was genotyped with the Affymetrix GeneChip Human Mapping 100 K array pair (Santa Clara, CA, USA), as described in the GeneChip Human Mapping 100 K Assay Manual. Genotype calling was performed using the BRLMM algorithm of the Affymetrix GeneChip Genotyping Analysis Software (GTYPE) version 4.1. Quality control of data was performed based on the following criteria: a sample call rate of >94%, a marker call rate of >90%, a Hardy–Weinberg equilibrium P-value of >0.001 and a minor allele frequency of >5%. In total, 352 samples from the Saami subjects were genotyped with the Affymetrix 100 K array. One subject was excluded because of low sample call rate. The total genotyping rate in the remaining samples was 0.9931. One additional sample was removed owing to an unintentional duplication event, and three further subjects were excluded owing to missing phenotypic information. After data quality control, 347 subjects and 83 381 SNPs were used for statistical analysis. Genotyping for SNP rs11928865 was performed using a Taqman assay (Applied Biosystems, Foster City, CA, USA) with a Lightcycler480 real-time PCR machine (Roche, Basel, Switzerland).
Association testing
Estimates of genome-wide pairwise identity-by-descent sharing, provided by PLINK,32 revealed a substantial degree of undocumented relatedness among the subjects. This was also reflected in an excess of SNPs that were not in Hardy–Weinberg equilibrium. The results of a linear regression for each of the three quantitative traits would, therefore, not be trustworthy as dependent data lead to underestimation of the standard errors for the regression coefficients, yielding P-values that are too small. In addition, population stratification may confound the results of the analysis. To account for the dependency of the data, as well as for population stratification, we analysed the data using a mixed model.33 With this approach, the covariance matrix of the phenotype is modelled as:
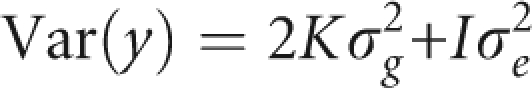 |
where y is the column vector with the phenotypes of the 347 subjects, K is a matrix with kinship coefficients estimated based on the genome-wide data, I is an identity matrix and σg2 and σe2 are the genetic and the residual variances, respectively. This was implemented using the MIXED procedure of SAS 9.1.3 within SAS macro, which was based on that used by Yu et al.33 The 2K matrix with the proportion of alleles shared identical-by-descent was estimated using PLINK.32 The model also corrected for origin. As samples originated from four municipalities, three dummy variables were included as covariates in the model. An additive genetic model was assumed for each SNP. As it was computationally infeasible to run this analysis for the entire set of SNPs, for each trait only the top 4000 SNPs (approximately 5% of the total number of SNPs passing quality control) resulting from an initial screening based on linear regression analysis (naive analysis) were considered for the mixed model analysis. To verify that no true association signals were lost by using this analytical strategy, we first examined how well the P-values of the naive analysis correlated with those of the mixed model analysis. For this purpose, we analysed 8000 randomly selected SNPs meeting the quality control standards, representing approximately 10% of the SNPs that passed the quality control, with the mixed model. Comparison of P-values between naive analysis and mixed-model results showed that no SNPs with highly significant P-values in the mixed-model analysis had a non-significant P-value in the naive analysis. A reciprocal relationship would be attributed to inflated test statistics. Therefore, for the data at hand, by analysing the top 5% (4000 SNPs) of a naive linear regression analysis, the chance of missing any SNP that would not be significant in the naive analysis, but highly significant in the mixed-model analysis seems very low (Supplementary Figure 2). Quantile–quantile plots in Supplementary Figure 2 show that residual population stratification or cryptic relatedness is of minor concern. Genomic control inflation factors varied from 1.02 to 1.08. The graph on the right of each panel in Supplementary Figure 2 further shows that P-values obtained from the naive analysis tend to be too low. For example, for PC1, the most extreme −log10(P-value) from the naive analysis is 4, whereas it is 3.5 for the mixed model.
Results
After quality control, 347 subjects and 83 381 SNPs were used for association testing. The quantitative traits tested were the first three PCs calculated from the log-transformed hearing threshold values, adjusted for age and sex. To account for the multiple levels of relatedness present in our data and potential population stratification, the associations between SNPs and the traits were tested using a mixed-model approach, based upon genome-wide kinship estimates. Owing to computational constraints, only the top 4000 SNPs (approximately 5% of the total number of SNPs passing quality control) resulting from an initial screening based on a linear regression analysis were used in the mixed-model analysis. In the Materials and methods section, we have outlined that, using this strategy, it is unlikely that strong association signals will be lost. For each of the three PCs, the top 25 SNPs resulting from this analysis are listed in Tables 2, 3, 4.
