

Abstract
The review (with 214 references cited) is devoted to moenomycins, the only known group of antibiotics that directly inhibit bacterial peptidoglycan glycosytransferases. Naturally occurring moenomycins and chemical and biological approaches to their derivatives are described. The biological properties of moenomycins and plausible mechanisms of bacterial resistance to them are also covered here, portraying a complete picture of the chemistry and biology of these fascinating natural products
1 Introduction
Moenomycin A (1, Fig. 1), the founding member of the moenomycin family of antibiotics, was first described in 1965.1-3 The majority of known moenomycin-type natural products were discovered by the end of the 1970s, although exact structures have been determined only for some members of the family.4,5 Moenomycins are classified as phosphoglycolipids based on their chemical composition. They contain 3-phosphoglyceric acid (3-PG), a unique structural element among bacterial secondary metabolites. The C2-hydroxy group of 3-PG is decorated with an unusual isoprenoid chain and the phosphate group is attached to the reducing end of a structurally complex pentasaccharide.
Fig. 1.

Moenomycin and its target. (A) Structures of bacterial cell wall-active antibiotics moenomycin A (1), penicllin G, and vancomycin. The building blocks of 1 are marked with capital letters A-H (red). (B) The extracellular steps of bacterial peptidoglycan biosynthesis and sites of antibiotic inhibition. Penicillin binding proteins (PBPs) are shown with both peptidoglycan glycosyltransferase (PGT) and transpeptidase (TP) domains. Black rectangles indicate the lipid carrier, undecaprenyl pyrophosphate. The sites of antibiotic action are indicated by red lines; the green arrow indicates the PGT reaction.
The moenomycins are potent antibiotics that operate through a common mechanism6: they directly inhibit peptidoglycan glycosyltransferases (PGTs) involved in the penultimate step of bacterial cell wall biosynthesis.7,8 The minimal inhibitory concentration (MIC) of 1 against various Gram-positive bacteria ranges from 1 ng/mL to 100 ng/mL.9 On a molar basis, 1 is 10-1000-fold more potent than vancomycin, a clinically useful glycopeptide that also targets a late stage of bacterial cell wall biosynthesis (Fig. 1).10 Despite their potency, interest in developing moenomycins as novel drugs dissipated because of their suboptimal pharmacokinetic properties8, 11 (discussed later in this review) combined with the availability in the 1960s and 70s of more tractable antibiotics. Nevertheless, moenomycins were successfully commercialized as animal growth promoters under the trademarks Flavomycin and Flavophospholipol (a natural mixture of structurally related phosphoglycolipids).12 The long-term consequences of antibiotic use in animal nutrition remain a subject of intense debate12, 13-15, since such a practice may eventually narrow down the options for treatment of human diseases.16 In 1985 all animal feed antibiotic additives were banned in Sweden17 and in 2006 this ban came into effect in the entire territory of the European Union.18 Flavomycin is still approved in the US and many other countries for cattle, swine and poultry. During the decades-long use of this product there have been no reports of animal microflora significantly resistant to moenomycin.12, 19
The unprecedented rise of multidrug resistant pathogens in recent years20 has revived interest in underexploited antibiotics such as 1.21,22 As a result, the molecular mechanisms of moenomycin action are now clearer, and better chemical and biological tools have been developed to generate novel phosphoglycolipids for various purposes. In the past, several reviews have covered the occurrence of moenomycin antibiotics in nature as well as their chemistry and biology.4,8,9,12,21,23-30 Some of these reviews are now outdated, while others do not provide a complete overview of the tripartite complexity of chemical manipulations, biosynthesis and the biological properties of moenomycins. The present review aims to provide an exhaustive and updated account of different aspects of the chemistry and biology of moenomycin family antibiotics. Although much effort has been put into chemical syntheses of building blocks of 1, such as monosaccharides, moenocinol, the C5N chromophore and analogs thereof 4, 31-35, these “building blocks” and lipidless oligosaccharide derivatives of 1 have no measurable antibiotic or PGT inhibitory activities. For this reason, the synthesis of only lipid-containing moenomycin derivatives and analogs having two or more sugar units will be covered here. The biosynthesis of 1 and prospects for isolation of novel phosphoglycolipids via genetics-guided approaches will be reviewed. Structure-activity relationship studies and a description of the antibiotic properties of moenomycins from the points of view of biochemistry, structural biology and microbiology will conclude this review.
2 Occurrence in nature
1 is major component of a mixture of phosphoglycolipids known as moenomycins (synonyms: flavomycin, bambermycins, flavophospholipol). Here we use the term “moenomycin” as a family name for all known phosphoglycolipid natural products. 1 is produced by at least four streptomycete strains, S. ghanaensis (ATCC14672), S. bambergiensis (ATCC13879), S. ederensis (ATCC15304) and S. geysiriensis (ATCC15303).1 The composition of the flavomycin complex produced by S. ghanaensis has been thoroughly studied.4 As a result, a number of phosphoglycolipids have been isolated and structures of moenomycins A 1, A12 2, C1 3, C3 4, C4 5 have been determined.36-43 All of these compounds contain a core BCEF tetrasaccharide attached to the moenocinol-phosphoglycerate moiety (Fig. 2). The structural diversity of the S. ghanaensis moenomycins stems from 5 variables. These include: the absence/presence of the branching glucose unit D; the structure of sugar units C and/or E, which can be either N-acetyl glucosamine or its 6-deoxy version, chinovosamine; the stereochemistry and further functionalization of position 4 of moenuronamide unit F (D-galacto versus D-gluco; there is an axial methyl group in the latter case); finally, in the majority of moenomycins the carboxylic acid group of galactopyranuronic acid unit B can be either linked to a 2-aminocyclopentane-1,3-dione (C5N) chromophore unit A, or converted into a carboxamide, or extended with glycine. (The latter modification, found in moenomycins G and H44 as well as some prasinomycins45, was inferred only from degradation studies and thus more rigorous structural analysis is required to confirm it). Four minor components of the flavomycin complex, 6-9, were also recently identified with the help of high-resolution tandem mass-spectrometry.46 Two of them (8 and 9) were subsequently rediscovered as the main products (nosokomycins A and B) of Streptomyces sp. K04-0144. In addition to 8 and 9, the strain also accumulates nosokomycins C 10 and D 11.5 The diversity of moenomycins is a testament to the important role of promiscuous catalysts and modularity in natural product biosynthesis.
Fig. 2.

Structures of naturally occuring moenomycins, as judged from MS and NMR experiments (except for 6 and 7, which are deduced only from MS analysis). The molecular mass of each compound (anion) is indicated (rightmost column).
Little is known about other members of the moenomycin family. Pholipomycin 12 (produced by S. lividoclavatus ATCC29865)38, 47, 48 and AC326-alpha 13 (from unidentified Actinomyces sp. AC326)49 are the only compounds that were structurally characterized. The latter resembles 1 in the oligosaccharide part but carries a C25 lipid isomer, diumycinol (Fig. 2), instead of the moenocinol lipid. This lipid was first identified in other phosphoglycolipid antibiotics, the diumycins (macarbomycins; produced by S. phaeochromogenes ATCC21346 and S. umbrinus ATCC15972), but this group of antibiotics has not been characterized beyond initial degradation studies.50-52 Other phosphoglycolipid antibiotics of Streptomyces origin, such as prasinomycins,45, 53 8036RP (quebemycin),54 11837RP,55 19402RP,56 ensanchomycin,57 prenomycin,58 are not characterized well enough to tell how they differ from the already described moenomycins. Teichomycin A1, a phosphoglycolipid antibiotic related to 1, is the only member of the family that has been isolated from a non-streptomycete strain, Actinoplanes teichomyceticus (ATCC31121, the producer of the clinically important lipoglycopeptide teicoplanin).59 The exact structure of teichomycin A1 remains unknown. All of the described phosphoglycolipids are very potent antibiotics, although those possessing unit F in the D-galacto configuration (2 and 3) are less active.9, 40
3 Moenomycins as a target of chemical synthesis and degradation experiments
1 poses a formidable synthetic challenge. Five different glycosidic linkages, including two beta linkages to C2 acetamido sugars, link five different carbohydrates, bristling with functional groups, to a lipid phosphoglycerate. The complexity of 1 has made it an excellent target for chemists. Derivatives and fragments of moenomycins, obtained through degradation and/or synthesis, have provided valuable insights into the pharmacology and mode of action of this family of PGT inhibitors.8, 21 As the glycosidic bonds of 1 have counterparts in other biologically relevant molecules, synthetic solutions developed in the course of chemical manipulations of 1 have applications beyond the moenomycin field.60
3.1 Di- and trisaccharide moenomycin analogs via chemical degradation and synthesis
Stepwise degradation of the decahydro-derivative of 1 (obtained through hydrogenation of 1 in methanolic solution61 afforded many moenomycin disaccharide (14, 19, 20)62, 63 and trisaccharide (24-26) analogs39, 40, 62 containing a saturated C25 lipid chain (Fig. 3). In addition, trisaccharide 30, containing the natural lipid, moenocinol, was generated via a Barry degradation.40
Fig. 3.

Di- and trisaccharide fragments and analogs of 1 obtained through degradation and chemical synthesis.
Except for 31 (Fig. 3) all synthetic moenomycin fragments were modelled after the proposed pharmacophore portion of 2, e.g., the lipidated C-E-F trisaccharide having the moenuronamide (F ring) in the galacto configuration. Unlike 1, 2 lacks the C4-methyl group in unit F, which simplifies chemical synthesis greatly, albeit at a cost of biological relevance, since 2 is less active than 1 (as discussed later in this review). A number of EF disaccharide analogs (15-18, 21-23) were prepared using different strategies.64-69 Scheme 1 shows one such route, which led to 16. The route features relatively mild glycosylation conditions and an efficient solution to introduce the carbamate into unit F. The resultant disaccharides differ mainly in the structure of the lipid chain and substituents at the C4 position of unit F (Fig. 3). Some of these disaccharides were used as precursors for the synthesis of trisaccharides, although the majority of 27-31 were prepared independently.39, 69-72 The diversity of synthetic approaches employed to make these molecules reflects the complexity of the target and the difficulty of making some of the glycosidic bonds stereoselectively using available glycosylation methodology.
Scheme 1.

Synthesis of 16
A solid phase glycosylation approach was reported for the construction of a disaccharide library mimicking the EF portion of 1,73 but containing simplified phospholipids. The sulfoxide glycosylation method was used to make the EF glycosidic bond, and the preconstructed disaccharide was attached to a photolinker resin. After derivatization of the disaccharides and their release from the resin, simplified phospholipid moieties (some of which lacked the carboxylate at the phospho-end, which is an essential part of moenomycin pharmacophore8, 74 were attached. A disaccharide library of 1300 members was created and several compounds (for example, representative member 32) were identified that inhibited bacterial growth at low microgram concentrations.75 Although some of these compounds were shown to interfere with the transglycosylation reaction, their mechanism(s) of PGT inhibition may be different from that of 1 since they contain functionalization at a position later discovered to be buried in the active site cleft,76-79 and additional targets of action were not excluded.80, 81
3.2 Moenomycin analogs with modified unit A
A number of synthetic moenomycin analogs were reported that carry various substituents instead of chromophore unit A, providing orthogonal reactive groups for further derivatization (Fig. 4). In one approach to such analogs, treatment of 1 with different aryl diazonium salts produced, via a Japp-Klingemann reaction, compounds that contain o-nitrobenzenediazonium (33) or various substituted triazole moieties (for example, 34-38).82-84 34, featuring a free thiol group, provided access to biotinylated and dansylated analogs 39-41.83, 85, 86 Other triazole derivatives include moenomycin dimers87 as well as analogs fused to an aminocoumarin moiety (42),88 various fluorescent labels (for example 43, carrying fluorescein isothiocyanate)85 and ampicillin (44).84 Another approach, based on thiouronium salt chemistry, also yielded a number of moenomycins with a modified unit A, exemplified by 45.89 Fundamental studies on cell wall biosynthesis were the main area of application of the A ring-modified moenomycins.88, 90-92 Fluorescein-labeled moenomycin was used for the development of a fluorescence anisotropy-based high-throughput assay for PGT inhibitors.87
Fig. 4.

