

Abstract
An intuitive normalization and gene identification method is proposed. After segmentation of the entire expression range into intensity intervals, the mean and standard deviation of the logarithm of expression ratios are calculated for each interval using the nearest neighbor genes. Genes with high differential expression are excluded from these calculations. For glass arrays, normalization is performed for each interval by using the mean of the logarithm of expression ratios in the interval. For nylon/plastic membranes, the average of the means of the logarithm of ratios across the intervals of higher intensities is used for normalization. Compared with other normalization methods, this method delivered the smallest normalization errors for 42 nylon/plastic arrays used to analyze cultured T cells and 22 Clostridium acetobutylicum glass arrays. For identifying differentially expressed genes, upper and lower boundaries are constructed for each interval by using the standard deviation of the expression ratio logarithms. When a C. acetobutylicum pSOL1 megaplasmid-deficient strain M5 was used, this method identified more “down-regulated” pSOL1 genes with fewer misidentifications in a comparative array analysis of M5 versus the parent strain. A comparison of quantitative RT-PCR results with different gene identification methods indicates that the proposed method is superior to other methods.
The DNA array technology is emerging as a powerful tool for large-scale gene expression analysis (1). Like any other measuring method, this technology contains inherent measurement errors. Because of variations in labeling, hybridization, spotting, or surface characteristics, expression intensities resulting from the same amount of mRNA can differ from experiment to experiment, and thus, a normalization method must be used to compare expression intensities obtained from two dyes or membranes. Normalization of array data involves two steps: (i) selection of genes to be used for normalization, and (ii) application of a mathematical operator or metric to normalize the data. Gene selection can include the entire gene set (global, G), housekeeping genes, rank-invariant genes, or genes with “constant expression.” Mathematical operators or metrics include expression-intensity mean (EM), expression-intensity median, expression-ratio mean (ERM), expression-ratio median (ERMD), mean of logarithm of expression ratio (LERM), median of logarithm of expression ratio (LERMD), expression-ratio probability density (ERPD), and linear or nonlinear regression. Any combination of one of these mathematical operators or metrics with a set of genes can be used. However, there are only a few normalization methods reported in the literature. Global normalization using the mean or median of expression intensities and normalization based on housekeeping genes are the most widely used methods (2–4). Other methods include an iterative method using ERPD based on housekeeping genes (5) and a LERMD method using only genes with constant expression levels (6, 7). Because of different labeling and detection efficiencies for various fluorescent dyes, glass arrays require nonlinear normalization protocols rather than a constant normalization factor (8, 9).
After normalization, data must be processed to identify differentially expressed genes before clustering (2). A frequently used method for selection of differentially expressed genes is based on setting an intensity threshold and a minimal fold change (typically 2- to 3-fold) to discard genes with low expression levels and insignificant fold differences, respectively (10). At high expression levels, the noise-to-signal-intensity ratio is small. A small change at very high expression levels might therefore be significant. On the other hand, at moderate expression levels, 2- or 3-fold changes may not be significant due to relatively higher noise-to-signal ratios. By using “calibration” experiments (4, 8) (identical samples labeled with both Cy3 and Cy5 dyes and hybridized on the same glass slide or labeled with radioactivity and hybridized on two separate membranes), Tien et al. (4) performed a segmental calculation of standard deviation (SD) or maximum/minimum fold changes, and developed masks for nondifferentially expressed genes. Statistical approaches have also been developed to identify differentially expressed genes. When a ratio probability density is used, upper and lower boundaries (masks) for differentially expressed genes can be derived at a given confidence level (5). Because such masks are intensity-dependent, Newton et al. (11) used a Gamma–Gamma Bernoulli model to develop the ratio probability density, and presented intensity-dependent gene-identification masks at various posterior odds based on hierarchical modeling. Such masks are wider at both low and high expression intensities. Using a Bayesian approach, Tseng et al. (8) developed a hierarchical Gaussian model for identification of differentially expressed genes. Another approach is application of an ANOVA model (12). However, no studies have compared the efficiencies of different normalization and gene-identification methods and their effects on transcriptional analysis.
We describe a superior array normalization and gene-identification method by segmenting the entire intensity range into a number of intensity intervals and determining the mean and SD of the logarithms of expression ratios (LERs) for each interval with the nearest neighbor nondifferentially expressed genes. Normalization is based on the mean of the LERs in each interval or in the intervals of higher expression intensities. For gene identification, upper and lower masks are constructed for each interval by using the interval SD of the LERs.