Table 2. Top 25 SNPs resulting from the mixed-model analysis for PC1.
| SNP | Chr. | Position | Alleles | Minor allele | MAF | Genotypesa | P HWEb | P Mixed | Gene regionc | Pos. Rel. gened |
|---|---|---|---|---|---|---|---|---|---|---|
| rs1413042 | 13 | 38 600 755 | C/T | C | 0.228916 | 24/104/204 | 0.0432 | 2.91E−05 | Similar to RIKEN cDNA 8030451K01 (LOC387921) | Downstream |
| rs1016111 | 6 | 14 121 677 | C/G | C | 0.367521 | 49/160/142 | 0.7308 | 5.86E−05 | CD83 antigen precursor (cell-surface protein HB15) | Upstream |
| rs1826882 | 15 | 19 989 036 | A/C | C | 0.113506 | 1/77/270 | 0.1024 | 9.41E−05 | / | / |
| rs854273 | 1 | 69 016 062 | G/T | T | 0.247863 | 18/138/195 | 0.3898 | 0.000115 | DEP domain containing-1 | Upstream |
| rs2825601 | 21 | 19 766 620 | A/G | A | 0.336182 | 39/158/154 | 0.9054 | 0.000134 | / | / |
| rs1988145 | 5 | 92 258 480 | A/C | A | 0.228228 | 25/102/206 | 0.0195 | 0.000144 | COUP transcription factor 1 (COUP-TF1) | Upstream |
| rs9311746 | 3 | 60 015 052 | C/T | T | 0.381766 | 50/168/133 | 0.8221 | 0.000144 | Bis(5′-adenosyl)-triphosphatase (EC 3.6.1.29) | Intron |
| rs161927 | 3 | 7 813 242 | A/G | G | 0.065407 | 3/39/302 | 0.1663 | 0.000149 | Metabotropic glutamate receptor-7 precursor (GRM7) | Downstream |
| rs1358520 | 2 | 181 148 326 | C/T | T | 0.195157 | 9/119/223 | 0.1738 | 0.000187 | Ubiquitin-conjugating enzyme E2 E3 (EC 6.3.2.19) | Upstream |
| rs2148418 | 13 | 38 564 233 | A/G | G | 0.215714 | 20/111/219 | 0.2675 | 0.000194 | Similar to RIKEN cDNA 8030451K01 (LOC387921) | Downstream |
| rs1661609 | 7 | 52 720 759 | A/T | T | 0.150000 | 7/91/252 | 0.8359 | 0.000208 | / | / |
| rs572619 | 11 | 115 738 853 | A/T | T | 0.361823 | 45/164/142 | 0.9080 | 0.000211 | BUD13 homologue | Downstream |
| rs10510777 | 3 | 55 212 778 | A/G | G | 0.472934 | 76/180/95 | 0.6684 | 0.000227 | Calcium channel, voltage-dependent, alpha-2/delta-3 subunit | Downstream |
| rs2182406 | 10 | 129 456 655 | C/T | T | 0.133903 | 8/78/265 | 0.4864 | 0.000231 | Transmembrane protein-12 | Downstream |
| rs9301169 | 13 | 106 353 066 | C/T | C | 0.250712 | 21/134/196 | 0.8870 | 0.000234 | / | / |
| rs1388654 | 1 | 68 985 603 | A/G | G | 0.295714 | 26/155/169 | 0.3039 | 0.000249 | DEP domain containing-1 | Upstream |
| rs3751591 | 15 | 49 394 002 | A/G | G | 0.148415 | 14/75/258 | 0.0100 | 0.000250 | Cytochrome P450 19A1 (EC 1.14.14.1) | Intron |
| rs7620394 | 3 | 55 206 368 | C/T | C | 0.304843 | 33/148/170 | 0.9003 | 0.000253 | Leucine-rich repeats and transmembrane domains-1 | Upstream |
| rs10503920 | 8 | 32 548 231 | A/G | G | 0.274929 | 34/125/192 | 0.0454 | 0.000259 | Neuregulin-1, sensory and motor neuron-derived factor isoform | Intron |
| rs10497334 | 2 | 168 549 782 | A/G | A | 0.081197 | 1/55/295 | 0.7146 | 0.000266 | STE20/SPS1-related, proline–alanine-rich protein kinase (EC 2.7.11.1) | Intron |
| rs7357631 | 9 | 99 568 141 | A/G | A | 0.497151 | 87/175/89 | 1.0000 | 0.000282 | Forkhead box protein-E1 (thyroid transcription factor-2) | Upstream |
| rs2055439 | 5 | 75 511 014 | A/G | A | 0.403458 | 61/158/128 | 0.3166 | 0.000292 | Synaptic vesicle glycoprotein-2C | Intron |
| rs10515629 | 5 | 148 875 068 | C/T | C | 0.196581 | 15/108/228 | 0.6132 | 0.000303 | Casein kinase-I isoform-alpha (EC 2.7.11.1) | Intron |
| rs2037344 | 8 | 121 088 918 | C/T | C | 0.348665 | 39/157/141 | 0.7191 | 0.000339 | DEP domain containing-6 | Intron |
| rs3912622 | 11 | 102 626 683 | A/C | C | 0.111748 | 8/62/279 | 0.0570 | 0.000355 | DYNC2H1 protein | Intron |
Abbreviations: Chr., chromosome; MAF, minor allele frequency; P HWE, P-value resulting after Hardy–Weinberg equilibrium testing; P Mixed, P-value resulting after mixed-model analysis (indicated in bold); Pos. Rel. Gene, position of the SNP relative to the gene listed in the previous column.