Synthetic moenomycins that resulted from modification or replacement of A ring.
3.3 Moenomycin analogs with a modified lipid portion
Besides the generation of bioactive moenomycins with saturated lipid chains via hydrogenation (e.g. trisaccharides 24-29, 31 and their pentasaccharide precursors,61 several other approaches were developed to replace moenocinol with various functional groups (Scheme 2). Ozonolysis in methanolic solution of 1 afforded aldehyde 46, lacking lipid and chromophore units, which was easily converted to primary alcohol 47.93 Different analogs with modified lipid chains were obtained either through indium-mediated Barbier-type reactions of 46 with allylic and benzylic halides (for example, 48-50),94 or reductive aminations of 46 (compounds 51-54).95
Scheme 2.

Moenomycins with modified lipid moiety.
An efficient strategy has been developed for the degradation of 1 to an intact pentasaccharide lactol carrying the ring, and the subsequent reconstruction of 1 from this pentasaccharide and synthetic moenocinyl glycerate.96 Access to the aforementioned lactol paves the way for thorough functional exploration and manipulation of 3-PG and lipid moieties of 1. As a proof of principle, novel nerylmoenomycin 55, containing a 10-carbon lipid, was synthesized using the developed chemistry. Several other derivatives 56-61 in which the phosphoglyceric acid moiety was altered were subsequently made74 in order to probe the role of the carboxylate and the negative charge on the phosphate.
3.4 Synthetic PGT inhibitors inspired by moenomycin
C-phosphonate disaccharides 62 and 63 were designed to mimic features of 1 and lipid II (the peptidoglycan precursor substrate used by PGTs). These rather simple compounds, remotely related to 1, showed modest (62) or no (63) PGT inhibition activity in vitro.97 Compound 64, which consists of the vancomycin aglycon and the disaccharide moiety of 32, was inspired by the concept that the lipid moiety of 1 could be replaced with another group that would deliver 1 to its PGT target at the cell membrane. The vancomycin aglycon binds D-Ala-D-Ala, which is found in the lipid II substrate of the PGTs. The antibacterial activity of this hybrid glycopeptide against several clinically relevant cocci was better than that of 32 itself or vancomycin,98 suggesting that it might be possible to replace the lipid portion of moenomycin, proposed to target the compound to bacterial membranes, with other targeting groups.
3.5 The total synthesis of moenomycin A
The synthetic compounds described above highlight many of the challenges involved in making analogs of 1, ranging from technical challenges (low yields, poor solubility of intermediates in suitable solvents, and deprotection issues) to conceptual ones (e.g., how to construct the glycosidic linkages efficiently and with stereochemical control). Only recently have these challenges been surmounted, culminating after decades of effort in the first total synthesis of 1 (Scheme 3). This achievement, in conjunction with the aforementioned moenomycin degradation-reconstruction approach, deploys a powerful toolkit for chemical synthesis of different types of moenomycin fragments and analogs.60, 99, 100
Scheme 3.

The total synthesis of 1.
The oligosaccharide portion of 1 was entirely constructed using sulfoxide glycosylation chemistry,101, 102 although multiple adjustments to and modifications of the standard procedure had to be made to stereoselectively generate several of the linkages.99 The pentasaccharide was built in a modular manner in which disaccharides BC and EF were produced first, then the CE bond was formed between the disaccharides to give the BCEF tetrasaccharide. The branching D ring sugar was then attached and the chromophore was installed on the pentasaccharide. Finally, using chemistry developed for the degradation and reconstruction of 1,96 moenocinyl-3PG was attached. The synthesis of the deprotected pentasaccharide-chromophore intermediate without the lipid takes 42 steps from known building blocks and the overall yield is 1%. This synthetic route provides access to key fragments of 1, including the EF, CEF, BCEF, and BCDEF saccharides. Structural units of 1 which are not essential for biological activity (e.g., rings A and D) or are suboptimal from a pharmacokinetic standpoint (e.g., the moenocinol lipid) are introduced at the end of the synthesis. Therefore, alternative lipids/chromophores can be readily attached to obtain molecules having potentially better pharmacological properties. Other strategies to make 1 have also been evaluated,60, 99 and more can be envisioned; however, the intuitive solution to utilize the EF disaccharide as a starting point for moenomycin oligosaccharide synthesis appears to offer the greatest freedom to chemically manipulate 1.
4 Biosynthesis of moenomycins
At the time of this writing, the only explored phosphoglycolipid biosynthetic pathway is that of S. ghanaensis ATCC14672. This strain is a model for biological studies on 1 and genetic manipulations of the bacterium began with the development of cloning vectors.103-105 S. bambergiensis, another producer of the flavomycin complex, was also a subject of early genetic experiments, which revealed the instability of moenomycin production upon the introduction of foreign plasmids.106-109 Apart from general endeavors into the biology of the producing strains, much of the early work on moenomycin biosynthesis relied on feeding experiments.110 These pioneering studies elucidated the metabolic origins of different parts of 1 and helped decipher the genetic basis of moenomycin biosynthesis.
4.1 Studies conducted prior to cloning of the moenomycin biosynthetic genes
Streptomycetes use both the non-mevalonate (also known as MEP or DOXP) and mevalonate pathways to make isopentenyl diphosphate units. The non-mevalonate pathway is used for housekeeping needs, while the mevalonate route is usually reserved for secondary metabolism.111 Nevertheless, tracer experiments revealed that the C25 chain of 1 is of non-mevalonate origin.112 The irregular structure of moenocinol suggests a unique mode of assembly, which was proposed to involve coupling of a C15 precursor to geranyl pyrophosphate, as depicted in Scheme 4.113, 114 The proposed mechanism involves attack by an internal carbon in the C15 chain, which sharply contrasts with the usual “head-to-head” and “head-to-tail” strategies to join isoprene units. UV-induced S. ghanaensis mutants were reported to produce moenomycins with shorter lipids, which were not rigorously characterized because of technical difficulties.115 These data provided initial evidence that moenocinol is made from C15 and C10 units, although the order of assembly of different parts of 1 remained unknown.
Scheme 4.

Proposed mechanism for rearrangement during biosynthesis of lipid portion of 1.
Feeding of radioactively labeled acetate to S. ghanaensis revealed the expected origin of the acetyl and carbamoyl groups of oligosaccharide moiety of 1.112 The C5N unit of 1, amide-linked to the B ring, was shown to originate from 5-aminolevulinic acid.110, 112 The latter is an essential metabolite that serves as a starting material in heme and corrin biosynthesis; in the case of moenomycins and several other C5N-containing antibiotics116 5-aminolevulinic acid was proposed to undergo an unusual cyclization to give a five-membered ring structure containing a free amine group.
4.2 Identification of S. ghanaensis genes for moenomycin biosynthesis
Moenomycins contain several structural features (e.g., the C6-deoxysugar chinovosamine, the carbamate, the prenyl chain) that lend themselves to cloning the underlying genetic determinants via the PCR-based approaches that have worked so well for identification of other classes of NPs.117 Although a number of candidate moenomycin biosynthetic genes were cloned, most of them were disqualified during further functional verification. A single exception was the 5-aminolevulinate synthase (ALS) gene moeC4, which was cloned from the S. ghanaensis genome118 using degenerate primers based on the reported sequences of ALS genes involved in the biosynthesis of the C5N-containing antibiotics asukamycin and ECO-02301.119, 120 Two other genes, moeA4 and moeB4, putatively involved in C5N formation and transfer, respectively, were found near moeC4 (Fig. 5), and the involvement of this three-gene cluster (referred to as moe cluster 2) in the production of 1 was verified via gene disruption. In particular, an S. ghanaensis strain deficient in the putative acyl-CoA ligase gene moeA4 accumulated 9. No other genes for the biosynthesis of 1 could be identified in the vicinity of moe cluster 2; furthermore, no other candidates cloned via PCR approaches led to the identification of additional moenomycin biosynthetic genes. Various other in vivo strategies were tested to identify possible biosynthetic genes, but none proved successful. Ultimately, in the first example of targeted natural product gene cluster discovery via whole genome sequencing, the S. ghanaensis genome was shotgun–sequenced and partially assembled to yield 1018 contigs that covered almost 80% of the chromosome.118 Scanning of the contigs for clustered glycosyltransferase (GT), sugar tailoring and prenyltransferase genes revealed the main moe cluster 1 (Fig. 5), which was verified by gene disruption and heterologous expression. For example, disruption of the moeM5 gene, proposed to encode a carbamoyltransferase, led to accumulation of descarbamoylated moenomycin 65, confirming the proposed role of MoeM5 in moenomycin biosynthesis. Additionally, expression of most of moe cluster 1 in S. lividans TK24 gave compound 66.118
Fig. 5.

Genetic organization of moe clusters 1 and 2. Red bidirectional arrows indicate moe genes that are likely to arise from duplication event.
The organization of the moe genes is notable for several reasons. First, the genes are split into two clusters, which reside in subtelomeric regions of the S. ghananesis chromosome (as judged from available sequence data at http://www.broadinstitute.org/annotation/genome/streptomyces_group/), a common, but not exclusive, location of actinobacterial secondary metabolic pathways.121 Notably, the main moe cluster 1 contains two genes, moeB5 and moeA5, that are highly homologous to moeA4 and moeC4, in moe cluster 2. However, moeB5 and moeA5 seem to be nonfunctional due to in-frame deletions. It is likely that moe cluster 2 is a result of translocation and divergence following the duplication of the ancestral moeB5-moeA5 segment.
The low sequence similarity of the majority of moe genes to other known secondary metabolism genes is a second salient feature of the moe pathway. It partially explains why PCR strategies failed to identify the moe genes. For example, of the five GTs in moe cluster 1, only MoeGT1 is predicted to contain a GT-B superfamily fold, a typical fold for antibiotic GTs.122 A third notable feature is that there are no pathway-specific regulatory or resistance genes within either of the moe clusters. Moreover, genes for the supply of activated sugars and isoprene units are absent as well. One has to assume that, except for chromophore unit A, all precursors of 1 are taken directly from primary metabolism and assembled with few modifications. Thus, the moenomycin pathway demonstrates a high degree of genetic and metabolic parsimony through extensive use of shared cellular resources. Finally, there is high degree of similarity between several Moe proteins, namely MoeF5 and MoeH5, MoeGT4 and MoeGT5, and MoeD5 and MoeJ5. Hence, duplications apparently played a role not only in the generation of the separate moe cluster 2, but also during evolution of the main moenomycin biosynthetic cluster.
4.3 Moenomycin biosynthetic logic and generation of novel derivatives
4.3.1 Step-by step description of sequence of reactions leading to moenomycin A
The biosynthetic pathway to 1 was inferred from metabolite analysis of S. lividans strains harboring different moe gene sets supplemented with in vitro reconstitution experiments.123 The sequence of reactions to generate 1 is summarized in Figures 6-8, taking into account that unit C can be either GlcNAc or its C6-deoxy derivative chinovosamine (due to the promiscuity of the respective GT). Fig. 6 also shows a shunt product 84, which accumulates as a result of deletion of moeK5 gene. Biosynthesis of 1 is proposed to be a seventeen-step process, which begins with formation of cis-farnesyl-3-PG 67 from 3-PG and trans-farnesyl pyrophosphate This reaction, with no currently known precedents in bacterial metabolism, is catalyzed by a TIM-barrel prenyltransferase MoeO5. Homologous TIM-barrel prenyltransferases are found in Archaea, but carry out a different enzymatic reaction and participate in the production of essential membrane lipids.124 Since the MoeO5 reaction product contains a cis-allylic ether bond, this enzyme apparently catalyzes double bond isomerization, a new feature for prenyltransferases that catalyze intermolecular coupling. 67 is then coupled to galacturonic acid by the GT MoeGT1, which uses as a substrate alpha-UDP-galacturonic acid provided from cellular pools of alpha-UDP-glucuronic acid by the epimerase MoeE5. The resulting monosaccharide 68, which retains the alpha configuration, is carboxyamidated by MoeF5, leading to 69. Both epimerization and carboxyamidation are crucial for the biosynthesis of 1, since moeE5 deletion blocks the first glycosylation and moeF5 deletion prevents the second, thus blocking the entire pathway. In contrast, methylation and carbamoylation of 68 or modification of other precursors or intermediates are not necessary to proceed from 68 to fully glycosylated moenomycins, such as 65 and 84. 84 accumulates when moeK5 is disrupted, implicating the encoded enzyme in methylation of the F ring. MoeK5 belongs to a small, unusual family of radical-SAM enzymes that are proposed to use methylcobalamin as a methyl group donor.125 Although feeding experiments had suggested an anionic methylation mechanism for unit F, which is common in nucleotide-sugar biosynthesis,110 ambiguity in the 13C-methionine labeling patterns126 and the absence of moe genes consistent with an anionic pathway support the hypothesis that MoeK5 employs a radical mechanism for methylation. Inversion of the C4 hydroxyl occurs concomitantly during the reaction. Further studies on MoeK5 will surely bring new insights into the highly unusual and pharmacologically relevant biotransformation at the F ring C4 hydroxyl.
Fig. 6.