Materials and Methods
Nylon and Plastic Arrays.
Thirty nylon arrays (Human Atlas 1.2, CLONTECH) containing 1,191 singly spotted genes, and 12 plastic arrays (Atlas Plastic Human 8K, CLONTECH), containing over 8,300 doubly spotted genes, were used to analyze samples of primary human T cells cultured at 5% or 20% oxygen tension (pO2) in serum-free medium (13). Gene-expression analysis was performed pairwise by using samples of 20% vs. 5% pO2 cultures initiated with the same donor cells. In addition, 6 nylon and 2 plastic calibration arrays were used to obtain a constant multiplier used for gene identification masks. RNA isolation, reverse transcription (RT), labeling (32P/33P), and hybridization were performed per manufacturer's instructions. Nylon or plastic arrays were exposed to GP/LE phosphor screens (Molecular Dynamics) for 24 or 72 h, respectively, and scanned by using a Storm PhosphorImager (Molecular Dynamics). imagequant (Molecular Dynamics) or ATLASIMAGE 2.01 (CLONTECH) was used for quantitation of signal intensities.
Glass Arrays.
cDNA arrays with triplicate spots representing 1,019 ORFs, approximately one-fourth of the Clostridium acetobutylicum ATCC 824 genome, were printed by using the TIGR protocol (14). The list of genes on the arrays and all primary array data used in this paper are available on our web site (http://www.chem-eng.northwestern.edu/faculty/papou.html). Three C. acetobutylicum strains were grown anaerobically in bioreactors (15): WT (ATCC and 824), 824(pSOS95del) (plasmid-control strain), and 824(pGroE1) (overexpresses GroESL operon genes using the thiolase promoter). Static flask cultures (400 ml) of the WT and M5 (lacks the pSOL1 megaplasmid; ref. 16) strains were also analyzed. Three sets of array experiments were performed, including eight slides of WT vs. 824(pSOS95del), six slides of WT vs. M5, and eight slides of 824(pSOS95del) vs. 824(pGroE1). Half of these slides were done with opposite labeling and were used for construction of a gene-identification mask based on ANOVA. In addition, three calibration glass arrays were used for setup of gene-identification masks. RNA was isolated by using the Trizol reagent (Invitrogen) with additional lysozyme treatment (15). Purified RNA was used to synthesize labeled cDNA in a hexamer-primed RT reaction (Roche Molecular Biochemicals) in the presence of Cy3-dUTP or Cy5-dUTP (Amersham Pharmacia) using Moloney murine leukemia virus reverse transcriptase (Promega). Slides were hybridized with probes overnight in a Corning hybridization chamber at 42°C, and were scanned with a GSI Lumonics ScanArray analyzer. Spot intensities were quantitated with QuantArray Microarray Analysis (Perkin–Elmer).
Quantitative RT-PCR (Q-RT-PCR).
For Q-RT-PCR, all RT reactions were performed by using the RETROscript kit (Ambion, Austin, TX). Each PCR contained 0.5 μl of RT reaction product, 1× SYBR Green PCR Master Mix (Applied Biosystems), and primers (Table 3, which is published as supporting information on the PNAS web site, www.pnas.org). All PCRs were run on the ABI PRISM 7700 Sequence Detection System with the following program: 10 min at 95°C, 35–40 cycles of 15 sec at 95°C and 1 min at the annealing temperature (Table 3). Three or six Q-RT-PCR replicates were performed for each gene in each T cell or C. acetobutylicum sample, respectively. The resulting average fold changes from these replicates were compared with a fixed value of one to determine the statistical significance (P < 0.05) of the fold changes using the single-sample t test (17).
Normalization and Gene Identification Method
Variations in several factors and processes of array analysis result in two types of errors, random and system errors, and affect the measured gene expression intensity. Random errors result from scanning errors and spot-to-spot variations (on the same array) in the amount of deposited cDNA and array-surface properties. These errors can generally be defined as noise with a mean of 0 across all spots in an array. System errors can be defined as those resulting from variations in array quality (array surface and printing, and amount of DNA spotted) as well as in sample preparation between two membranes or dyes (array storage, RNA amount used, reverse transcription and labeling, hybridization, and washing).
Let x* and y* be the true intensities of a nondifferentially expressed gene on two membranes or of two dyes. True intensities should be free of random errors. Thus, the difference between x* and y* is caused by system errors only. Practically, one will never know these true values. What is known are measurements that contain random errors (noise). If no such random errors were present, normalization would be accomplished by using the ratio of true intensities directly, namely λ(x, y) = x*/y*.