The number of homozygotes for the minor allele/the number of heterozygotes/the number of homozygotes for the major allele.
The Hardy–Weinberg equilibrium P-value should be larger than 0.001 for the SNP genotyping to be valid.
‘/' indicates that this SNP was not located within a gene region.
‘Upstream' or ‘Downstream' was based on the Affymetrix annotation.
Table 3. Top 25 SNPs resulting from the mixed model analysis for PC2.
| SNP | Chr. | Position | Alleles | Minor allele | MAF | Genotypesa | P HWEb | P mixed | Gene regionc | Pos. Rel. gened |
|---|---|---|---|---|---|---|---|---|---|---|
| rs10499138 | 6 | 128 533 875 | A/T | A | 0.253561 | 30/118/203 | 0.0473 | 1.70E−06 | Receptor-type tyrosine-prot phosphatase kappa precursor (EC 3.1.3.48) | Intron |
| rs10491923 | 9 | 76 139 000 | A/G | A | 0.365672 | 49/147/139 | 0.3460 | 2.79E−05 | Nuclear receptor ROR-beta (nuclear receptor RZR-beta) | Upstream |
| rs10499136 | 6 | 128 419 828 | A/C | C | 0.296296 | 36/136/179 | 0.2002 | 3.58E−05 | Receptor-type tyrosine-prot phosphatase-kappa precursor (EC 3.1.3.48) | Intron |
| rs9308868 | 2 | 103 727 852 | C/T | C | 0.268895 | 25/135/184 | 1.0000 | 6.45E−05 | U3 small nucleolar RNA | Upstream |
| rs1555167 | 6 | 62 465 026 | G/T | T | 0.398860 | 57/166/128 | 0.8239 | 9.62E−05 | KH dom-containing, RNA-binding signal transduction-associated prot-2 | Intron |
| rs9294311 | 6 | 62 470 112 | C/T | T | 0.398860 | 57/166/128 | 0.8239 | 9.62E−05 | KH dom-containing, RNA-binding, signal transduction-associated prot-2 | Intron |
| rs9294314 | 6 | 62 470 480 | A/G | G | 0.398860 | 57/166/128 | 0.8239 | 9.62E−05 | KH dom-containing, RNA-binding, signal transduction-associated prot-2 | Intron |
| rs10485162 | 6 | 128 639 684 | C/T | T | 0.145299 | 12/78/261 | 0.0530 | 0.000122 | Receptor-type tyrosine-prot phosphatase-kappa precursor (EC 3.1.3.48) | Intron |
| rs514878 | 5 | 73 705 460 | A/G | A | 0.058405 | 2/37/312 | 0.3294 | 0.000132 | Ectoderm-neural cortex 1 protein (ENC-1) | Downstream |
| rs10520939 | 5 | 27 970 143 | C/T | T | 0.082813 | 5/43/272 | 0.0516 | 0.000149 | U6 spliceosomal RNA | Downstream |
| rs1201494 | 6 | 62 547 842 | A/G | A | 0.398571 | 56/167/127 | 0.9116 | 0.000165 | KH dom-containing, RNA-binding, signal transduction-associated prot-2 | Intron |
| rs304408 | 18 | 1 095 249 | A/G | G | 0.245588 | 28/111/201 | 0.0288 | 0.000175 | Putative C18 or f2 variant-3 | Downstream |
| rs768166 | 10 | 8 087 556 | A/C | C | 0.228198 | 18/121/205 | 1.0000 | 0.000175 | Transcription initiation factor TFIID subunit-3 | Intron |
| rs304409 | 18 | 1 096 342 | A/G | A | 0.246356 | 25/119/199 | 0.2439 | 0.000180 | Pituitary adenylate cyclase-activating polypeptide precursor (PACAP) | Downstream |
| rs4936417 | 11 | 117 409 046 | A/G | G | 0.200000 | 20/100/230 | 0.0460 | 0.000202 | Eukaryotic-type signal recognition particle RNA | Upstream |
| rs4386936 | 8 | 135 859 109 | A/C | C | 0.219373 | 15/124/212 | 0.6414 | 0.000221 | Zinc finger prot 406 (Protein ZFAT) | Upstream |
| rs211004 | 5 | 161 577 376 | C/T | T | 0.370640 | 53/149/142 | 0.2033 | 0.000222 | Gamma-aminobutyric-acid receptor gamma-2 subunit precursor | Downstream |
| rs522914 | 6 | 62 592 457 | A/T | A | 0.400285 | 57/167/127 | 0.9115 | 0.000224 | KH dom-containing, RNA-binding, signal transduction-associated prot-2 | Intron |
| rs565795 | 6 | 62 597 708 | A/G | G | 0.400285 | 57/167/127 | 0.9115 | 0.000224 | KH dom-containing, RNA-binding, signal transduction-associated prot-2 | Intron |
| rs265360 | 6 | 129 579 569 | A/G | A | 0.272727 | 33/120/188 | 0.0404 | 0.000247 | Laminin-alpha-2 chain precursor (laminin M-chain) | Intron |
| rs9312176 | 4 | 67 882 411 | A/G | A | 0.456522 | 74/167/104 | 0.6647 | 0.000254 | Similar to interferon-induced transmembrane prot-3 | Downstream |
