

Abstract
Neuronal intermediate filament inclusion disease (NIFID) is a frontotemporal lobar degeneration (FTLD) characterized by frontotemporal dementia (FTD), pyramidal and extrapyramidal signs. The disease is histologically characterized by the presence of abnormal neuronal cytoplasmic inclusions (NCIs) which contain α-internexin and other neuronal intermediate filament (IF) proteins. Gigaxonin (GAN) is a cytoskeletal regulating protein and the genetic cause of giant axonal neuropathy. Since the immunoreactive profile of NCIs in NIFID is similar to that observed in brain sections from GanΔex1/Δex1 mice, we speculated that GAN could be a candidate gene causing NIFID. Therefore, we performed a mutation analysis of GAN in NIFID patients. Although the NCIs of NIFID and GanΔex1/Δex1 mice were immunohistochemically similar, no GAN variant was identified in DNA obtained from well-characterized cases of NIFID.
Keywords: Neuronal intermediate filament inclusion disease, α-Internexin, Gigaxonin, Mutation analysis
1. Introduction
NIFID is a progressive, early-onset neurodegenerative disease leading to patient death within 3–5 years after clinical onset of symptoms. NIFID is easily distinguished from other FTLD entities by the abundant presence of pathological NCIs containing mainly α-internexin as well as other type IV IF (Cairns et al., 2004). So far, the screening for mutations in potential candidate genes such as type IV IF genes as well as NUDEL and SOD1 failed to reveal a genetic defect linked to NIFID (Momeni et al., 2006). Mutations in the gene encoding gigaxonin, a cytoskeletal regulating protein, cause giant axonal neuropathy (GAN) which is an early-onset neurodegenerative disease affecting both central and peripheral nervous system (Bomont et al., 2000). GAN is basically an autosomal recessive disorder with mutations in the gigaxonin gene affecting both alleles. Nonetheless, there is a report suggesting that heterozygous carriers of some gigaxonin mutations might exhibit mild neurological symptoms (Kuhlenbaumer et al., 2002). Our laboratory