Assume that in the neighborhood of (x*, y*) there are K nondifferentially expressed genes with true expression intensities (x*i, y*i ), i = 1, … , K, where the neighborhood is defined by the space spanned by the K non-differentially expressed genes closest to (x*, y*). The measured intensities, (xi, yi), after prefiltering (as described in the supporting text, which is published on the PNAS web site) for these K genes contain random errors (ɛx,i, ɛy,i). Thus, we can write that xi = x*i + ex,i and yi = y*i + ey,i. To treat up-regulated and down-regulated genes equally, natural LER is used. The logarithm of the normalization factor of two membranes or dyes can be estimated using the K nearest neighbors of (x*, y*) (18):
 |
1 |
If the number K is large enough, one can rewrite this equation as
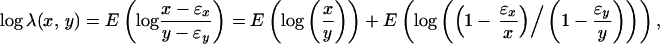 |
2 |
where E{⋅} represents the mean of the argument. When this normalization factor is used, the normalized expression ȳ in the second membrane or dye to the expression in the first membrane or dye can be written as log ȳ = log y + log λ(x, y) or log ȳ = λ(x, y)y.
Random Errors of Two Different Membranes or Dyes.
The first term on the right side of Eq. 2 aims to eliminate system errors, while the second term describes the effects of the noise-to-signal ratios on normalization. The normalization objective is to capture and eliminate system errors in the presence of random errors (noise). In membrane array experiments, samples are hybridized on two separate arrays. Random errors in two different arrays should be independent of one another, and thus, ɛx and ɛy may have opposite signs, which may lead to a large variation in y/x = (y* + ɛy)/(x* + ɛx), especially at low intensities. A typical distribution of LER against the logarithmic mean intensity in a nylon/plastic array is shown in Fig. 1 Ia and IIa. Expression ratios have a wide spread at lower intensities and gradually converge with increasing intensities. For random errors in two membranes, one can write
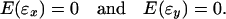 |
3 |
In glass arrays, samples are labeled with two different dyes, and in contrast to membrane arrays, are hybridized on the same slide; thus, both samples compete for binding with the cDNA of each array spot. Therefore, spot variations, which result in random errors, affect the two dyes simultaneously. Because random errors contain measurement errors in addition to spot variations, they are expected to be both related because of same spot variations and random because different measurement errors. As a result, variations in expression ratios are nearly uniformly distributed across the whole intensity range (Fig. 1IIIa), and one may write that y/x = (1 + ɛ0)y*/x*, where ɛ0 is a random signal with a mean of zero. The noise-to-signal relationship between the two channels can then be written as
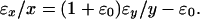 |
4 |
Figure 1.

Normalization results. (a and b) Original expression ratios (a) and normalized expression ratios (b) for nylon (I), plastic (II), and glass (III) arrays are shown.
Normalization of Calibration Arrays.
For membrane arrays at higher intensities, ɛx ≪ x* and ɛy ≪ y*. Considering Eqs. 2 and 3, the normalization factor between two plastic or nylon membranes can then be obtained by using genes with higher expression intensities:
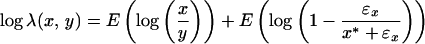 |
5 |
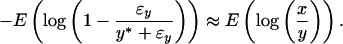 |
For glass arrays, assuming that ɛ0 ≪ 1, one can derive the following expression from Eqs. 2 and 4,
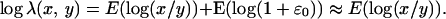 |
6 |
Normalization of Two Different Samples.
In Eq. 5 or 6, the genes used for normalization are only those that are nondifferentially expressed. In an array experiment, typically only a fraction of genes have altered expression when comparing two samples. Nevertheless, highly differentially expressed genes (outliers) may considerably affect the mean of the LERs, and therefore should be removed before normalization. One can use the increase or decrease in SD of a group of n + 1 data points to identify outliers. When equidistantly distributed, one can write
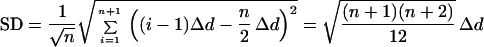 |
7 |
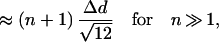 |
where Δd is the distance between two data points. Therefore, the SD increases approximately linearly with increasing number of data points. As a point with a larger distance to the neighboring point is added, the SD increases at a greater rate. To eliminate genes with strong differential expression, we first calculate the LER mean and SD for a set of genes, and then remove the gene with the LER the farthest from the LER mean from the set of genes. This calculation and removal procedure is repeated until only a few genes are left (e.g., five). By doing so, we obtain the SD profile as a function of the number of data points (genes) (Fig. 2). Data with a uniform or normal distribution (without tails) result in a relatively constant increase in SD as the number of data points increases, whereas data points in the tails of a normal distribution and strongly expressed genes cause a sharp increase in the SD. These highly expressed genes are excluded, and the LER mean derived from the remaining nearest neighbor genes is used for normalization.