Early steps of moenomycin biosynthesis. Solid arrows indicate the order of moenomycin assembly. Dashed line indicates product 84 accumulated as a result of loss of moeK5 gene function. Structural components shown in red indicate the positions of new moieties in the intermediates.
Fig. 8.

Final steps of moenomycin biosynthesis. The sequence of reactions leading from pentasaccharide precursors to 1 is proposed, but may involve other intermediates and/or enzymes.
Methylation occurs at the monosaccharide stage, as inferred from metabolite analysis, and the methylated monosaccharide 70 is then converted to disaccharide 71 (Fig. 6), which subsequently enters one of the two alternative glycosylation branches (Fig. 7). The trisaccharide stage (72 or 75/76) appears to be the earliest point when the C15 lipid moiety can be converted into the C25 moenocinol chain (compounds 73 or 77/78) and carbamoylation occurs (compounds 74 or 79/80). The prenyltransferase MoeN5 is responsible for formation of the C25 chain, catalyzing the unusual rearrangement of its central part, possibly via the mechanism shown in Scheme 4. Expression of moe cluster 1 in the absence of moeN5 leads to accumulation of compounds 85-88. The two glycosylation branches converge at the tetrasaccharide stage (81/82; Fig. 7), and MoeGT2 transfers the last sugar, galacturonic acid, leading to pentasaccharide 8/83 (Fig. 8).
Fig. 7.

Sequence of reactions leading from disaccharide precursor 71 to tetrasaccharide 81/82. Structural components shown in red, green and blue indicate the positions of new moieties in the intermediates.
Attachment of the chromophore to a pentasaccharide precursor of 1 is still poorly understood. Both moe gene knockouts and biochemical studies on other C5N-containing molecules suggest that the concerted action of the PLP-dependent aminolevulinate synthase MoeC4 and the putative acyl-CoA ligase MoeA4 lead to the C5N unit (Fig. 8). Then the putative amide synthase MoeB4 would catalyze attachment of the chromophore to 8/83 through attack of the C5N amine on an activated acid to form the amide.127 However, an additional gene, moeH5, has been shown to be important for chromophore attachment. MoeH5, a close homologue of the amidotransferase MoeF5, converts 8/83 into amides 9/65, adding an unexpected twist on the way to 1.123 From available genetic data it is likely that moeH5 and at least two moe cluster 2 genes (moeA4 and moeC4) are needed to produce 1 from 8/77 or 9/65, although the participation of other enzymes (e.g., MoeF5, MoeB4) and intermediates cannot be excluded (Fig.8). Accumulation of 9/65 as shunt products would be the most parsimonious explanation of the last stage of moenomycin biosynthesis, since it does not involve additional steps/enzymes on the way from 8/83 to 1. Biochemical assays of MoeB4 and MoeH5 are needed to illuminate this intriguing step of moenomycin biosynthesis.
The work described above establishes that 1, and, by extension, the entire class of phosphoglycolipid antibiotics, is produced via sequential glycosylation of a prenyl-3PG precursor. Overall, the moenomycin pathway is an excellent example of a highly convergent and flexible biosynthetic strategy. Out of the 6 sugar tailoring reactions, only two are obligatory; also, the order of glycosylation of disaccharide 65 is optional, as is production of the full-length C25 lipid moiety. Moreover, the GTs MoeGT4 and MoeGT5 can transfer either GlcNAc or chinovosamine, leading to production of pholipomycin 12 and moenomycin C3 4 in addition to 1. The entire spectrum of moenomycins produced by S. ghanaensis can be explained as a result of either unbalanced/absent expression of certain moe genes or the substrate promiscuity of glycosyltransferases and B ring tailoring enzymes (Fig. 6-8). Different moenomycin analogs can be readily accessed through combinatorial expression of moe genes, as evidenced by production of 72 and 79-82.77, 123
4.3.2 Regulatory aspects of moenomycin production
In actinomycetes, an intricate web of nutritional and genetic factors determines when and how much of a secondary metabolite is produced.128, 129 Little is known about either of these factors with regard to production of 1, which is extremely low (less than 1 mg l−1; shaken flask fermentation) in the wild type (ATCC14672) strain. Moenomycin production is dramatically decreased in presence of high (3-5%) concentrations of glucose in solid and liquid media,130 suggesting that this pathway is subject to catabolite repression. Also, an intensive oxygen supply and relatively high temperatures (e.g., 37 °C) improve and accelerate moenomycin production under conditions of submerged fermentations.112, 130
Since biosynthesis of 1 heavily relies on abundant cellular metabolites, there is probably no need for pathway-specific regulatory genes, which usually serve to coordinate the production of exotic building blocks and their subsequent assembly. Of known streptomycete regulatory systems, moe cluster 1 only contains elements of a bldA-based “switch”.118 In S. coelicolor, bldA encodes leucyl tRNAUUA, which in mature form is accumulated in significant quantities only in late stationary phase,131 effectively limiting the expression of all TTA+ genes.132 The TTA codon, the rarest one in streptomycetes, is embedded in genes associated with secondary metabolism and other auxiliary functions.133 In the moe clusters, moeA5, moeO5, and moeR5 contain 1 TTA codon while moeE5 contains two. By analogy to other studied precedents,134 increased accumulation of the mature tRNAUUA late in growth of S. ghanaensis may impact moenomycin production, directly enabling the translation of UUA+ moe transcripts, or triggering the expression of bldA-dependent transcriptional activator(s) of moe genes. The transcriptional regulators of the moe genes, evidently unclustered with the structural ones, have yet to be discovered. Efficient expression of moe cluster 1 in various streptomycetes118, 135 suggests that the S. ghanaensis genome does not contain moe pathway-specific regulator(s). Such regulators, often belonging to the OmpR-like SARP (Streptomyces Antibiotic Regulatory Proteins) family, are crucial for production of many antibiotics.128, 136 At the same time, the production of moenomycins varies significantly in different heterologous hosts,135 which may be a result of differences in the abundance of primary metabolites137 and/or pleiotropic regulatory circuits that govern secondary metabolism. Consistent with the idea of involvement of global regulators in the biosynthesis of 1, the S. coelicolor ppGpp synthetase gene relA has been shown to improve moenomycin production by native and heterologous hosts.135 More studies are needed to understand what limits moenomycin production by wild type and heterologous strains and whether there is some coordination of expression of moe clusters 1 and 2. Initial experimental evidence shows that manipulation of moe gene dosage may be used to increase antibiotic titers,135, 138 encouraging the search for other regulators of this unorthodox metabolic pathway.
4.3.3 Genome mining as a tool to discover new moenomycin-like biosynthetic pathways
The discovery of the moe genes was greatly facilitated by advances in genome sequencing technologies,118 which have led to the exponential growth of microbial genome databases. There are now more than 30 draft or complete actinomycete genomes, offering an exciting opportunity to search for novel natural products through the lense of structural genomics.139 For example, the capacity of a given microorganism to produce a polyketide compound can be quickly assessed through in silico screening of its genome for conserved polyketide synthase genes. Further classification and analysis of flanking genomic segments usually provides enough information to determine clusters that direct the production of novel molecules. Likewise, genome mining for MoeO5 homologues clustered with glycosyltransferase(s) can be used to find new phosphoglycolipid metabolic pathways. Indeed, our analysis of completely sequenced bacterial genomes has revealed three moeO5 homologues located in the vicinity of glycosyltransferase genes (Fig. 9). One such gene cluster is located on megaplasmid pSCL4 from Streptomyces clavuligerus ATCC27064, best known as an industrially important producer of clavulanic acid and cephamycin C.140 Both the organization and sequences of the moe genes and their S. clavuligerus counterparts are strikingly similar, pointing to the possibility of horizontal transfer of moe genes between streptomycetes. Curiously, there are no TTA codons in any of the moe genes from S. clavuligerus. As judged from gene content, moe cluster of S. clavuligerus could direct the biosynthesis of 12 or a derivative without the A ring. Two other clusters are present in the related enthomopathogenic gamma-proteobacteria Photorhabdus luminescens and P. asymbiotica. In these cases, the identified gene clusters are similar to each other but they bear only a modest resemblance to moe cluster 1 of S. ghanaensis. Their genetic capacity appears to be sufficient to control the production of a phosphoglycolipid containing 4 (P. asymbiotica) or 5 (P. luminescens) sugars (Fig. 9). As many more bacterial genomes will be available, it is likely that additional moenomycin-type gene clusters will be identified through genome mining. Their investigation will ultimately broaden our understanding of natural diversity, evolution and ecological roles of phosphoglycolipid biosynthesis.
Fig. 9.