created a mouse deficient for GAN by disruption of exon 1 as a model for the human disease (Dequen et al., 2008). Of particular interest was the observation that the GanΔex1/Δex1 mice develop NCIs in the brain containing α-internexin similar to those NCIs reported in human NIFID. In view of the striking resemblance in NCIs immunoreactive profiles found in both GanΔex1/Δex1 mice and human NIFID, we have examined whether GAN was involved as a genetic component of NIFID.
2. Material and methods
Immunohistochemistry (IHC) was undertaken on 6-μm-thick sections prepared from formalin-fixed (human NIFID-1 and -2) paraffin wax-embedded tissue blocks as previously described (Cairns et al., 2004). For GanΔex1/Δex1 mice, brain sections were processed for IHC (Dequen et al., 2008).
Primer pairs are described in Tables S2 and S3. The corresponding amplicon size and used for PCR amplification as follows: 95 °C, 30 s; 58 °C, 1 min; 72 °C, 2 min; 40 cycles with Taq polymerase (Qiagen). Half of the 50 μl reactions were used for automated DNA sequencing.
3. Results and discussion
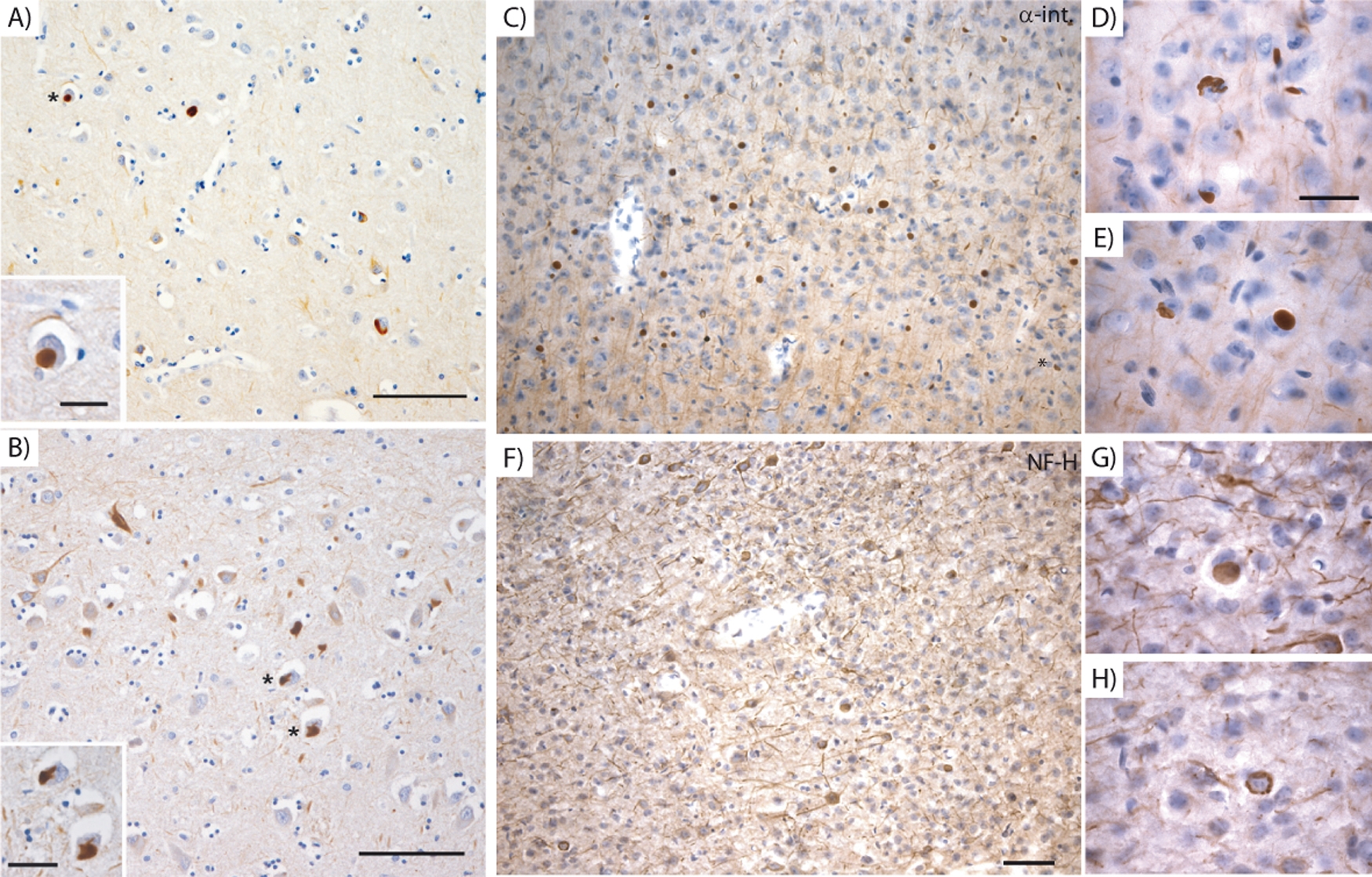
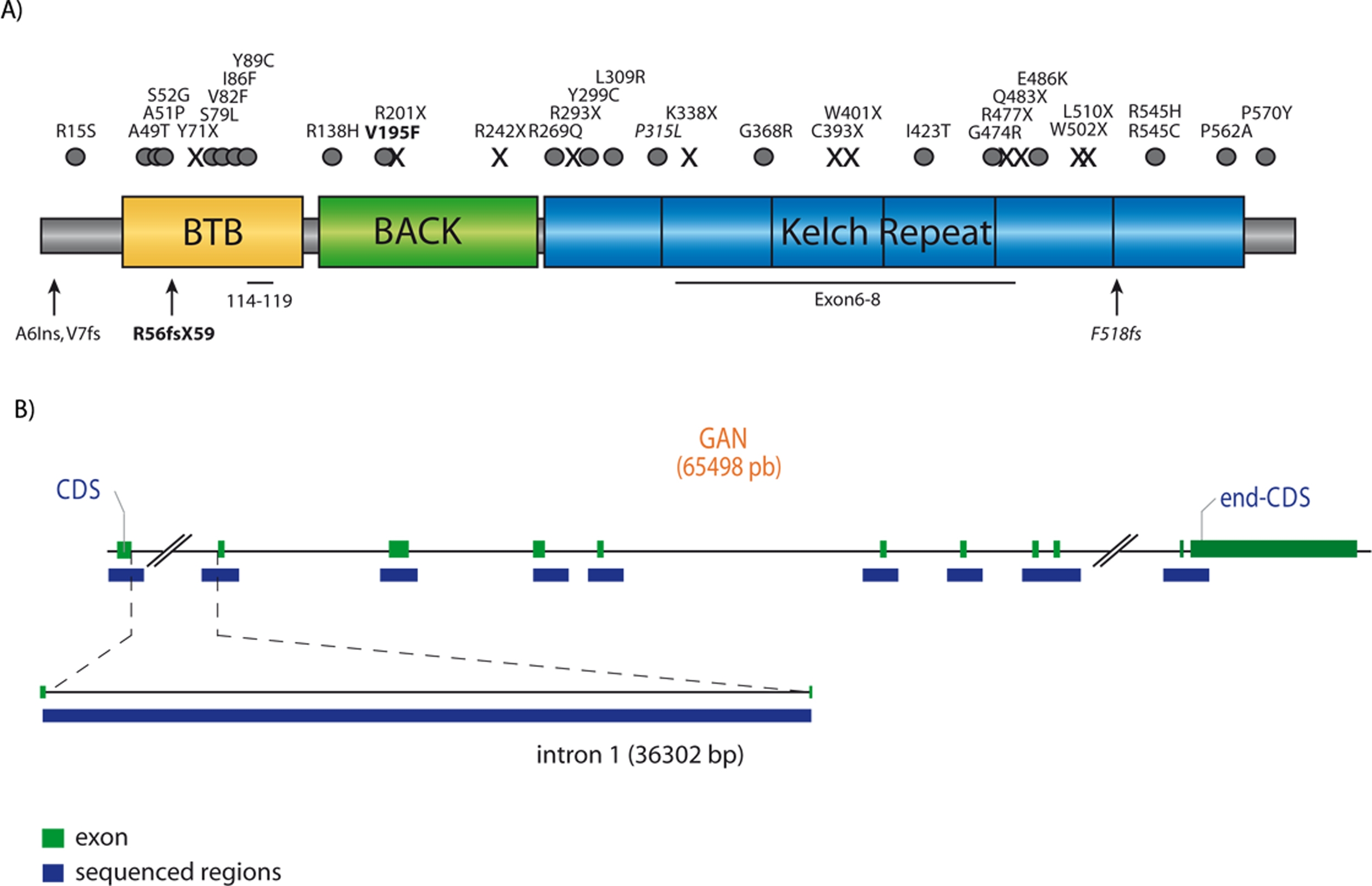
Brain sections from GanΔex1/Δex1 mice treated with antibodies against α-internexin showed a comparable immunostaining pattern to that seen in human NIFID, with abundant α-internexin NCIs in the cerebral cortex (Supp. Fig. 1A–C and F). The inclusions were also similar in diameter and density (cf. values Supp. Table 1). These data suggested that defects in GAN may be related to the NCIs phenotype seen in the human NIFID and prompted us to consider GAN as a potential genetic candidate for mutation analysis in cases of NIFID. DNA was extracted from brain samples of two NIFID sporadic patients (Supp. Table 2). Primers were designed to target the GAN entire coding sequence, including exon–intron junctions as well as intron 1 (Supp. Fig. 2A; Supp. Tables 3 and 4). Analyses of amplified sequences for GAN mutations revealed no insertion, deletion or substitution for both patients (Supp. Fig. 2B). Thus, we conclude