Figure 2.

Identification of strongly expressed genes using the SD profiles as a function of the number of data points from a uniform (diamonds) or normal (thin line) distribution, or an array data set (thick line). Vertical lines separate the highly expressed genes or data points in the tails of a normal distribution from the rest of genes or data points.
Discretization of Intensities and Normalization Based on a Moving-Gene Neighborhood.
As shown, the validity of Eq. 5 is restricted to higher expression intensities. Thus, we can only use non-differentially expressed genes at higher expression intensities to normalize two different membrane arrays. The expression ratio to intensity relationship between two dyes in a glass array is slide-dependent and nonlinear (Fig. 1IIIa), and such nonlinearity cannot be corrected by a global normalization factor. Thus, we propose a piecewise normalization to account for this nonlinear relationship. To distinguish the higher from the lower expression in the case of membrane arrays or to obtain a piecewise normalization for glass arrays, the whole range of the natural logarithmic mean intensity was discretized into M equidistant intervals. K nondifferentially expressed genes around the middle of each interval were then used to determine the LER mean and its SD in the interval. The M and K values were determined by minimizing the total normalization errors, as discussed below. The normalization factor between two membrane arrays was evaluated based on the average of the LER means over the intervals of higher intensities (e.g., >5× background intensity). For glass arrays, the normalization of two dyes was performed by using the LER mean in each interval. This piecewise normalization factor across all intensity intervals was used to model the slide-dependent nonlinear relationship between the expression ratios and intensities in glass arrays.
Masks for Identifying Differentially Expressed Genes.
After normalization, the normalized LERs should be distributed around zero. Because the SD may vary from interval to interval and from array to array, the normalized LERs of various intensity intervals in separate arrays are distributed differently. Therefore, masks for identification of differentially expressed genes obtained from one array experiment cannot be applied to other arrays (4). The distribution of the LERs can be standardized by dividing the LER by the corresponding SD. Similarity can be found for various intervals of different arrays (Figs. 5 and 6, which are published as supporting information on the PNAS web site). The standardized probability densities are normal-like, but with heavier tails (Fig. 7, which is published as supporting information on the PNAS web site) and the confidence level cannot be accurately determined based on normal distributions and t statistics (12). Instead, the percentile method was used to estimate the confidence level. Because of the similarity of the standardized probability densities in various intervals and samples, one can apply the results from one interval to any other interval or array. Using the percentile method, the SDj of LERs in an interval j along with a constant multiplier can be used to set up the upper and lower mask borders, ϑ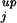 = γSDj and ϑ
= γSDj and ϑ = −γSDj, for that interval. The constant multiplier γ is the number of SDs required to ensure, with a certain confidence level (e.g., 95%), that genes with expression ratios falling between the upper and lower mask borders are not differentially expressed. In other words, we can state with 95% confidence that genes lying outside of the mask are differentially expressed. Because the standardized probability densities in different intervals are similar, γ is independent of the interval index j. To determine γ, a number of calibration array experiments and the percentile method were used. After normalization, any deviations of the LERs of the calibration arrays from zero are caused by noise. Ideally, γ should be chosen such that all genes are located inside the masks. However, because of significant noise in the data, an extremely large γ was needed to produce a mask that included all genes. For example, γ = 4.3, 5.1, and 3.9 were necessary for three pairs of nylon calibration arrays, one pair of plastic calibration arrays, and three calibration glass arrays, respectively. Hence, γ was chosen such that a maximum of 5% of all genes lie outside the mask for all calibration experiments. For nylon, plastic, and glass arrays, γ = 2.1, 2.3, and 2.0, respectively. More stringent masks can be constructed by increasing the value of γ. As in the case of normalization, the determination of the SD was performed after excluding highly differentially expressed genes. It should be noted that an improvement in the similarity of standardized LER probability densities will make masks more robust and accurate, and such an improvement can be achieved by a better standardization or discretization protocol.