| rs1544044 | 20 | 14 459 168 | C/T | T | 0.465318 | 72/178/96 | 0.5892 | 0.000257 | OTTHUMP00000030317 | Intron |
| rs10493937 | 1 | 101 115 238 | A/T | A | 0.472934 | 85/162/104 | 0.1650 | 0.000267 | Exostosin-like-2 (EC 2.4.1.223) | Intron |
| rs1358815 | 6 | 57 471 407 | G/T | G | 0.464183 | 73/178/98 | 0.6680 | 0.000274 | DNA primase large subunit (EC 2.7.7.-) | Intron |
| rs6489314 | 12 | 1 627 431 | A/C | A | 0.341642 | 47/139/155 | 0.0916 | 0.000300 | Adiponectin receptor prot-2 | Upstream |
Abbreviations: Chr., chromosome; dom, domain; MAF, minor allele frequency; P HWE, P-value resulting after Hardy–Weinberg equilibrium testing; P Mixed, P-value resulting mixed-model analysis (indicated in bold); Pos. Rel. Gene, position of the SNP relative to the gene listed in the previous column; prot, protein.
The number of homozygotes for the minor allele/the number of heterozygotes/the number of homozygotes for the major allele.
The Hardy–Weinberg equilibrium P-value should be larger than 0.001 for the SNP genotyping to be valid.
‘/' indicates that this SNP was not located within a gene region.
‘Upstream' or ‘Downstream' was based on the Affymetrix annotation.
Table 4. Top 25 SNPs resulting from the mixed model analysis for PC3.
| SNP | Chr. | Position | Alleles | Minor allele | MAF | Genotypesa | P HWEb | P mixed | Gene regionc | Pos. Rel. gened |
|---|---|---|---|---|---|---|---|---|---|---|
| rs457717 | 5 | 75 956 728 | C/T | T | 0.251429 | 26/124/200 | 0.2589 | 3.55E−07 | Ras GTPase-activating-like prot IQGAP2 | Intron |
| rs1697845 | 5 | 75 958 260 | C/T | C | 0.293447 | 37/132/182 | 0.0935 | 1.63E−05 | Ras GTPase-activating-like prot IQGAP2 | Intron |
| rs989414 | 2 | 151 552 224 | C/T | T | 0.061960 | 3/37/307 | 0.1326 | 3.72E−05 | CDNA FLJ45645 fis, clone CTONG2003517 | Downstream |
| rs9310194 | 3 | 70 713 297 | A/T | A | 0.334770 | 35/163/150 | 0.3994 | 4.96E−05 | Microphthalmia-associated transcription factor | Downstream |
| rs10514104 | 5 | 76 843 891 | A/G | G | 0.113506 | 4/71/273 | 1.0000 | 5.08E−05 | / | / |
| rs7913244 | 10 | 10 224 371 | A/T | T | 0.102857 | 5/62/283 | 0.3902 | 5.69E−05 | / | / |
| rs2299641 | 11 | 17 397 566 | C/G | G | 0.200000 | 14/112/224 | 1.0000 | 7.90E−05 | ATP-binding cassette transporter subfamily-C member-8 | Intron |
| rs10494078 | 1 | 108 157 301 | C/T | T | 0.472303 | 72/180/91 | 0.3860 | 9.16E−05 | Prot vav-3 | Intron |
| rs4636135 | 7 | 103 406 504 | A/G | A | 0.246418 | 19/134/196 | 0.6650 | 0.000100 | Reelin prec (EC 3.4.21.-) | Intron |
| rs6539455 | 12 | 107 744 032 | C/G | G | 0.115607 | 9/62/275 | 0.0310 | 0.000101 | Prot phosphatase Slingshot homologue-1 (EC 3.1.3.48) | Intron |
| rs10497072 | 2 | 151 581 781 | A/G | A | 0.058405 | 1/39/311 | 1.0000 | 0.000135 | Rho-related GTP-binding prot RhoE (Rho family GTPase-3) | Upstream |
| rs4664883 | 2 | 151 583 181 | A/C | A | 0.058405 | 1/39/311 | 1.0000 | 0.000135 | CDNA FLJ45645 fis, clone CTONG2003517 | Downstream |
| rs1500237 | 5 | 98 643 065 | C/T | T | 0.205128 | 17/110/224 | 0.5116 | 0.000145 | Y RNA | Upstream |
| rs6433104 | 2 | 151 559 112 | C/T | T | 0.058908 | 1/39/308 | 1.0000 | 0.000151 | Rho-related GTP-binding prot RhoE (Rho family GTPase-3) | Upstream |
| rs10483426 | 14 | 32 670 589 | C/T | T | 0.452991 | 79/160/112 | 0.1326 | 0.000163 | Neuronal PAS domain-containing prot-3 (neuronal PAS3) | Intron |
| rs1940484 | 11 | 98 447 082 | C/T | C | 0.475714 | 82/169/99 | 0.5920 | 0.000164 | / | / |
| rs9301099 | 13 | 105 483 574 | A/G | G | 0.169516 | 14/91/246 | 0.1321 | 0.000183 | / | / |
| rs2527756 | 8 | 5 390 145 | C/T | C | 0.188571 | 10/112/228 | 0.4856 | 0.000199 | / | / |