Gene clusters that potentially may direct the production of phosphoglycolipid secondary metabolites.
5 Biological and structural determinants of activity of moenomycins
A great deal of the research reviewed above was motivated by the desire to understand which parts of 1 are necessary for bioactivity and how to alleviate its poor pharmacokinetic properties. Here we detail what is known about effects of moenomycins on bacterial cells, how the structures of different moenomycins relate to their antibiotic and PGT inhibitory properties, and what can be learned from the recent structural studies on PGT-moenomycin co-complexes. Since the problem of resistance arises for all antibiotics immediately after their first practical use, it is of paramount importance to understand antibiotic resistance mechanisms if we want to counter or slow down their propagation. With this in mind, we also summarize here the relatively scarce data on moenomycin resistance in streptomycetes, human pathogens and animal microflora.
5.1 Biological properties of moenomycins
5.1.1 Studies on pure bacterial cultures: mode of action, spectrum of activity, factors that influence potency
All known moenomycins display similar potency and spectrum of antibiotic activity as judged from MIC measurements.9, 49, 141, 142 They are typically active in the submicrogram per mL range, against gram-positive bacteria, including various methicillin- and vancomycin-resistant cocci,49, 80, 96, 141 although intrinsically moenomycin-resistant strains are well known.143-145 At 100-1000-fold higher concentrations, moenomycins elicit antibiotic effects against mycobacteria and gram-negative bacteria,9, 141, 146 (Table 1) suggesting that the physical barrier created by thick cell wall or outer membrane of enterobacteria effectively shields the PGTs from 1. Consistent with this idea, E. coli strains with increased outer membrane permeability due to mutations or chemical treatment are significantly more susceptible to 1 (Table 1).66, 80, 147, 148 Certain strains of gram-negative genera, including Neisseria, Helicobacter, Brucella, Pasteurella and Pseudomonas, are relatively sensitive to moenomycins,11, 146, 149-151 pointing to the differences in permeability of the outer membrane of different species. Basic pH (>8.0), addition of serum, whole blood or starch to moenomycin-containing media strongly (10-1000-fold) suppresses its antibiotic activity.11, 143, 146 The effect of media supplements might stem from non-specific capture of phosphoglycolipids on the surface of blood cell membranes or serum proteins.88, 152 Also, the MIC values for antibiotics and moenomycins in particular are 2-5-fold higher when rich media are used153, perhaps because bacteria have more resources to adapt themselves to stress conditions. 1 displays dose-dependent activity against E. coli. It is bacteriostatic at low concentrations (1 μg/ml) while increased doses (10 μg/ml and higher) cause cell lysis and death of actively dividing cells.80, 147, 154 A special case of lysis of non-growing relA mutants was also reported.155 Under conditions that prevent lysis (addition of Mg2+), 1 was still able to kill E. coli. In contrast, 1 had only bacteriostatic effects on cocci.80, 156 Only a small fraction of total peptidoglycan biosynthesis can be inhibited by 1 in Staphylococcus and Enterococcus, and yet it is sufficient to kill them.80 Since PGTs are known to be a part of multiprotein complexes involved in cell wall biogenesis and cytokinesis,29, 157, 158 inhibition of one PGT, even a non-essential PGT, may disrupt the function of an essential protein complex, leading to cell death. As the physiological roles and repertoire of PGTs vary across species,27, 159 it is not too surprising to see differential effects of 1 on bacteria.87
Table 1.
MIC a values (mcg/ml−1) of natural moenomycins against selected Gram-positive and Gram-negative bacteria
| Microorganism | Phosphoglycolipid compound |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 12 | 13 | Macb | Prac | Teid | 803e | |
|
Staphylococcus aureus (SG511, ATCC29213, or FDA 209P) |
0.016- 0.025 |
0.195 | 0.391 | 0.10 | 0.05 | 0.09 | 0.06- 0.25 |
0.05 | 0.15 | 0.05 | 0.01 |
| Staph. epidermitis ATCC12228 | 0.025 | -i | - | - | - | - | - | - | - | - | - |
| Streptococcus pyogenes (A77) | 0.001 | 0.002 | 0.002 | >0.01 | >0.01 | - | 0.25 | - | 0.24 | - | - |
| Strep. pneumoniae | 2 | - | - | - | - | - | 4 | - | - | 0.5 | - |
| Strep. pneumoniae R6 pbp2af | 0.13 | - | - | - | - | - | - | - | - | - | - |
| Enterococcus cloacae | 1.56 | - | - | 1.56 | 0.78 | - | - | - | - | - | - |
| Ent. faecalis GC2243 (PipR) g | - | - | - | - | - | - | 32 | - | - | - | - |
| Ent. faecalis ATCC29212 | 0.063 | - | - | - | - | - | 0.12 | - | - | - | - |
| Ent. faecium ATCC49624 | >200 | - | - | - | - | - | 0.06 | - | - | - | - |
| Mycobacterium phlei | - | - | - | - | - | - | - | 12.5 | - | - | - |
| M. tuberculosis (H37RV) | >66 | - | - | - | - | - | - | - | - | >100 | - |
| Bifidobacterium | >128 | - | - | - | - | - | - | - | - | - | - |
| Corynebacterium pyogenes | 2.5 | - | - | - | - | - | - | - | - | - | - |
| Bacillus subtilis PC1219 | 0.1 | - | 25 | - | - | - | - | - | - | - | 30 |
| Bacillus anthracis | - | - | - | - | - | - | - | 0.025 | - | - | - |
| Escherichia coli K12 | 8-62 | 50 | 50 | 25 | 25 | 12.5 | >64 | 50 | 37 | 10 | 35 |
| E. coli BAS849 or BE100 h | 0.025 | - | - | - | - | - | - | - | - | - | - |
| Clostridium perfringens | 256 | - | - | - | - | - | - | - | - | - | - |
| Neisseria meningitidis | 2.4 | - | - | - | - | - | - | - | - | - | - |
| N. gonorrhea | - | - | - | - | - | - | - | - | - | - | 0.6 |
| Pseudomonas sp. SC-8452 | >62 | - | - | 50 | 25 | 25 | - | - | 50 | - | 45 |
| P. aeruginosa 1771m | 3.125 | 3.125 | 3.13 | 1.56 | 1.56 | - | - | - | - | - | - |
| Listeria monocytogenes | 0.04 | - | - | - | - | - | - | - | - | - | - |
A strict comparison of the MICs should not be made since the experimental conditions were not the same.9, 23, 38, 40, 49, 80, 141, 143, 146
Macarbomycin.
Prasinomycin.
Teichomycin A1.
803RP.
Piperacillin-resistant strain.
Disruption of pbp2a gene.
imp (increased membrane permeability) strains.
Not determined.
5.1.2 Animal studies
In mouse streptococcal infection models, 1, prasinomycin and teichomycin A1 demonstrated excellent curative effects (1-20 mg/kg range) upon subcutaneous administration.11, 141, 149 Equally excellent efficacy (CD50 = 0.25 mg/kg) was reported for macarbomycin against S. aureus infection,160 while pholipomycin 12 was significantly less efficient.146 The latter may be due to the use of different S. aureus strains and workup conditions (for example, 12 was administered simultaneously or after the infection, while in most of the studies moenomycins are usually injected or fed before). The LD50 for 1 and 12 is within a range of 600-800 mg/kg (intravenous injection),11, 146 which exceeds 10-100 times the curative or protective doses. Except for hemolytic activity at high doses, there were no side effects of 1 on animal physiology upon systemic administration.11, 161 The phosphoglycolipids have also been shown to have exceptionally long prophylactic activity, lasting 2-8 weeks depending on the antibiotic, dose, and infection model used.11, 149, 160 This prophylactic effect is attributed to the fact that moenomycins bind to the components of blood and then excrete slowly (with a half-life of 9 days in mice9) over a long period of time. Moenomycins show almost no curative or protective effects and no toxicity via oral route,11, 149 reflecting their extremely low absorption from gastrointestinal tract. Moenomycin is remarkably resistant to digestive enzymes and excretes almost completely in an unchanged form.1, 162
As a feed additive, moenomycins (flavomycin) have been used for all kinds of agriculturally important animals.12 Flavomycin supplementation leads to weight gain as well increased wool and milk protein production.162, 163 Flavomycin did not promote the growth of gnotobiotic chickens,164 implying that its effects in animal are a result of an influence on gut microflora. Flavomycin is thought to suppress the so called hyper-ammonia producing bacteria (Desulfomonas, Fusobacterium and others) as well as opportunistic Gram positive pathogens, thereby increasing the amount of aminoacids available to the animal and alleviating the immune burden and ammonia-associated toxicity.163, 165-168 These studies also revealed highly complex and sometimes unpredictable interactions between the antibiotic, bacteria and a host. For example, although 1 was not effective against the gram-negative Clostridium perfringens according to MIC tests,169 it reduced intestinal populations of this pathogen in broilers.170 Similarly, 1 decreased the number of salmonellae in calves and pigs,165,171 but not in experimentally infected broiler chickens.172 Hence, multiple microbe- and host-specific factors appear to determine the therapeutic value of 1 when given orally. This is also illustrated by intriguing observation of selective activity of moenomycins - both in laboratory and field studies - against enterococci and E. coli carrying conjugative antibiotic resistance plasmids (so called donor strains, capable of transferring the plasmid to recipient strains).12, 173-178 Conjugative transfer requires major remodeling of cell wall, which may facilitate the access of 1 to its PGT targets. This could explain earlier observations that 1 decreased the number of R plasmid-containing bacteria in animals.165, 176 Nevertheless, not all animal-based studies agree that 1 specifically kills donor strains,179 and therefore as-yet-unknown “resistance-curative” mechanisms may be in play. For instance, besides PGTs moenomycins are known to affect the first two glycosylations of dolichol phosphate, which are catalyzed by GTs, that, like the PGTs, utilize lipid acceptor substrates.180 Perhaps there are other as-yet-unknown targets for moenomycins, more accessible than PGTs, in certain Gram-negative bacteria, which render them sensitive to 1.
5.2 Relationships between the structure of moenomycins and their biological activity
5.2.1 Impact of the lipid-3PG moiety
Despite impressive antibiotic activity and low toxicity, 1 has poor physical properties that undermine its clinical value. 1 is a very stable181 detergent-like molecule (with a cmc of 0.5 mM at pH 6.8) that tends to aggregate in aqueous solutions and partition into membranes.88, 152, 182, 183 As a consequence, 1 has a very long half-life in the bloodstream, some hemolytic activity and it is not orally bioavailable. The long lipid chain of 1 is suggested to underlie these undesired effects,12, 96 although this never been directly tested. The issue of suboptimal pharmacology prompted the comparison of biological activities of different moenomycins described in sections 2-4 using microbiological and biochemical assays. Hydrogenation of moenocinol (e.g., 24 versus 30) and its isomerization to diumycinol (13) were shown to be the only lipid modifications neutral with respect to antibiotic activity of moenomycins (Fig. 10).49,62 There is a positive correlation between the length of lipid chain of moenomycins and their potency. Delipidated analog 47, neryl-moenomycin 55 and compounds 48 and 49 containing short aliphatic chains are all devoid of antibacterial activity.7, 96 Of note, 55 has submicromolar PGT inhibitory activity, while 47 is 300 times less active, suggesting that the lipid does not just help deliver the pharmacophore to membranes, but also interacts specifically with the PGTs; otherwise, both the delipidated analog 47 and neryl-containing 55 would have comparable activity in vitro. C15 moenomycins 85-88 and compound 50 (containing a C17 branched aliphatic chain) showed low antimicrobial activity (1-10% of that of 1).94, 95, 123 Both the phosphoryl and carboxyl groups of 3PG are important for activity, and it has been established that the charge of the carboxylate is critical for target binding, as evident from analysis of synthetic compounds 56-61.74 Oxidation of moenocinol and its replacement with long (C13-C17) aliphatic chains or other lipid-like groups (51-54) via reductive amination kill the antibiotic properties of 1.90, 94, 95, 184 Unfortunately, there are no pharmacokinetic data on compounds 48-55 and 85-88 to verify that moenomycins with shorter lipids have a shorter half-life.
Fig. 10.