= −γSDj, for that interval. The constant multiplier γ is the number of SDs required to ensure, with a certain confidence level (e.g., 95%), that genes with expression ratios falling between the upper and lower mask borders are not differentially expressed. In other words, we can state with 95% confidence that genes lying outside of the mask are differentially expressed. Because the standardized probability densities in different intervals are similar, γ is independent of the interval index j. To determine γ, a number of calibration array experiments and the percentile method were used. After normalization, any deviations of the LERs of the calibration arrays from zero are caused by noise. Ideally, γ should be chosen such that all genes are located inside the masks. However, because of significant noise in the data, an extremely large γ was needed to produce a mask that included all genes. For example, γ = 4.3, 5.1, and 3.9 were necessary for three pairs of nylon calibration arrays, one pair of plastic calibration arrays, and three calibration glass arrays, respectively. Hence, γ was chosen such that a maximum of 5% of all genes lie outside the mask for all calibration experiments. For nylon, plastic, and glass arrays, γ = 2.1, 2.3, and 2.0, respectively. More stringent masks can be constructed by increasing the value of γ. As in the case of normalization, the determination of the SD was performed after excluding highly differentially expressed genes. It should be noted that an improvement in the similarity of standardized LER probability densities will make masks more robust and accurate, and such an improvement can be achieved by a better standardization or discretization protocol.
Results and Discussion
Normalization of Array Data.
Fifteen pairs of nylon arrays and six pairs of plastic arrays hybridized with T cell samples (20% vs. 5% pO2) were used for normalization (Fig. 8, which is published as supporting information on the PNAS web site). Normalization of glass array data were performed with 22 glass arrays cohybridized with various pairs of C. acetobutylicum samples. Three representative examples are depicted in Fig. 1. After normalization, the LERs were distributed around zero for all three array types (Figs. 1 and 5a).
If a normalization method is perfect, the ratio of the normalized expression ȳ to the expression x should be close (because of random errors) to 1. To compare different normalization methods, the following criterion is proposed based on the normalization error:
 |
8 |
where n is the total number of genes, and p is the number of membrane pairs or arrays. The smaller Jnorm_error, the better the normalization. Although most Jnorm_error values range between 0 and 1, poor normalization methods will result in values >1 (see below).
The proposed normalization method is referred to as the segmental nearest neighbor LER mean method (SNN-LERM). The search for the optimal number K of nearest-neighbor genes and the optimal number M of intervals was performed by normalizing the array data with varying M and K values. If K is small, an interval is underrepresented by the K nearest neighbor genes and the normalization will not be very accurate (Fig. 9a, which is published as supporting information on the PNAS web site). At K = 0, the resulting normalization corresponds to the case of no normalization. If K is large and approaches the total number of genes n, the quality of SNN-LERM is approximately that of the global LER mean (G-LERM) normalization. A small M will result in a normalization method neglecting the ratio-intensity nonlinear relationship (as in global normalization). As M increases, the normalization quality first improves, but eventually plateaus as M increases further (Fig. 9b). The optimal M and K values for nylon, plastic, or glass arrays, which were calculated by minimizing Jnorm_error, were 20, 45, 25 and 250, 300, 200, respectively.
For all three forms of DNA arrays, SNN-LERM produces the lowest normalization error (Fig. 3). For membrane arrays, the proposed method is only slightly better in terms of the total normalization error, when compared with other median and LER methods (Fig. 3 a and b). For pairwise (between the corresponding 5% and 20% pO2 T cell samples) normalization, the difference between normalization and no normalization was small, because the samples were processed at the same batch, and thus system errors were small. Often, different membrane arrays are compared with a single array (e.g., array hybridized with a global RNA pool; ref. 19). In this case, system errors are much greater (e.g., because of various batches of sample processing), and hence, compared with the no normalization case, the normalization error is reduced considerably (>45%) when using the proposed method (Fig. 3b). For glass arrays, normalization based on SNN-LERM was far better than normalization based on any global method. The improvement in normalization using SNN-LERM was also considerable (>11%) compared with other methods such as rank-invariant nonlinear regression (Fig. 3c). We also examined the SNN-LER median method and found it to give results similar to those obtained using SNN-LERM (Fig. 3). Among various normalization methods, we demonstrated that the proposed method delivers superior results.
Figure 3.

Comparison of different normalization methods for 15 pairs (30 arrays) of nylon arrays (pairwise normalization) (a), 30 nylon arrays (all normalized to the first array) (b), and 22 glass arrays (c). NN, no normalization.
Identification of Differentially Expressed Genes.