| rs10512077 | 9 | 80 741 480 | A/G | A | 0.417379 | 67/159/125 | 0.2269 | 0.000201 | / | / |
| rs42064 | 7 | 95 557 193 | A/T | A | 0.319088 | 42/140/169 | 0.1399 | 0.000210 | Cytoplasmic dynein-1 intermediate chain-1 | Intron |
| rs1363556 | 5 | 144 886 649 | C/T | T | 0.498457 | 90/143/91 | 0.0351 | 0.000216 | 7SK RNA | Upstream |
| rs1260764 | 1 | 64 316 010 | G/T | G | 0.176638 | 12/100/239 | 0.7133 | 0.000221 | Tyrosine-prot kinase transmembrane receptor ROR1 prec (EC 2.7.10.1) | Intron |
| rs1260772 | 1 | 64 313 964 | A/G | G | 0.176638 | 12/100/239 | 0.7133 | 0.000221 | Tyrosine-prot kinase transmembrane receptor ROR1 prec (EC 2.7.10.1) | Intron |
| rs1559776 | 15 | 55 746 920 | C/T | T | 0.150997 | 7/92/252 | 0.8356 | 0.000236 | GRINL1A combined prot isoforms-1 | Intron |
| rs4809590 | 20 | 44 518 234 | C/T | T | 0.102564 | 7/58/286 | 0.0732 | 0.000253 | / | / |
Abbreviations: Chr., chromosome; MAF, minor allele frequency; P HWE, P-value resulting after Hardy–Weinberg equilibrium testing; P Mixed, P-value resulting after mixed-model analysis (indicated in bold); Pos. Rel. Gene, position of the SNP relative to the gene listed in the previous column; prot, protein; prec, precursor.
The number of homozygotes for the minor allele/the number of heterozygotes/the number of homozygotes for the major allele.
The Hardy–Weinberg equilibrium P-value should be larger than 0.001 for the SNP genotyping to be valid.
‘/' indicates that this SNP was not located within a gene region.
‘Upstream' or ‘Downstream' was based on the Affymetrix annotation.
P-values are indicated in bold.
If, in agreement with other GWAS,34 we used a P-value cut-off of 5 × 10−7 to declare genome-wide significance, the top-ranked SNP for PC3 (rs457717; Table 4) survived this significance threshold (P-value=3.55 × 10−7). This SNP is located in an intron of the IQ motif-containing GTPase-activating-like protein (IQGAP2). The second-ranked SNP (rs1697845) is also positioned in IQGAP2, and is highly correlated with the first (D′=1; r2=0.965), indicating that this signal is not a genotyping artefact.
For the other two traits, PC1 and PC2, none of the SNPs reached this level of significance (Tables 2 and 3). Intriguingly, however, the eighth-ranked SNP for PC1 (rs161927; Table 2; P-value 0.000149) was positioned 40 kb downstream from the metabotropic GRM7 gene (Supplementary Figure 3). We have previously reported an association of a common variant in the GRM7 gene, rs11928865, with hearing ability based on a pooling-based GWAS of a European sample set.9 Therefore, we also genotyped this SNP in the Saami population and analysed these data using the mixed-model approach. This resulted in a P-value of 0.045. However, the A-allele was associated with an elevated hearing threshold in the Saami sample set, while in the European sample set, the associated allele was T.9 One of the two top-scoring GRM7 SNPs resulting from a GWAS of a Finnish sample set,9 rs779701, was present on the 100 K Affymetrix array. However, in the Saami sample set, this SNP did not result in a significant association (P-value of 0.735). The LD pattern within the GRM7 region deduced for the Saami (Supplementary Figure 3D) was similar to that observed in the HapMap for the European population (Supplementary Figure 3C). No obvious biological meaning related to the inner ear could be deduced for the gene regions of the seven higher-ranked SNPs for PC1.
For PC2, no strong biological candidates were present among the top 25 SNPs (Table 3). The presence of six SNPs from the same gene (KH domain-containing, RNA-binding, signal transduction-associated protein-2; KHDRBS2) in Table 3 is due to the fact that all of these SNPs reside in the same region of high LD.