Summary of SAR studies around MmA scaffold. Functional groups of oligosaccharide-3PG moiety of 1 crucial for antibiotic activity are shown in dark orange. Grey shade indicates minimal in vivo pharmacophore 1 of MmA (30); another recently identified minimal pharmacophore 2 (72) contains D ring (shaded in yellow) instead of C ring. Key modifications of lipid and chromophore parts of MmA are listed on both sides of 1 and their relative antistaphylococcal activity is mentioned.
5.2.2 Impact of oligosaccharide-chromophore portion
Compound 9 lacking the A ring is 10 times less active than 1, which is considered a moderate decrease due to the immense potency of the parent molecule.84 It assumed that the chromophore is not essential for the interaction between PGT and 1, although it may provide additional contacts to the enzyme, making 1 a tighter binder. Compounds containing various fluorophores, tags or ampicillin instead of the A ring were shown to have, at best, 1-10% potency of 1 (33-39, 41, 43-45),83, 85, 86, 91 and some of them were virtually inactive (40, 42).86, 88, 89 As a general trend, compounds having less bulky substituents or those attached via long linker have higher potency.
As the MIC values of natural moenomycins 1-7 do not differ more than tenfold (Table 1),38-40 we can conclude that the D ring, the 6-deoxy group on the C ring and the F ring 4-C-methyl are not critical for bioactivity. The latter functionality goes invariably with equatorial hydroxyl and the exact role(s) of either of these groups in activity of moenomycins remain vague.64-67, 70 In any case, since removal of both groups (21) leads to complete inactivation,67 and 2 and its trisaccharide analogs 25-27 are much less active than 1, proper “decoration” of the F ring C4 carbon adds antibiotic potency.40, 70,71 The C-E-F trisaccharides 30/24 and the E-F disaccharide 14 were long thought to represent the minimal in vivo and in vitro pharmacophores, respectively.40, 66, 92 The structural elements of 30 and 14 critical for activity are shown on Fig. 10; their modification/removal ablated the inhibitory properties of moenomycins in vivo and in vitro.8, 21, 61-63, 68, 71, 93, 185 Recently, however, the structurally simpler D-E-F trisaccharide 72 was shown to possess antibiotic properties comparable to that of 24.77 It is possible that the flexibility of D-E glycoside bond allows 72 to adopt a conformation where the D ring interacts with the PGT target, compensating for the loss of the C ring NHAc group.
The minimal requirements for efficient PGT inhibitors are now relatively well understood with regard to the carbohydrate moieties of 1. In contrast, much has to be explored to understand the requirements for the “optimal” lipid – e.g., one that supports good antibacterial activity and improves pharmacokinetics. Such explorations are now feasible and are worthwhile since some fragments of 1 having shorter lipids have biological activity.
5.3 Biochemical and structural insights into PGT inhibition by moenomycins
5.3.1 Biochemical studies
With the exception of a few intracellular parasites, all bacteria make peptidoglycan (PG) and most contain multiple PGTs that are similar structurally and functionally. Generally, the more complex the morphological features of the bacteria are, the richer the suite of PG-metabolizing proteins, including PGTs.159 PGTs fall into two groups. One group comprises the monofunctional transglycosylases, which consist of a cytoplasmic tail, a transmembrane helix and a PGT domain. The other group contains these features and also has a C-terminal transpeptidase (TP) domain (Fig. 11A). These PGTs tare referred to as class A penicillin binding proteins (PBPs), reflecting the history of their discovery through covalent interaction with beta-lactams.159 Deletion of all known PGT genes in B. subtilis and E. faecalis did not prevent peptidoglycan biosynthesis,186, 187 pointing to the existence of as-yet unknown synthetic transglycosylases that evidently bear no similarity to the described ones. Biochemical and genetic experiments show that different PGTs within an organism play unique roles at certain stages of bacterial growth, such as elongation or septation.159, 188 Sequence analysis revealed 5 aminoacid motifs conserved across the entire PGT family (Fig. 11A-B),189 suggesting that all PGTs function through the same catalytic mechanism. Nevertheless, different PGTs differ in their affinity for 1, pH optima, metal and detergent requirements, and they produce PG strands of different average length.87, 159, 190, 191 PGTs utilize as a substrate a unique pyrophospho-lipid linked disaccharide, Lipid II, and are categorized into GT family 51. They produce glycan chains by the addition of Lipid II (acceptor) to the reducing end of the donor molecule, which is Lipid II during first reaction cycle or the growing polymer in subsequent cycles (Fig. 1).192 It has been established that PGTs are processive polymerases,191, 193 and the donor site lipid length plays a major role in the processivity.194 Substantial biochemical evidence, summarized by Sauvage and coauthors,159 shows that 1 directly inhibits PGTs of different origin, although not all PGTs are sensitive to it.186, 187, 195, 196 The binding of 1 to PGT is reversible87, 90 but is not competitive with respect to Lipid II.7 The dissociation constants for PGT binding and IC50 for its inhibition by 1 are typically in the nanomolar range.7, 84, 197 There is some evidence that transmembrane domain of PGTs enhances the affinity of binding of 1 to the enzyme.87
Fig. 11.
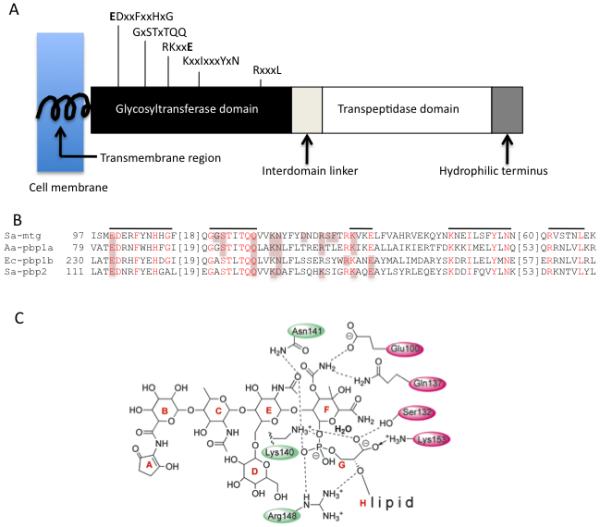
PGT structure and inhibition. A. Topology diagram depicting the arrangement of domains for a typical class A bifunctional pencillin-binding protein. The conserved residues of the PGT domain (signature motifs) are shown above the diagram. This domain arrangement is conserved in the monofunctional PGTs, but the linker region and TP domain are absent B. Multiple sequence alignment of a portion of PGT domains of S. aureus Mtg (Sa-mtg), PBP2 (Sa-pbp2), A. aeolicus PBP1a (Aa-pbp1a) and E. coli PBP1b (Ec-pbp1b). Five signature motifs are shown in red and lined above the alignment. For the sake of clarity, two aminoacid stretches are not shown in the alignment (marked with square brackets). Residues forming potential interactions with moenomycin pharmacophore are highlighted. C. A schematic view of interaction between pharmacophore (E-F-G-H units) and PGT. Signature motif aminoacids that contact 1 in at least 3 out of 4 structures are marked with filled magenta ovals; numbering is taken from 1:Mtg cocomplex.78 Other conservative residues are indicated by green ovals.
5.3.2. Structural studies
Recently crystal structures of four different PGTs bound to moenomycin have been solved.76-79 Two PGTs are from S. aureus (PBP2 and Mtg) and two others are from the Gram-negative bacteria Aquifex aeolicus (PBP1a) and E. coli (PBP1b). All these PGTs have an identical bilobal α-helical fold that resembles that of phage λ lysozyme. The active site cleft is between the two lobes, and the 5 PGT signature amino acid motifs shown in Fig. 11A-B help form the active site. The pentasaccharide portion of moenomycin extends along the active site cleft. A number of invariant and catalytically important amino acid residues from the first three motifs form direct or water-mediated hydrogen bonds to the E, F rings and the 3-PG unit of the inhibitor (Fig. 11C). No or few contacts are observed between the PGTs and the A, B, and D rings. Contacts to the moenocinol chain are poorly defined due to weak electron density, but it can be inferred from the position of the phosphoglycerate in conjunction with electron density for the first 15 carbons of moenocinol that the lipid chain funnels down from the active site through a hydrophobic groove in the smaller lobe towards the membrane (between motifs 2 and 3; Fig.11B).78 The smaller lobe is proposed to be partially embedded in the lipid bilayer (Fig. 12A). In agreement with SAR studies, the cocomplex structures suggest that the specificity of moenomycin binding is largely determined by interactions of the EF disaccharide-phosphoglycerate with side chains in the active site; other interactions with the distal portion of the pentasaccharide in the cleft, combined with hydrophobic interactions between the lipid chain and the enzyme, provide affinity.
Fig. 12.

Structural details of interaction between moenomycin and PGT and implications for PGT mechanism. A. Close-up view of surface representation of A. aeolicus Pbp1a co-complexed with neryl-moenomycin (55). The highly conserved residues of the 5 PGT motifs (colored magenta) are located in the active site cleft where 55 binds. Enzyme consists of a smaller lobe (closer to membrane) and a larger lobe. The lipid tail portion points down towards the proposed membrane interface. The 3-PG and pentasaccharide moieties extend upwards along the long groove. B. Working model for PGT-catalyzed reaction. Growing chain occupies donor substrate site and reacts with incoming lipid II unit (acceptor). The outer helix and the loop in the smaller lobe of PGT are mobile and help transfer the reaction product to donor site for next round of polymerization. Through binding to the donor site, moenomycins may prevent the initiation of polymerization or elongation of the first product, lipid IV (tetrasaccharide).
The aforementioned cocomplexes provide a structural basis for the design of PGT inhibitors. That being said, there are some differences in the inferred contacts between the proteins and the 3-PG unit of 1 among the different structures. The most notable differences relate to how the carboxylate of the phosphoglycerate binds. In one structure, 1:PBP2, a glutamate side chain is near the carboxylate of 3-PG,76 while in two other structures, A. aeolicus PBP1A and S. aureus Mtg, conserved positively charged groups bond to the carboxylate.77,78. In the fourth structure, E. coli PBP1b, there are no side chains near the carboxylate.79 A study to probe the role of the carboxylate in binding shows that the negative charge, and not simply the hydrogen bonding network, is critical, supporting the importance of the positively charged side chains observed in two of the complexes in binding moenomycin.74 Obtaining oral bioavailability with moenomycin-based scaffolds given the importance of both the phosphate and the carboxylate charges in binding to PGTs will be a challenge.
An interesting feature of the PGT structures that have been reported is a highly mobile region of the smaller lobe (the so called outer helix, between motifs 1 and 2). This region, captured in different conformations and degrees of disorder in different cocomplexes, is proposed to play an important role in processive polymerization.29, 77, 198 This region in PGT structures containing moenomycin is typically more ordered than in structures that do not contain moenomycin. An exception is the A. aeolicus apo enzyme structure, which resembles the four moenomycin-bound structures. The moenomycins are proposed to mimic the growing glycan chain, and they likely bind in the donor substrate site of the PGTs (donor substrate; Fig. 12A-B), but the existence of both open (outer helix disordered) and closed (outer helix ordered) conformations of the PGTs suggest that the mode of inhibition is not simply based on mimicry of the growing glycan strand. The structural data suggest a possible scenario in which PGTs have an open, active conformation to which substrates bind and a closed, inactive conformation to which the moenomycins bind. Establishing whether the moenomycins interfere with chain initiation or chain elongation, and whether inhibition is competitive or not with respect to the growing glycan strand, is essential for understanding how to design PGT inhibitors. Knowing the precise mechanism of PGT inhibition by 1 would inform about optimal functional characteristics we should seek in PGT inhibitors emerging from all kinds of screens.87, 199 Structures of PGTs bound to substrate analogs, combined with deeper biochemical dissection of the PGT reaction, should help illuminate one of the last “white spots” in PG biosynthesis.
5.4 Resistance to moenomycins
There are no reports on animal19, 200 or human201 isolates significantly resistant to 1. Many Enterococcus faecium strains are reported to be naturally resistant to 1 (Table 1), although such conclusions are often influenced by both experimental conditions and the definition of what to consider resistant.202 No significant natural cross-resistance has been revealed between 1 and other clinically useful classes of antibiotics and no plasmid-borne moenomycin resistance determinants have been detected.9, 11, 200 It is possible to raise mutations that confer resistance to both moenomycins and other classes of cell wall-active antibiotics.49, 148, 203, 204 For example, null mutations in bamB (formerly yfgL) suppress outer membrane membrane defects in E. coli due to mutations in lptD (formerly imp). This suppression leads to increased resistance to vancomycin as well as 1.205 That changes in membrane makeup or charge could affect resistance to 1 and glycopeptides is supported by studies on Gram-positive bacteria. Particularly, reduced content of lysyl-phosphatidylglycerol in S. aureus membranes led in reduced susceptibility to 1 and vancomycin.203 Recent work on the ruminal microbe Prevotella bryantii also suggests that increased availability of undecaprenylpyrophosphate may account for resistance to 1, as well as to vancomycin and bacitracin.206
The aforementioned studies revealed rather non-specific mechanisms of defense against 1. Currently, no dedicated moenomycin resistance mechanisms have been described, although isolation of cocci highly resistant to 1 was reported by several groups.11, 149 Mutations in PGTs that would abolish its binding of 1 have not yet been identified despite repeated efforts.148, 203-205 Since a number of amino acids that contact 1 are also essential for catalysis (vide supra), mutations that would decrease/abolish moenomycin binding may also undermine PGT activity. If so, occurrence of such mutations would not be advantageous for a cell, thus slowing down the resistance development.207 Cleavage or modification of 1 would be viable resistance mechanism. Indeed, a Bacillus strain DSM4675 (isolated from contaminated moenomycin fermentation broth) was shown to cleave the phosphoglycosidic bond of 1.208 No other moenomycin-modifying bacteria have been described so far; given that 1 is excreted intact from animal body, degradation of 1 does not seem to be widely represented at least in mammalian and avian microflorae.
Gene clusters for moenomycin production from S. ghanaensis and S. clavuligerus do not carry resistance genes, posing a question of how producers of 1 avoid suicide. Analysis of a big collection of phylogenetically distant actinomycetes, many of which are proven moenomycin-nonproducers (such as S. coelicolor209), revealed their uniformly high resistance to 1 (B. Ostash unpublished data). The only exception is S. albus J1074, but moenomycin-resistant clones of this strain occur at relatively high frequency.135 Therefore, resistance to 1 appears to be a general trait of actinobacteria (see also Table 1), which may stem from presence of PGT having low affinity to 1, or peculiarities of their cell wall organization.
6 Outlook
Completion of the total synthesis of 160, 96 discovery of the moenomycin biosynthetic pathway118, 123 and structural studies on PGT inhibition by 174, 76-79 are the most significant achievements in the field of moenomycin research over the last 5 years. They open the door to exploration of a wider set of problems ranging from fundamental ones to more applied, such as urgent need for new antibiotics. Below we will summarize those that, in our opinion, are the most important.
There is undoubted progress in our ability to generate novel moenomycins. In fact, number of synthetic and genetically engineered derivatives and fragments of 1 (even excluding very specialized 1-based chemical probes, such as 33-45) now greatly surpasses that of natural parent compounds. Nevertheless, many moenomycins are not yet chemically characterized,4, 9 implying that their true natural diversity is still an open question. On a wider scope, it is not known how widely moenomycin-like biosynthetic pathways are represented in Nature. There is a number of in vivo and in vitro assays capable of detecting cell wall inhibitors in biological samples,21, 199, 210-213 although none of them is moenomycin-specific. In this respect, discovery of S. ghanaensis regulatory proteins that use phosphoglycolipids as ligands may prove useful not only for strain improvement but also as biosensor elements for screening purposes. In silico genome mining and PCR-based screening for conserved moe genes represent other, “gene-centric”, approaches to address this issue.
The stage is now set for integration of synthetic and biological chemistries to produce phosphoglycolipids. Either synthetic analogs would be modified enzymatically, or biosynthetic intermediates may serve as a starting point for chemical derivatizations. The scope of structures accessible through a former strategy will ultimately hinge on the degree of substrate promiscuity of Moe enzymes. Initial studies demonstrate relatively high modularity and flexibility of moenomycin biosynthetic machinery,118, 123 showing the promise for aforementioned “mix-and-match” experiments. Biochemical studies of Moe proteins as well as discovery of new phosphoglycolipid biosyntheses should be pursued, as they could provide valuable tools for chemoenzymatic design of moenomycins.
It will be interesting to find out whether other secondary pathways exist that possess parsimonious genetic architecture similar to that of moe cluster 1. Such pathways may be a special case of evolution, or represent primordial types of secondary metabolism (e.g., one that branched from core metabolism early in evolutionary history of the host organism). It is important to understand a minimal set of genetic features that define generally nonessential (secondary) metabolic pathway and control its expression; such a knowledge will help improve annotation of secondary metabolome and subsequent efforts to activate cryptic gene clusters.
One of the most striking features of moenomycins is that, being mainly active against Gram-positive bacteria in laboratory tests, their principal value as an animal growth promoter is thought to come from inhibition of Gram-negative microflora. It is difficult to explain this observation using current data. However, recent studies on human microbiome214 show how little we know about animal prokaryotic community; more extensive application of metagenomic techniques should help uncover true picture of interactions between bacteria and antibacterial agents in digestive tract. These studies will also help to understand better the evolution of moenomycin resistance in animal. Comparing these data with the rates and types of resistance mutations in pure cultures would help organize any potential moenomycin-based therapy in the most rational way.
After decades of use, moenomycins are now prohibited as animal feed additive in Europe, clearing the way for their development as a drug to treat human infections. However, their poor pharmacokinetics has to be improved first. C25 carbon chain of 1 is thought to be a reason for its exceedingly long half-life in mammalian body; in the same time, it also determines high antibacterial potency of 1. Therefore, although lipid modification (truncation) seems to be natural way to improve moenomycins, one has to come up with idea of how to compensate the loss of activity. As judged from SAR and structural studies, only small portion of 1 (e.g., E, F rings, 3-PG and, possibly, C10 part of moenocinol) makes essential contacts to PGT; other moieties provide high binding affinity. Hence, it may be feasible to prepare analogs with shifted “accents” - ones that lack poor physical properties of 1 (e.g., through lipid truncation), but do show reasonable antibiotic activity (through modification of A, B, C, D rings). In this respect, more studies are also required to uncover the exact mechanism of PGT inhibition by 1. This will aid in more precise modeling and virtual screening of novel moenomycins.
Ability of 1 and its fragments to inhibit wide variety of PGTs in vitro and ability of various natural moenomycins to inhibit Gram-positive and G-negative bacteria in animals is well documented. Hence, there is no principal barrier to development of a broad-spectrum antibiotic based on PGT inhibition. Deeper chemical and biological exploration of phosphoglycolipids may turn up such an antibiotic. Now, when powerfull tools are available to model, generate and assay novel moenomycins, we anticipate that progress in the areas outlined above will further boost the efforts to develop moenomycin-based molecules for human use.
Fig. 13.