After SNN-LERM normalization, 15 pairs of nylon arrays hybridized with samples taken from four sets of T cell cultures (20% vs. 5% pO2) and nine C. acetobutylicum glass arrays were used to compare different gene-identification methods. The proposed method of using SNN genes and LER SD (SNN-LERSD) delivered dynamic upper and lower mask boundaries, in contrast to the rigid minimal fold change with an intensity threshold (MF&T) and the expression ratio probability density (ERPD) methods (Fig. 4). Unlike the mask contours (CHM) of Newton et al. (11), SNN-LERSD masks for membrane arrays are broader at lower intensities and narrower at higher intensities, allowing identification of differentially expressed genes of both higher-fold changes at lower intensities and lower-fold changes at higher intensities (Fig. 4a). Successful identification includes properly identifying (i) differentially expressed genes and (ii) nondifferentially expressed genes, while avoiding misidentification of (iii) nondifferentially expressed genes as differentially expressed, (iv) differentially expressed genes as nondifferentially expressed, and (v) significantly up-regulated genes as down-regulated or vice versa. Let us assume that ndi and nnd genes are known to be differentially and nondifferentially expressed, respectively. The total identification error Jiden_error may be defined as
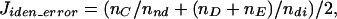 |
9 |
where nC to nE are the numbers of genes in the above defined categories iii–v, respectively. Jiden_error values range from 0 to 1, and ideally should be equal to 0.
Figure 4.

Comparison of different gene identification methods for a T cell pair hybridized on plastic arrays (a) and a C. acetobutylicum 824(pSOS95del)-824(pGroE1) pair cohybridized on a glass array (b). ○, Array data; ♦ and ■, identified by Q-RT-PCR as up-regulated and nondifferentially expressed, respectively.
A robust method for assessing the absolute and relative accuracy of the proposed gene-identification method can be based on the gene-expression analysis of the megaplasmid (pSOL1) deficient C. acetobutylicum strain M5 relative to WT (Table 1). Strain M5 is isogenic to WT but lacking the pSOL1 plasmid. Up to 178 genes, which are expressed with a broad range of levels in WT, therefore, have null expression in M5 and can be used for method validation. All 178 ORFs of pSOL1 are included on the C. acetobutylicum glass slides. Thus, these 178 pSOL1 genes are expected to be classified as “down-regulated” (if expressed in WT samples) or nondifferentially expressed (if not expressed in WT samples), when M5 samples are cohybridized with WT. Traditional methods such as MF&T and ERPD identified a number of “down-regulated” pSOL1 genes, but at the same time misidentified a few pSOL1 genes as “up-regulated”. Several misidentifications were made when using CHM, and even more when using ANOVA. SNN-LERSD properly identified the largest number of “down-regulated” pSOL1 genes and at the same time had no misidentifications of “up-regulated” pSOL1 genes. The strategy of using the C. acetobutylicum pSOL1 genes to validate gene-identification methods is unique in the DNA-array literature.
Table 1.
Comparison of different methods for identification of C. acetobutylicum pSOL1 genes in three sample pairs (six slides) of M5-WT experiments
| Gene identification method | No. of pSOL1 genes identified
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Up-regulated
|
Down-regulated
|
Nondifferentially expressed
|
|||||||
| I | II | III | I | II | III | I | II | III | |
| MF&T (Mf = 2.5, Th = 500) | 1 | 0 | 2 | 31 | 18 | 17 | 146 | 160 | 159 |
| ERPD (95%) | 1 | 0 | 3 | 36 | 12 | 23 | 141 | 165 | 152 |
| ANOVA (95%) | 8 | 5 | 6 | 40 | 23 | 19 | 130 | 144 | 153 |
| CHM (Po = 100:10) | 2 | 3 | 4 | 35 | 14 | 26 | 141 | 161 | 148 |
| SNN-LERSD (95%) | 0 | 0 | 0 | 44 | 38 | 27 | 134 | 140 | 151 |
Mf, minimal fold change; Th, threshold; Po, posterior odds; 95%, confidence level. Roman numerals indicate time points.