that the NIFID phenotype in these two patients is not caused by GAN mutations. Nonetheless, NIFID is a disease of variable clinical phenotypes and the possibility of GAN mutations in a subset of patients with NIFID cannot be entirely excluded. More studies are needed to clarify whether both diseases share a common defective pathway leading to the formation of α-internexin inclusions.
Supplementary Material
{kind=link}
{kind=link}
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.neurobiolaging.2009.08.018.
References
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tuysuz B, Landrieu P, Hentati F, Koenig M. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat. Genet. 2000;26:370–374. doi: 10.1038/81701. [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Uryu K, Bigio EH, Mackenzie IR, Gearing M, Duyckaerts C, Yokoo H, Nakazato Y, Jaros E, Perry RH, Arnold SE, Lee VM, Trojanowski JQ. alpha-Internexin aggregates are abundant in neuronal intermediate filament inclusion disease (NIFID) but rare in other neurodegenerative diseases. Acta Neuropathol. (Berl.) 2004;108:213–223. doi: 10.1007/s00401-004-0882-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dequen F, Bomont P, Gowing G, Cleveland DW, Julien JP. Modest loss of peripheral axons, muscle atrophy and formation of brain inclusions in mice with targeted deletion of gigaxonin exon 1. J. Neurochem. 2008;107:253–264. doi: 10.1111/j.1471-4159.2008.05601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlenbaumer G, Young P, Oberwittler C, Hunermund G, Schirmacher A, Domschke K, Ringelstein B, Stogbauer F. Giant axonal neuropathy (GAN): case report and two novel mutations in the gigaxonin gene. Neurology. 2002;58:1273–1276. doi: 10.1212/wnl.58.8.1273. [DOI] [PubMed] [Google Scholar]
- Momeni P, Cairns NJ, Perry RH, Bigio EH, Gearing M, Singleton AB, Hardy J. Mutation analysis of patients with neuronal intermediate filament inclusion disease (NIFID) Neurobiol. Aging. 2006;27:778 e771–778 e776. doi: 10.1016/j.neurobiolaging.2005.03.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.