Acknowledgements
We thank Dr. D. Perlstein for the help with figures. Work in the laboratory of Prof. S. Walker was supported by NIH grant 5R01GM076710, NERCE grant AI057159; SPARC Award 2740563. B. O. is supported by grant from the Ukrainian Ministry of Science and Education (Bg-01F).
References
- 1.Wallhausser KH, Nesemann G, Prave P, Steigler A. Antimicrob. Agents Chemother. 1965;5:734. A. [PubMed] [Google Scholar]
- 2.Huber G, Schacht U, Weidenmuller HL, Schmidt-Thome J, Duphorn J, Tschesche R. Antimicrob. Agents Chemother. 1965;5:737. [PubMed] [Google Scholar]
- 3.Lenoir D, Tschesche R, Wucherpfennig W, Huber G, Weidenmuller HL. Antimicrob. Agents Chemother. 1969;9:144. [PubMed] [Google Scholar]
- 4.Welzel P. Chem. Rev. 2005;105:4610. doi: 10.1021/cr040634e. [DOI] [PubMed] [Google Scholar]
- 5.Uchida R, Iwatsuki M, Kim Y-P, Omura S, Tomoda H. J. Antibiot. 2010;63:157. doi: 10.1038/ja.2010.10. [DOI] [PubMed] [Google Scholar]
- 6.van Heijenoort Y, van Heijenoort J. FEBS Lett. 1980;110:241. doi: 10.1016/0014-5793(80)80082-2. [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Walker D, Sun B, Hu Y, Walker S, Kahne D. Proc. Natl. Acad. Sci. USA. 2003;100:5658. doi: 10.1073/pnas.0931492100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halliday J, McKeveney, Muldon C, Rajarathan P, Meutermans W. Biochem. Pharmacol. 2006;71:957. doi: 10.1016/j.bcp.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 9.Huber G. In: Antibiotics. Hahn FE, editor. Springer-Verlag; Berlin/Heidelberg: 1979. pp. 135–153. [Google Scholar]
- 10.Kahne D, Leimkuhler C, Lu W, Walsh CT. Chem. Rev. 2005;105:425. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 11.Wasielewski E, Muschaweck R, Schutze E. Antimicrob. Agents Chemother. 1965;5:743. [PubMed] [Google Scholar]
- 12.Pfaller MA. Diagn. Microbiol. Infect. Dis. 2006;56:115. doi: 10.1016/j.diagmicrobio.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 13.Dealy J, Moeller MW. J. Anim. Sci. 1977;45:1239. doi: 10.2527/jas1977.4561239x. [DOI] [PubMed] [Google Scholar]
- 14.Edwards JR, McEwan NR, Wallace RJ. J. Appl. Microbiol. 2008;104:1617. doi: 10.1111/j.1365-2672.2007.03689.x. [DOI] [PubMed] [Google Scholar]
- 15.Cassone M, Giordano A. Expert Rev. Anti Infect. 2009;7:637. doi: 10.1586/eri.09.50. [DOI] [PubMed] [Google Scholar]
- 16.Martinez JL. Science. 2008;321:365. doi: 10.1126/science.1159483. [DOI] [PubMed] [Google Scholar]
- 17.Hammerum M, Heuer OE, Emborg H-D, Skjot L, Jensen VF, Rogues A-M, Skov RL, Agerso Y, Brandt CT, Seyfarth AM, Muller A, Hovgaard K, Ajufo J, Bager F, Aarestrup FM, Frimodt-Moller N, Wegener HC, Monnet DL. Emerg. Infect. Dis. 2007;13:1632. doi: 10.3201/eid1311.070421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallo P, Fabbrocino S, Serpe L, Fiori M, Civitareale C, Stacchini P. Rapid Comm. Mass Spectrom. 2010;24:1017. doi: 10.1002/rcm.4478. [DOI] [PubMed] [Google Scholar]
- 19. [accessed Mar 2010];Danish Integrated Antimicrobial Resistance Monitoring and Research Program. DANMAP-97. doi: 10.3201/eid1311.070421. Available from http://www.danmap.org/pdffiles/danmap_1996_uk.pdf. [DOI] [PMC free article] [PubMed]
- 20.Chastre J. Clin. Microbiol. Infect. 2008;14(Suppl. 3):3. doi: 10.1111/j.1469-0691.2008.01958.x. [DOI] [PubMed] [Google Scholar]
- 21.Ostash, Walker S. Curr. Opin. Chem. Biol. 2005;9:459. doi: 10.1016/j.cbpa.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 22.Walsh T, Wright GD. Curr. Opin. Microbiol. 2009;12:473. doi: 10.1016/j.mib.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Slusarchyk WA. Biotechnol. Bioeng. 1971;13:399. doi: 10.1002/bit.260130307. [DOI] [PubMed] [Google Scholar]
- 24.Crawford LM. In: Antibiotics, sulphonamides, and public health. Jukes TH, Dupont JL, Crawford LM, editors. CRC Press; Boca Raton: 1984. pp. 351–354. [Google Scholar]
- 25.Welzel P. In: Antibiotic and antiviral compounds – chemical synthesis and modifications. Krohn K, Kirst H, Maas H, editors. VCH, Weinheim; 1993. pp. 144–149. [Google Scholar]
- 26.Goldman RC, Gange D. Curr. Med. Chem. 2000;7:801. doi: 10.2174/0929867003374651. [DOI] [PubMed] [Google Scholar]
- 27.van Heijenoort J. Glycobiology. 2001;11:25R. doi: 10.1093/glycob/11.3.25r. [DOI] [PubMed] [Google Scholar]
- 28.Di Guilmi M, Dessen A, Dideberg O, Vernet T. Curr. Pharm. Biotechnol. 2002;3:63. doi: 10.2174/1389201023378436. [DOI] [PubMed] [Google Scholar]
- 29.Lovering L, Gretes M, Strynadka NCJ. Curr. Opin. Struct. Biol. 2008;18:534. doi: 10.1016/j.sbi.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 30.Ostash, Doud E, Fedorenko V. Biol. Chem. 2010;391:499. doi: 10.1515/BC.2010.053. [DOI] [PubMed] [Google Scholar]
- 31.Coates RM, Johnson MW. J. Org. Chem. 1980;45:2685. [Google Scholar]
- 32.Kocienski PJ, Todd M. J. Chem. Soc. Perkin Trans. 1983;1:1777. [Google Scholar]
- 33.Kakarla R, Ghosh M, Anderson JA, Dulina RG, Sofia MJ. Tetrahedron Lett. 1999;40:5. [Google Scholar]
- 34.Ghosh M, Kakarla R, Sofia MJ. Tetrahedron Lett. 1999;40:4511. [Google Scholar]
- 35.Hilt G, Treutwein J. Chem. Commun. 2009:1395. doi: 10.1039/b822023a. [DOI] [PubMed] [Google Scholar]
- 36.Welzel P, Witteler F-J, Muller D, Riemer W. Angew. Chem. Int. Ed. Engl. 1981;20:121. [Google Scholar]
- 37.Kurz M, Guba W, Vertesy L. Eur. J. Biochem. 1998;252:500. doi: 10.1046/j.1432-1327.1998.2520500.x. [DOI] [PubMed] [Google Scholar]
- 38.Scherkenbeck J, Hiltmann A, Hobert K, Bankova W, Siegels T, Kaiser M, Muller D, Veith H, Fehlhaber H-W, Siebert G, Markus A, Limbert M, Huber G, Bottger D, Stark A, Takahashi S, van Heijenoort Y, van Heijenoort J, Welzel P. Tetrahedron. 1993;49:3091. [Google Scholar]
- 39.Hessler-Klintz M, Hobert K, Biallass A, Siegels T, Hiegemann M, Maulshagen A, Muller D, Welzel P, Huber G, Bottger D, Markus A, Siebert G, Stark A, Fehlhaber H-W, van Heijenoort Y, van Heijenoort J. Tetrahedron. 1993;49:7667. [Google Scholar]
- 40.Donnerstag, Marzian S, Muller D, Welzel P, Bottger D, Stark A, Fehlhaber H-W, Markus A, van Heijenoort Y, van Heijenoort J. Tetrahedron. 1995;51:1931. [Google Scholar]
- 41.Donnerstag, Hennig L, Findeisen M, Welzel P. Magn. Reson. Chem. 1996;34:1031. [Google Scholar]
- 42.Hennig L, Findeisen M, Welzel P, Haessner R. Magn. Reson. Chem. 1998;36:615. [Google Scholar]
- 43.Kurz M, Guba W, Vertesy L. Eur. J. Biochem. 1998;252:500. doi: 10.1046/j.1432-1327.1998.2520500.x. [DOI] [PubMed] [Google Scholar]
- 44.Schacht U, Huber G. J. Antibiot. 1969;22:597. [PubMed] [Google Scholar]
- 45.Weisenborn FL, Bouchard JL, Smith D, Pansy F, Maestrone G, Miraglia G, Meyers E. Nature. 1967;213:1092. doi: 10.1038/2131092a0. [DOI] [PubMed] [Google Scholar]
- 46.Zehl M, Pittenauer E, Rizzi A, Allmaier G. J. Am. Soc. Mass. Spectrom. 2006;17:1081. doi: 10.1016/j.jasms.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 47.Arai M, Nakayama R, Yoshida K, Takeuchi M, Teramoto S, Torikata A. J. Antibiot. 1977;30:1055. doi: 10.7164/antibiotics.30.1055. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi S, Serita K, Arai M, Seto H, Furihata K, Otake N. Tetrahedron Lett. 1983;24:499. [Google Scholar]
- 49.He H, Shen B, Korshalla J, Siegel MM, Carter GT. J. Antibiot. 2000;53:191. doi: 10.7164/antibiotics.53.191. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi S, Okanishi A, Utahara R, Nitta K, Maeda K, Umezawa H. J. Antibiot. 1970;23:48. doi: 10.7164/antibiotics.23.48. [DOI] [PubMed] [Google Scholar]
- 51.Slusarchyk WA, Osband JA, Weisenborn FL. J. Am. Chem. Soc. 1970;92:4486. doi: 10.1021/ja00717a073. [DOI] [PubMed] [Google Scholar]
- 52.Sattler, Kreuzig F. J. Antibiot. 1975;28:200. doi: 10.7164/antibiotics.28.200. [DOI] [PubMed] [Google Scholar]
- 53.US Pat., 3 493 653. 1970
- 54.So. African Pat., 65/6204. 1966
- 55.Belgian Pat., 653 168. 1965
- 56.Netherlands Pat., 68 02093. 1968
- 57.US Pat., 3 891 754. 1973
- 58.US Pat., 3 891 753. 1974
- 59.Bardone MR, Paternoster M, Coronelli C. J. Antibiot. 1978;31:170. doi: 10.7164/antibiotics.31.170. [DOI] [PubMed] [Google Scholar]
- 60.Taylor JG, Li X, Oberthur M, Zhu W, Kahne D. J. Am. Chem. Soc. 2006;128:15084. doi: 10.1021/ja065907x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Welzel P, Wietfeld B, Kunisch F, Schubert T, Hobert K, Duddeck H, Muller D. Tetrahedron. 1983;39:1583. [Google Scholar]
- 62.Welzel P, Kunisch F, Kruggel F, Stein H, Scherkenbeck J, Hiltmann A, Duddeck H, Muller D, Maggio JE, Fehlhaber H-W, Seibert G, van Heijenoort Y, van Heijenoort J. Tetrahedron. 1987;43:585. [Google Scholar]