We also used T cell samples hybridized on nylon arrays to validate different gene-identification methods. Q-RT-PCR results for 10 genes from 15 pairs of 20% vs. 5% pO2 cultures (2 failed, leaving 148 RT-PCR measurements) were used to determine ndi and nnd (Table 2). Although fold changes obtained from methods such as Northern analysis and Q-RT-PCR are often used to validate array data (3, 7, 20), there is no established way for comparing fold changes obtained from such methods to array fold changes. To determine the differential status of genes based on Q-RT-PCR results, we first selected genes with their fold changes of replicate Q-RT-PCR results statistically significantly >1. Among those genes, genes with fold changes >1.5 were considered as differentially expressed. We chose the 1.5 value as a conservative benchmark to test the power of the various gene-identification methods. Based on this criterion, we obtained that ndi = 34 and nnd = 114. For various identification methods, different sets of nC to nE were determined, and the corresponding total identification errors were calculated. SNN-LERSD delivered the smallest identification error. With decreasing fold change and threshold in MF&T and increasing posterior odds in CHM, these two methods may be able to correctly identify more genes are differentially or nondifferentially expressed genes but at the cost of increasing misidentifications. EPRD, SNN-LERSD, and CHM (with posterior odds of 100:5) were similar in classifying differentially expressed genes. In identifying nondifferentially expressed genes, SNN-LERSD was, however, superior to EPRD and CHM by making far fewer misidentifications. Similar conclusions are obtained when a higher-fold change, such as 2, is used for selecting genes as differentially expressed by Q-RT-PCR, although the number of genes that meet this criterion is smaller, that thus, the ability to discriminate among the various gene-identification method is reduced. In summary, for plastic/membrane arrays, though our proposed normalization method performed only slightly better than other normalization methods (Fig. 3 a and b), the proposed overall (normalization and gene identification) method produces significant improvements (Table 2).
Table 2.
Comparison of Q-RT-PCR results for 10 genes in 15 pairs of 20% vs. 5% pO2 T cell cultures with different gene-identification methods
| Result with Q-RT-PCR | No. of genes identified by array analysis using
|
|||||
|---|---|---|---|---|---|---|
| ERPD (95%) | MF&T (Mf = 3; Th = 1,000) | MF&T (Mf = 2.2; Th = 500) | CHM (Po = 100:10) | CHM (Po = 100:5) | SNN-LERSD (95%) | |
| Differentially expressed (ndi = 34) | ||||||
| Differentially expressed | 14 | 4 | 10 | 9 | 16 | 15 |
| Nondifferentially expressed | 18 | 30 | 23 | 24 | 15 | 18 |
| Oppositely differentially expressed | 2 | 0 | 1 | 1 | 3 | 1 |
| Nondifferentially expressed (nnd = 114) | ||||||
| Nondifferentially expressed | 99 | 111 | 105 | 108 | 76 | 105 |
| Differentially expressed | 22 | 6 | 11 | 24 | 55 | 11 |
| Jiden_error | 0.36 | 0.45 | 0.39 | 0.39 | 0.43 | 0.32 |
Abbreviations are as in Table 1.
To further test the accuracy of SNN-LERSD, in C. acetobutylicum 824(pSOS95del)-824(pGroE1) glass array experiments, three genes identified by SNN-LERSD but not by other methods as differentially expressed and one gene identified by other methods (ERPD; 95% confidence level; MF&T with the minimal fold of 2.5 and threshold intensity of 500; CHM with posterior odds at 100:10) but not by SNN-LERSD (95% confidence level) as differentially expressed were chosen for Q-RT-PCR verification (two such genes are identified in Fig. 4b). Three genes identified by SNN-LERSD as differentially expressed were assayed as differentially expressed by Q-RT-PCR (average LERs of these three genes: 2, 1.4, 1.1). The array expression ratios of these genes were relative small (average LERs: 0.77, 0.83, 0.71), and only SNN-LERSD was able to identify them correctly. The Q-RT-PCR LER for the gene identified by SNN-LERSD as nondifferentially expressed was −0.26. In contrast to other methods, SNN-LERSD was able to discard this gene from being classified as differentially expressed (Fig. 4b).
Although Q-RT-PCR results demonstrate that the proposed gene-identification method is superior to other methods, it should be noted that neither Q-RT-PCR nor DNA-array analysis is error free. The discrepancy in the number of differentially expressed genes identified by Q-RT-PCR and DNA-array analysis may be due to the fact that array fold changes are smaller compared with Northern analysis or Q-RT-PCR (3, 7). Thus, a large number of genes assayed by Q-RT-PCR as differentially expressed with a fold change of 1.5 are judged as nondifferentially expressed by array analysis (Table 2). As a result, array analysis is conservative in predicting differentially expressed genes compared with Q-RT-PCR. At the same time, when using SNN-LERSD, array analysis produces few false positives.
Supplementary Material
Acknowledgments
We thank Dr. N. Jafari of the Center for Genetic Medicine, Northwestern University, and L. T. Huang and J. Beamish for assistance. We acknowledge the use of Keck Biophysics and Center for Genetic Medicine facilities at Northwestern University. This work was supported by National Science Foundation Grant BES-9905669, National Institutes of Health Grant R01-GM65476, and a Whitaker Foundation Graduate Fellowship (to H.H.).