Discussion
We conducted a genome-wide association scan for ARHI traits derived from audiometric data in the genetically isolated Finnish Saami population. We summarised the information contained in each audiogram by the first three components obtained through PC analysis. Previously, we have shown that these traits have significant heritabilities.8 Using these audiometric measures, we retain biologically important information on the shape of the audiogram that would be lost when simple PTAs or individual frequencies are analysed. Indeed, premortem-derived audiometric patterns have been correlated to pathophysiological findings in human temporal bones.30, 31 Nelson et al35 studied individuals with downward-sloping audiograms and reported that the severity of hearing loss, based on audiometric thresholds, was highly associated with a decrease in stria vascularis volume and degeneration of outer and inner hair cell and ganglion cell populations. The slope of the audiogram was significantly associated with the extent of ganglion cell degeneration.
Our association scan resulted in two SNPs in or near genes that are reasonable biological candidates and that are candidates for further research. The top-ranked SNP, rs457717 (P-value 3.55 × 10−7), that reached genome-wide significance, was associated with PC3 and was localised in an intron of the IQ motif-containing, GTPase-activating-like protein (IQGAP2). IQGAP2 is a member of the Ras superfamily of GTPases. This class of proteins regulates a wide variety of cellular signalling pathways. IQGAP2 is expressed inside the cochlea36 and has been implicated in cadherin-mediated cell adhesion.37 As cadherin-23 (CDH23) plays an important role in the sensory hair cells,38 and mutations in CDH23 are responsible for syndromic and non-syndromic autosomal recessive deafness in humans39, 40 and age-related hearing loss in inbred mouse strains,41 this warrants further research. Intriguingly, the SNP rs161927 (P-value 0.000149), that ranked eighth for PC1, was positioned immediately downstream from the metabotropic GRM7 gene. A previous GWAS of a European and a Finnish sample set, which was based on an age- and gender-corrected Z-score highly correlated with our PC1,9, 11, 28, 42 already suggested a role for GRM7 in ARHI.9 GRM7 encodes a metabotropic glutamate receptor that can be activated by -glutamate, the primary excitatory neurotransmitter in the cochlear hair cell. GRM7 expression was observed in the inner and outer hair cells, and in the spiral ganglion nerve cell bodies of the inner ear.9 We have hypothesised that certain GRM7 variants may cause increased susceptibility to glutamate excitotoxicity, which may consequently lead to the development of ARHI. However, variants that were significant in our previous study did not replicate in our current study. The LD structure within the GRM7 region indicates that these are independent signals. These results, therefore, are consistent with allelic heterogeneity, which is a plausible scenario given the likely absence of any selective pressure for this disorder. Independent studies are needed to investigate the role of common variants in GRM7 in ARHI.
This study was conducted involving the genetically isolated Saami population. A number of studies provide evidence that the extent of LD is much higher in the Saami as compared to that in other European populations.20, 21, 22, 23 This would provide an important power advantage, as higher LD leads to better genomic coverage. It has been proposed that association studies based on a map of modest marker density within a population isolate such as the Saami would provide a first rough identification of the region associated with a particular complex disease. This first demarcation of the region should then preferably be refined by association studies of an outbred population using a denser map.22 We have published elsewhere the results of an elaborate genome-wide evaluation of the relative power for GWAS in the Saami.43
A possible limitation of this study is the limited sample size. Recent studies indicate that several thousand subjects need to be recruited in order to obtain sufficient power to detect the effect sizes typically expected for complex phenotypes (with each contributing variant explaining as little as 0.1–1.3% of the variation),44, 45 although successful GWAS have been conducted using only 96 cases and 50 controls.46 Obviously, large sample sizes can never be recruited in small isolated populations such as the Saami. A specific difficulty of working with isolated populations became apparent in the current study. Owing to the fact that isolated populations are often derived from a limited number of founders and that inbreeding is sometimes inevitable, an increased relatedness is observed within such populations. As a consequence, classical statistical methods that assume unrelatedness, cannot be applied to analyse an association study performed in a population isolate. In our exploratory phase, we investigated several approaches, including a naive linear regression analysis and a PC-based correction (EIGENSTRAT47). However, for the latter approach, the basic assumptions were violated and the inbreeding and relatedness present in our Saami sample created spurious axes of variation.48 Mixed models offered a solution. In this approach, we estimated the genetic relationships between the study subjects based on genome-wide data and subsequently accounted for them in the analysis.33
In conclusion, we present the results of a first GWAS performed in the genetically isolated Saami population. We identified IQGAP2 as a candidate ARHI gene. Independent replication is needed to confirm this lead. In addition, we found further evidence implicating GRM7 in ARHI.
Acknowledgments
We express our most sincere gratitude to all the Saami volunteers who participated in this study. JRH thanks Gael Pressoir and Jianming Yu from the Ed Buckler Lab for providing help with the SAS macro. This study was funded by the European Community (5th Framework project QLRT-2001-00331), the University of Antwerp (TOP project), the Research Foundation – Flanders (FWO grant G.0163.09), and the State of Arizona. JRH is a fellow of the Research Foundation – Flanders (FWO).