- 63.Fehlhaber H-W, Girg M, Seibert G, Hobert K, Welzel P, van Heijenoort Y, van Heijenoort J. Tetrahedron. 1990;46:1557. [Google Scholar]
- 64.Hohgardt H, Dietrich W, Kuhne H, Muller D, Grzelak D, Welzel P. Tetrahedron. 1988;44:5771. [Google Scholar]
- 65.Moller U, Hobert K, Donnerstag A, Wagner P, Muller D, Fehlhaber H-W, Markus A, Welzel P. Tetrahedron. 1993;49:1635. [Google Scholar]
- 66.El-Abadla N, Lampilas M, Hennig L, Findeisen M, Welzel P, Muller D, Markus A, van Heijenoort J. Tetrahedron. 1999;55:699. [Google Scholar]
- 67.Riedel S, Donnerstag A, Hennig L, Welzel P, Richter J, Hobert K, Muller D, van Heijenoort J. Tetrahedron. 1999;55:1921. [Google Scholar]
- 68.Ferse F-T, Floeder K, Hennig L, Findeisen M, Welzel P, Muller D, van Heijenoort J. Tetrahedron. 1999;55:3749. [Google Scholar]
- 69.Yang G, Mansourova M, Hennig L, Findeisen M, Oehme R, Giesa S, Welzel P. Helv. Chim. Acta. 2004;87:1807. [Google Scholar]
- 70.Luning J, Moller U, Muller D, Welzel P, Markus A, van Heijenoort Y, van Heijenoort J. Tetrahedron. 1993;49:10587. [Google Scholar]
- 71.Range G, Krahmer R, Welzel P, Muller D, Herrmann GH, Kragl U, Wandrey C, Markus A, van Heijenoort Y, van Heijenoort J. Tetrahedron. 1997;53:1695. [Google Scholar]
- 72.Ritzeler O, Hennig L, Findeisen M, Welzel P, Muller D. Tetrahedron. 1997;53:1665. [Google Scholar]
- 73.Silva, Wang H, Allanson NM, Jain RK, Sofia M. J. Org. Chem. 1999;64:5926. [Google Scholar]
- 74.Fuse S, Tsukamoto H, Yuan Y, Wang T-SA, Zhang Y, Bolla M, Walker S, Kahne D. ACS Chem. Biol. 2010 doi: 10.1021/cb100048q. DOI: 10.1021/cb800078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sofia M, Allanson NM, Hatzenbuhler NT, Jain RK, Kakarla R, Kogan N, Liang R, Liu D, Silva DJ, Wang H, Gange D, Anderson J, Chen A, Chi F, Dulina R, Huang B, Kamau M, Wang C, Baizman E, Branstrom A, Bristol N, Goldman R, Han K, Longley C, Midha S, Axelrod HR. J. Med. Chem. 1999;42:3193. doi: 10.1021/jm990212a. [DOI] [PubMed] [Google Scholar]
- 76.Lovering L, de Castro LH, Lim D, Strynadka NCJ. Science. 2007;315:1402. doi: 10.1126/science.1136611. [DOI] [PubMed] [Google Scholar]
- 77.Yuan Y, Fuse S, Ostash B, Sliz P, Kahne D, Walker S. ACS Chem. Biol. 2008;3:429. doi: 10.1021/cb800078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heaslet H, Shaw B, Mistry A, Miller AA. J. Struct. Biol. 2009;167:129. doi: 10.1016/j.jsb.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 79.Sung M-T, Lai Y-T, Huang C-Y, Chou L-Y, Shih H-W, Cheng W-C, Wong C-H, Ma C. Proc. Natl. Acad. Sci. USA. 2009;106:8824. doi: 10.1073/pnas.0904030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baizman R, Branstrom AA, Longley CB, Allanson N, Sofia MJ, Gange D, Goldman RC. Microbiology. 2000;146:3129. doi: 10.1099/00221287-146-12-3129. [DOI] [PubMed] [Google Scholar]
- 81.Goldman RC, Baizman ER, Branstrom AA, Longley CB. Bioorg. Med. Chem. 2000;10:2251. doi: 10.1016/s0960-894x(00)00443-1. [DOI] [PubMed] [Google Scholar]
- 82.Kempin U, Hennig L, Muller D, Markus A, Welzel P. Tetrahedron Lett. 1996;37:5087. [Google Scholar]
- 83.Kempin U, Hennig L, Knoll D, Welzel P, Muller D, Markus A, van Heijenoort J. Tetrahedron. 1997;53:17669. [Google Scholar]
- 84.Ruhl T, Daghish M, Buchynskyy A, Barche K, Volke D, Stembera K, Kempin U, Knoll D, Hennig L, Findeisen M, Oehme R, Giesa S, Ayala J, Welzel P. Bioorg. Med. Chem. 2003;11:2965. doi: 10.1016/s0968-0896(03)00187-1. [DOI] [PubMed] [Google Scholar]
- 85.Buchynskyy, Kempin U, Vogel S, Hennig L, Findeisen M, Muller D, Giesa S, Knoll H, Welzel P. Eur. J. Org. Chem. 2002:1149. [Google Scholar]
- 86.Li X. Ph.D. Thesis. Harvard University; 2006. [Google Scholar]
- 87.Cheng T-JR, Sung M-T, Liao H-Y, Chang Y-F, Chen C-W, Huang C-Y, Chou L-Y, Wu Y-D, Chen Y-H, Cheng Y-SE, Wong C-H, Ma C, Cheng W-C. Proc. Natl. Acad. Sci. USA. 2008;105:431. doi: 10.1073/pnas.0710868105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anikin, Buchynskyy A, Kempin U, Stembera K, Welzel P, Lantzsch G. Angew. Chem. Int. Ed. 1999;38:3703. doi: 10.1002/(sici)1521-3773(19991216)38:24<3703::aid-anie3703>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 89.Buchynskyy, Stembera K, Hennig L, Findeisen M, Giesa S, Welzel P. Eur. J. Org. Chem. 2002:1163. [Google Scholar]
- 90.Stembera K, Vogel S, Buchynskyy A, Ayala J, Welzel P. ChemBioChem. 2002;3:559. doi: 10.1002/1439-7633(20020603)3:6<559::AID-CBIC559>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 91.Ruhl T, Volke D, Stembera K, Hatanaka Y, Hennig H, Schumer F, Welzel P. Chem. Commun. 2002;15:1630. doi: 10.1039/b204007g. [DOI] [PubMed] [Google Scholar]
- 92.Stembera K, Buchynskyy A, Vogel S, Knoll D, Osman AA, Ayala JA, Welzel P. ChemBioChem. 2002;3:332. doi: 10.1002/1439-7633(20020402)3:4<332::AID-CBIC332>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 93.Vogel S, Buchynskyy A, Stembera K, Richter K, Hennig L, Muller D, Welzel P, Maquin F, Bonhomme C, Lampilas M. Bioorg. Med. Chem. 2000;10:1963. doi: 10.1016/s0960-894x(00)00377-2. [DOI] [PubMed] [Google Scholar]
- 94.Vogel S, Stembera K, Hennig L, Findeisen M, Giesa S, Welzel P, Lampilas M. Tetrahedron. 2001;57:4139. [Google Scholar]
- 95.Vogel S, Stembera K, Hennig L, Findeisen M, Giesa S, Welzel P, Tillier C, Bonhomme C, Lampilas M. Tetrahedron. 2001;57:4147. [Google Scholar]
- 96.Adachi M, Zhang Y, Leimkuhler C, Sun B, LaTour JV, Kahne DE. J. Am. Chem. Soc. 2006;128:14012. doi: 10.1021/ja065905c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Garneau S, Qiao L, Chen L, Walker S, Vederas JC. Bioorg. Med.Chem. 2004;12:6473. doi: 10.1016/j.bmc.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 98.Sun, Chen Z, Eggert US, Shaw SJ, LaTour JV, Kahne D. J. Am. Chem. Soc. 2001;123:12722. doi: 10.1021/ja0166693. [DOI] [PubMed] [Google Scholar]
- 99.Taylor JG. Ph.D. Thesis. Harvard University; 2006. [Google Scholar]
- 100.Welzel P. Angew. Chem. Int. Ed. 2007;46:4825. doi: 10.1002/anie.200700765. [DOI] [PubMed] [Google Scholar]
- 101.Kahne, Walker S, Cheng Y, Van Engen D. J. Am. Chem. Soc. 1989;111:6881. [Google Scholar]
- 102.Gildersleeve J, Pascal RAJ, Kahne D. J. Am. Chem. Soc. 1998;120:5961. [Google Scholar]
- 103.Wohlleben W, Pühler A. Arch. Microbiol. 1987;148:298. doi: 10.1007/BF00456708. [DOI] [PubMed] [Google Scholar]
- 104.Muth G, Wohlleben W, Pühler A. Mol. Gen. Genet. 1988;211:424. doi: 10.1007/BF00425695. [DOI] [PubMed] [Google Scholar]
- 105.Labes G, Bibb M, Wohlleben W. Microbiology. 1997;143:1503. doi: 10.1099/00221287-143-5-1503. [DOI] [PubMed] [Google Scholar]
- 106.Zotchev SB, Rozenfel’d SM, Zhdanov VG. Antibiot. Khimioter. 1988;33:200. [PubMed] [Google Scholar]
- 107.Zotchev SB, Orekhov AV, Rozenfel’d SM, Zhdanov VG. Antibiot. Khimioter. 1989;34:180. [PubMed] [Google Scholar]
- 108.Zotchev SB, Beritashvili DR, Rozenfel’d SM, Zhdanov VG, Orekhov AV. Dokl. Akad. Nauk SSSR. 1990;313:203. [PubMed] [Google Scholar]
- 109.Zotchev SB, Schrempf H, Hutchinson CR. J. Bacteriol. 1995;177:4809. doi: 10.1128/jb.177.16.4809-4812.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schuricht U, Endler K, Hennig L, Findeisen M, Welzel P. J. Prakt. Chem. 2000;342:761. [Google Scholar]
- 111.Dairi T. J. Antibiot. 2005;58:227. doi: 10.1038/ja.2005.27. [DOI] [PubMed] [Google Scholar]
- 112.Endler K, Schuricht U, Hennig L, Welzel P, Holst U, Aretz W, Bottger D, Huber G. Tetrahedron Lett. 1998;39:13. [Google Scholar]
- 113.Schuricht U, Hennig L, Findeisen M, Welzel P, Arigoni D. Tetrahedron Lett. 2001;42:3835. [Google Scholar]
- 114.Neundorf, Kohler C, Hennig L, Findeisen M, Arigoni D, Welzel P. ChemBioChem. 2003;4:1201. doi: 10.1002/cbic.200300622. [DOI] [PubMed] [Google Scholar]
- 115.Subramaniam-Niehaus, Schneider T, Metzger JW, Wohlleben W. Z. Naturforsch. C. 1997;52:217. doi: 10.1515/znc-1997-3-413. [DOI] [PubMed] [Google Scholar]
- 116.Nakagawa, Wu T-S, Keller PJ, Lee JP, Omura S, Floss HG. J. Chem. Soc. Chem. Commun. 1985:519. [Google Scholar]
- 117.Weber T, Welzel K, Pelzer S, Vente A, Wohlleben W. J. Biotechnol. 2003;106:221. doi: 10.1016/j.jbiotec.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 118.Ostash B, Saghatelian A, Walker S. Chem. Biol. 2007;14:257. doi: 10.1016/j.chembiol.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Petricek M, Petrickova K, Havlicek L, Felsberg J. J. Bacteriol. 2006;188:5113. doi: 10.1128/JB.01919-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McAlpine JB, Bachmann BO, Piraee M, Tremblay S, Alarco AM, Zazopoulos E, Farnet CM. J. Nat. Prod. 2005;68:493. doi: 10.1021/np0401664. [DOI] [PubMed] [Google Scholar]