Abbreviations
- CHM
contour based on hierarchical modeling
- ERM
expression-ratio mean
- ERMD
expression-ratio median
- ERPD
expression-ratio probability density
- LER
logarithm of expression ratio
- LERM
mean of logarithm of expression ratio
- MF&T
minimal fold change with an intensity threshold
- Q-RT-PCR
quantitative RT-PCR
- SNN
segmental nearest neighbor
References
- 1.Schena M, Shalon D, Heller R, Chai A, Brown P O, Davis R W. Proc Natl Acad Sci USA. 1996;93:10614–10619. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iyer V R, Eisen M B, Ross D T, Schuler G, Moore T, Lee G C F, Trent J M, Staudt L M, Hudson H, Jr, Boguski M S, et al. Science. 1999;283:83–87. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- 4.Tsien C L, Libermann T A, Gu X, Kohane I S. Pacific Symposium on Biocomputing. 2001;6:496–507. doi: 10.1142/9789814447362_0048. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Dougherty E R, Bittner M L. J Biomed Optics. 1997;2:364–374. doi: 10.1117/12.281504. [DOI] [PubMed] [Google Scholar]
- 6.Beissbarth T, Fellenberg K, Brors B, Arribas-Part R, Boer J M, Hauser N C, Scheideler M, Hoheisel J D, Schuetz G, Poustka A, et al. Bioinformatics. 2000;16:1014–1022. doi: 10.1093/bioinformatics/16.11.1014. [DOI] [PubMed] [Google Scholar]
- 7.Steidl U, Kronenwett R, Rohr U P, Fenk R, Kliszewski S, Maercker C, Neubert P, Aivado M, Koch J, Modlich O, et al. Blood. 2002;99:2037–2044. doi: 10.1182/blood.v99.6.2037. [DOI] [PubMed] [Google Scholar]
- 8.Tseng G C, Oh M K, Rohlin L, Liao J C, Wong W H. Nucleic Acids Res. 2001;29:2549–2557. doi: 10.1093/nar/29.12.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Y H, Dudoit S, Luu P, Lin D M, Peng V, Ngai J, Speed T P. Nucleic Acids Res. 2002;30:4–15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kao L C, Tulac S, Lobo S, Imani B, Yang J P, Germeyer A, Osteen K, Taylor R N, Lessey B A, Giudice L C. Endocrinology. 2002;143:2119–2138. doi: 10.1210/endo.143.6.8885. [DOI] [PubMed] [Google Scholar]
- 11.Newton M A, Kendziorski C M, Richmond C S, Blattner F R, Tsui K W. J Comput Biol. 2001;1:37–52. doi: 10.1089/106652701300099074. [DOI] [PubMed] [Google Scholar]
- 12.Kerr M K, Martin M, Churchill G A. J Comput Biol. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- 13.Haddad H, Papoutsakis E T. Cytotherapy. 2001;3:435–447. doi: 10.1080/146532401317248045. [DOI] [PubMed] [Google Scholar]
- 14.Hedge P, Qi R, Abernathy K, Gay C, Dharap S, Gaspard R, Hughes J E, Snesrud E, Lee N, Quackenbush J. BioTechniques. 2000;29:548–562. doi: 10.2144/00293bi01. [DOI] [PubMed] [Google Scholar]
- 15.Harris L M, Welker N E, Papoutsakis E T. J Bacteriol. 2002;184:3586–3597. doi: 10.1128/JB.184.13.3586-3597.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cornillot E, Nair R V, Papoutsakis E T, Soucaille P. J Bacteriol. 1997;179:5442–5447. doi: 10.1128/jb.179.17.5442-5447.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang H, Miller W M, Papoutsakis E T. Stem Cells. 2002;20:320–328. doi: 10.1634/stemcells.20-4-320. [DOI] [PubMed] [Google Scholar]
- 18.Cover D, Hart P. Proc IEEE Trans Inform Theory. 1967;11:21–27. [Google Scholar]
- 19.Garber M E, Troyanskaya O G, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, Rijn M, Rosen G D, Perou C M, Whyte R I, et al. Proc Natl Acad Sci USA. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajeevan M S, Vernon S D, Taysavang N, Unger E R. J Mol Diag. 2001;3:26–31. doi: 10.1016/S1525-1578(10)60646-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.