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Davis A.Prevalence of hearing impairmentin Davis A (ed): Hearing in Adults London: Whurr Publishers Ltd; 199443–321. [Google Scholar]
- Gates GA, Mills JH. Presbycusis. Lancet. 2005;366:1111–1120. doi: 10.1016/S0140-6736(05)67423-5. [DOI] [PubMed] [Google Scholar]
- Christensen K, Frederiksen H, Hoffman HJ. Genetic and environmental influences on self-reported reduced hearing in the old and oldest old. J Am Geriatr Soc. 2001;49:1512–1517. doi: 10.1046/j.1532-5415.2001.4911245.x. [DOI] [PubMed] [Google Scholar]
- Gates GA, Couropmitree NN, Myers RH. Genetic associations in age-related hearing thresholds. Arch Otolaryngol Head Neck Surg. 1999;125:654–659. doi: 10.1001/archotol.125.6.654. [DOI] [PubMed] [Google Scholar]
- Karlsson KK, Harris JR, Svartengren M. Description and primary results from an audiometric study of male twins. Ear Hear. 1997;18:114–120. doi: 10.1097/00003446-199704000-00003. [DOI] [PubMed] [Google Scholar]
- DeStefano AL, Gates GA, Heard-Costa N, Myers RH, Baldwin CT. Genomewide linkage analysis to presbycusis in the Framingham Heart Study. Arch Otolaryngol Head Neck Surg. 2003;129:285–289. doi: 10.1001/archotol.129.3.285. [DOI] [PubMed] [Google Scholar]
- Garringer HJ, Pankratz ND, Nichols WC, Reed T. Hearing impairment susceptibility in elderly men and the DFNA18 locus. Arch Otolaryngol Head Neck Surg. 2006;132:506–510. doi: 10.1001/archotol.132.5.506. [DOI] [PubMed] [Google Scholar]
- Huyghe JR, Van Laer L, Hendrickx JJ, et al. Genome-wide SNP-based linkage scan identifies a locus on 8q24 for an age-related hearing impairment trait. Am J Hum Genet. 2008;83:401–407. doi: 10.1016/j.ajhg.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RA, Van Laer L, Huentelman MJ, et al. GRM7 variants confer susceptibility to age-related hearing impairment. Hum Mol Genet. 2009;18:785–796. doi: 10.1093/hmg/ddn402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eyken E, Van Camp G, Van Laer L. The complexity of age-related hearing impairment: contributing environmental and genetic factors. Audiol Neurootol. 2007;12:345–358. doi: 10.1159/000106478. [DOI] [PubMed] [Google Scholar]
- Van Laer L, Van Eyken E, Fransen E, et al. The grainyhead like 2 gene (GRHL2), alias TFCP2L3, is associated with age-related hearing impairment. Hum Mol Genet. 2008;17:159–169. doi: 10.1093/hmg/ddm292. [DOI] [PubMed] [Google Scholar]
- Bai U, Seidman MD, Hinojosa R, Quirk WS. Mitochondrial DNA deletions associated with aging and possibly presbycusis: a human archival temporal bone study. Am J Otol. 1997;18:449–453. [PubMed] [Google Scholar]
- Fischel-Ghodsian N, Bykhovskaya Y, Taylor K, et al. Temporal bone analysis of patients with presbycusis reveals high frequency of mitochondrial mutations. Hear Res. 1997;110:147–154. doi: 10.1016/s0378-5955(97)00077-4. [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Piazza A. Human genomic diversity in Europe: a summary of recent research and prospects for the future. Eur J Hum Genet. 1993;1:3–18. doi: 10.1159/000472383. [DOI] [PubMed] [Google Scholar]
- Lahermo P, Sajantila A, Sistonen P, et al. The genetic relationship between the Finns and the Finnish Saami (Lapps): analysis of nuclear DNA and mtDNA. Am J Hum Genet. 1996;58:1309–1322. [PMC free article] [PubMed] [Google Scholar]
- Lahermo P, Savontaus ML, Sistonen P, et al. Y chromosomal polymorphisms reveal founding lineages in the Finns and the Saami. Eur J Hum Genet. 1999;7:447–458. doi: 10.1038/sj.ejhg.5200316. [DOI] [PubMed] [Google Scholar]
- Sajantila A, Lahermo P, Anttinen T, et al. Genes and languages in Europe: an analysis of mitochondrial lineages. Genome Res. 1995;5:42–52. doi: 10.1101/gr.5.1.42. [DOI] [PubMed] [Google Scholar]
- Tambets K, Rootsi S, Kivisild T, et al. The western and eastern roots of the Saami – the story of genetic ‘outliers' told by mitochondrial DNA and Y chromosomes. Am J Hum Genet. 2004;74:661–682. doi: 10.1086/383203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingman M, Gyllensten U. A recent genetic link between Saami and the Volga–Ural region of Russia. Eur J Hum Genet. 2007;15:115–120. doi: 10.1038/sj.ejhg.5201712. [DOI] [PubMed] [Google Scholar]
- Johansson A, Vavruch-Nilsson V, Cox DR, Frazer KA, Gyllensten U. Evaluation of the SNP tagging approach in an independent population sample – array-based SNP discovery in Saami. Hum Genet. 2007;122:141–150. doi: 10.1007/s00439-007-0379-2. [DOI] [PubMed] [Google Scholar]
- Johansson A, Vavruch-Nilsson V, Edin-Liljegren A, Sjolander P, Gyllensten U. Linkage disequilibrium between microsatellite markers in the Swedish Saami relative to a worldwide selection of populations. Hum Genet. 2005;116:105–113. doi: 10.1007/s00439-004-1213-8. [DOI] [PubMed] [Google Scholar]
- Kaessmann H, Zollner S, Gustafsson AC, et al. Extensive linkage disequilibrium in small human populations in Eurasia. Am J Hum Genet. 2002;70:673–685. doi: 10.1086/339258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laan M, Paabo S. Demographic history and linkage disequilibrium in human populations. Nat Genet. 1997;17:435–438. doi: 10.1038/ng1297-435. [DOI] [PubMed] [Google Scholar]
- Bonnen PE, Pe'er I, Plenge RM, et al. Evaluating potential for whole-genome studies in Kosrae, an isolated population in Micronesia. Nat Genet. 2006;38:214–217. doi: 10.1038/ng1712. [DOI] [PubMed] [Google Scholar]
- Service S, DeYoung J, Karayiorgou M, et al. Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat Genet. 2006;38:556–560. doi: 10.1038/ng1770. [DOI] [PubMed] [Google Scholar]
- Slatkin M. Linkage disequilibrium in growing and stable populations. Genetics. 1994;137:331–336. doi: 10.1093/genetics/137.1.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger JD, Zollner S, Laan M, Paabo S. Mapping genes through the use of linkage disequilibrium generated by genetic drift: ‘drift mapping' in small populations with no demographic expansion. Hum Hered. 1998;48:138–154. doi: 10.1159/000022794. [DOI] [PubMed] [Google Scholar]
- Van Eyken E, Van Laer L, Fransen E, et al. KCNQ4: a gene for age-related hearing impairment. Hum Mutat. 2006;27:1007–1016. doi: 10.1002/humu.20375. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Wichern DW. Applied Multivariate Statistical Analysis. New Jersey: Prentice Hall; 2002. [Google Scholar]
- Nelson EG, Hinojosa R. Presbycusis: a human temporal bone study of individuals with downward sloping audiometric patterns of hearing loss and review of the literature. Laryngoscope. 2006;116:1–12. doi: 10.1097/01.mlg.0000236089.44566.62. [DOI] [PubMed] [Google Scholar]
- Schuknecht HF, Gacek MR. Cochlear pathology in presbycusis. Ann Otol Rhinol Laryngol. 1993;102:1–16. doi: 10.1177/00034894931020S101. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Pressoir G, Briggs WH, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38:203–208. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson EG, Hinojosa R. Presbycusis: a human temporal bone study of individuals with flat audiometric patterns of hearing loss using a new method to quantify stria vascularis volume. Laryngoscope. 2003;113:1672–1686. doi: 10.1097/00005537-200310000-00006. [DOI] [PubMed] [Google Scholar]
- Robertson NG, Khetarpal U, Gutierrez-Espeleta GA, Bieber FR, Morton CC. Isolation of novel and known genes from a human fetal cochlear cDNA library using subtractive hybridization and differential screening. Genomics. 1994;23:42–50. doi: 10.1006/geno.1994.1457. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Abe H, Mabuchi I. IQGAP2 is required for the cadherin-mediated cell-to-cell adhesion in Xenopus laevis embryos. Dev Biol. 2007;308:485–493. doi: 10.1016/j.ydbio.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Kazmierczak P, Sakaguchi H, Tokita J, et al. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature. 2007;449:87–91. doi: 10.1038/nature06091. [DOI] [PubMed] [Google Scholar]
- Bolz H, von Brederlow B, Ramirez A, et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet. 2001;27:108–112. doi: 10.1038/83667. [DOI] [PubMed] [Google Scholar]
- Bork JM, Peters LM, Riazuddin S, et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001;68:26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet. 2003;35:21–23. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eyken E, Van Camp G, Fransen E, et al. The N-acetyltransferase 2 polymorphism NAT2*6A is a causative factor for age related hearing impairment. J Med Genet. 2007;44:570–578. doi: 10.1136/jmg.2007.049205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyghe JR, Fransen E, Hannula S, et al. Genome-wide SNP analysis reveals no gain in power for association studies of common variants in the Finnish Saami Eur J Hum Genet 2009. e-pub ahead of print]. [DOI] [PMC free article] [PubMed]
- Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- Weedon MN, Lango H, Lindgren CM, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet. 2008;40:575–583. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.