- 121.Nett M, Ikeda H, Moore BS. Nat. Prod. Rep. 2009;26:1362. doi: 10.1039/b817069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hu Y, Walker S. Chem. Biol. 2002;9:1287. doi: 10.1016/s1074-5521(02)00295-8. [DOI] [PubMed] [Google Scholar]
- 123.Ostash, Doud EH, Lin C, Ostash I, Perlstein DL, Fuse S, Wolpert M, Kahne D, Walker S. Biochemistry. 2009;48:8830. doi: 10.1021/bi901018q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Payandeh J, Fujihashi M, Gillon W, Pai EF. J. Biol. Chem. 2006;281:6070. doi: 10.1074/jbc.M509377200. [DOI] [PubMed] [Google Scholar]
- 125.Woodyer RD, Li GY, Zhao HM, van der Donk WA. Chem. Commun. 2007:359. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- 126.Kuzuyama T, Hidaka T, Kamigri K, Imai S, Seto H. J. Antibiot. 1992;45:1812. doi: 10.7164/antibiotics.45.1812. [DOI] [PubMed] [Google Scholar]
- 127.Znahg W, Bolla M, Kahne D, Walsh CT. J. Am. Chem. Soc. 2010;132:6402. doi: 10.1021/ja1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bibb MJ. Curr. Opin. Microbiol. 2005;8:208. doi: 10.1016/j.mib.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 129.van Wezel GP, McKenzie NL, Nodwell JR. Meth. Enzymol. 2009;458:117. doi: 10.1016/S0076-6879(09)04805-8. [DOI] [PubMed] [Google Scholar]
- 130.Makitrinskyy R, Dumych Ye., Ostash B, Fedorenko V. Visn. Lviv. Univ. Ser. Biol. 2010;52:21. [Google Scholar]
- 131.Leskiw BK, Mah R, Lawlor EJ, Chater KF. J. Bacteriol. 1993;175:1995. doi: 10.1128/jb.175.7.1995-2005.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chater KF. Phil. Trans. R. Soc. 2006;361:761. doi: 10.1098/rstb.2005.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zaburannyy N, Ostash B, Fedorenko V. Bioinformatics. 2009;25:2432. doi: 10.1093/bioinformatics/btp402. [DOI] [PubMed] [Google Scholar]
- 134.Chater KF, Chandra G. J. Microbiol. 2008;46:1. doi: 10.1007/s12275-007-0233-1. [DOI] [PubMed] [Google Scholar]
- 135.Makitrinskyy R, Yu Rebets, Ostash B, Zaburanyi N, Rabyk M, Walker S, Fedorenko V. J. Ind. Microbiol. Biotechnol. 2010;37:559. doi: 10.1007/s10295-010-0701-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hertweck C, Luzhetskyy A, Yu Rebets, Bechthold A. Nat. Prod. Rep. 2007;24:162. doi: 10.1039/b507395m. [DOI] [PubMed] [Google Scholar]
- 137.Hodgson D. Adv. Microb. Physiol. 2000;42:47. doi: 10.1016/s0065-2911(00)42003-5. [DOI] [PubMed] [Google Scholar]
- 138.Ostash B, Makitrinskyy R, Walker S, Fedorenko V. Plasmid. 2009;61:171. doi: 10.1016/j.plasmid.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Walsh CT, Fischbach MA. J. Am. Chem. Soc. 2010;132:2469. doi: 10.1021/ja909118a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Medema MH, Trefzer A, Kovalchuk A, van de Berg M, Muller U, Heijne W, Wu L, Alam MT, Ronning CM, Nierman WC, Bovenberg RAL, Breitling R, Takano E. Genome Biol. Evol. 2010;2:212. doi: 10.1093/gbe/evq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Parenti F, Beretta G, Berti M, Arioli V. J. Antibiot. 1978;31:276. doi: 10.7164/antibiotics.31.276. [DOI] [PubMed] [Google Scholar]
- 142.Uchida R, Iwatsuki M, Kim Y-P, Ohte S, Omura S, Tomoda H. J. Antibiot. 2010;63:151. doi: 10.1038/ja.2010.9. [DOI] [PubMed] [Google Scholar]
- 143.Butaye P, Devriese LA, Haesebrouck F. J. Clin. Microbiol. 1998;36:1907. doi: 10.1128/jcm.36.7.1907-1911.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Butaye P, Devriese LA, Haesebrouck F. Antimicrob. Agents Chemother. 1999;43:2569. doi: 10.1128/aac.43.10.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Butaye P, Van Damme K, Devriese LA, Van Damme L, Baele M, Lauwers S, Haesebrouck F. J. Clin. Microbiol. 2000;54:181. doi: 10.1016/s0168-1605(99)00198-1. [DOI] [PubMed] [Google Scholar]
- 146.Torikata, Yoshikawa H, Katayama T, Arai M, Nakahara M, Kitano N. J. Antibiot. 1977;30:1060. doi: 10.7164/antibiotics.30.1060. [DOI] [PubMed] [Google Scholar]
- 147.van Heijenoort Y, Leduc M, Singer H, van Heijenoort J. J. Gen. Microbiol. 1987;133:667. doi: 10.1099/00221287-133-3-667. [DOI] [PubMed] [Google Scholar]
- 148.Eggert US, Ruiz N, Falcone BV, Branstrom AA, Goldman RC, Silhavy TJ, Kahne D. Science. 2001;294:361. doi: 10.1126/science.1063611. [DOI] [PubMed] [Google Scholar]
- 149.Meyers E, Miraglia GJ, Smith DA, Basch HI, Pansy FE, Trejo WH, Donovick R. Appl. Microbiol. 1968;16:603. doi: 10.1128/am.16.4.603-608.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.US Pat., 6 077 830. 2000
- 151.US Pat., 6 242 424. 2001
- 152.Volke F, Waschipky R, Pampel A, Donnerstag A, Lantzsch G, Pfeiffer H, Richter W, Klose G, Welzel P. Chem. Phys. Lipids. 1997;85:115. doi: 10.1016/s0009-3084(96)02649-7. [DOI] [PubMed] [Google Scholar]
- 153.Butaye P, Devriese LA, Haesebrouck F. J. Antimicrob. Chemother. 2000;46:713. doi: 10.1093/jac/46.5.713. [DOI] [PubMed] [Google Scholar]
- 154.Leduc M, Kasra R, van Heijenoort J. J. Bacteriol. 1982;152:26. doi: 10.1128/jb.152.1.26-34.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kusser W, Ishiguro EE. J. Bacteriol. 1985;164:861. doi: 10.1128/jb.164.2.861-865.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kariyama R. J. Antibiot. 1982;35:1700. doi: 10.7164/antibiotics.35.1700. [DOI] [PubMed] [Google Scholar]
- 157.Vollmer W, von Rechenberg M, Holtje J-V. J. Biol. Chem. 1999;274:6726. doi: 10.1074/jbc.274.10.6726. [DOI] [PubMed] [Google Scholar]
- 158.Vollmer W, Bertsche U. Biochim. Biophys. Acta. 2008;1778:1714. doi: 10.1016/j.bbamem.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 159.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. FEMS Microbiol. Rev. 2008;32:234. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 160.Takahashi S, Nitta K, Honjo S, Cho F, Umezawa H. J. Antibiot. 1973;36:513. doi: 10.7164/antibiotics.26.513. [DOI] [PubMed] [Google Scholar]
- 161.Frese E, Blobel H. Z. Veterinarmed. 1973;B20:46. [PubMed] [Google Scholar]
- 162.Bauer F, Bost G. Antimicrob. Agents Chemother. 1965:749. [PubMed] [Google Scholar]
- 163.Edwards JE, Bequette BJ, McKain N, McEwan NR, Wallace RJ. British J. Nutr. 2005;94:64. doi: 10.1079/bjn20051444. [DOI] [PubMed] [Google Scholar]
- 164.Reidel G, Reiter H, Losch U. Z. Tierphysiol. 1974;32:328. [PubMed] [Google Scholar]
- 165.Dealy J, Moeller MW. J. Anim. Sci. 1977;44:734. doi: 10.2527/jas1977.445734x. [DOI] [PubMed] [Google Scholar]
- 166.Hagsten, Grant RJ, Meade RJ, O’Kelley R. J. Anim. Sci. 1980;50:484. doi: 10.2527/jas1980.503484x. [DOI] [PubMed] [Google Scholar]
- 167.Russell B, Strobel HJ, Chen G. Appl. Environ. Microbiol. 1988;54:872. doi: 10.1128/aem.54.4.872-877.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Edwards E, McEwan NR, McKain N, Walker N, Wallace RJ. Microbiology. 2005;151:717. doi: 10.1099/mic.0.27602-0. [DOI] [PubMed] [Google Scholar]
- 169.Devriese A, Daube G, Hommez J, Haesebrouck F. J. Appl. Bacteriol. 1993;75:55. doi: 10.1111/j.1365-2672.1993.tb03407.x. [DOI] [PubMed] [Google Scholar]
- 170.Bolder M, Wagenaar JA, Putirulan FF, Veldman KT, Sommer M. Poult. Sci. 1999;78:1681. doi: 10.1093/ps/78.12.1681. [DOI] [PubMed] [Google Scholar]
- 171.Dealy J, Moeller MW. J. Anim. Sci. 1976;42:1331. doi: 10.2527/jas1976.4251331x. [DOI] [PubMed] [Google Scholar]
- 172.George BA, Fagerberg DJ, Quarles CL, Fenton JM, McKinley GA. Am. J. Vet. Res. 1982;43:299. [PubMed] [Google Scholar]
- 173.Mitsuhashi S, Iyobe S, Hashimoto H, Umezawa H. J. Antibiot. 1970;23:319. doi: 10.7164/antibiotics.23.319. [DOI] [PubMed] [Google Scholar]
- 174.Iyobe S, Mitsuhashi S, Umezawa H. J. Bacteriol. 1971;108:946. doi: 10.1128/jb.108.2.946-947.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Brana H, Hubacek J, Konig J. Folia Microbiol. 1973;13:257. doi: 10.1007/BF02872865. [DOI] [PubMed] [Google Scholar]
- 176.George BA, Fagerberg DJ. Am. J. Vet. Res. 1984;45:2336. [PubMed] [Google Scholar]
- 177.Riedl S, Ohlsen K, Werner G, Witte W, Hacker J. Antimicrob. Agents Chemother. 2000;44:3189. doi: 10.1128/aac.44.11.3189-3192.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.van den Bogaard E, Hazen M, Hoyer M, Oostenbach P, Stobberingh EE. Antimicrob. Agents Chemother. 2002;46:110. doi: 10.1128/AAC.46.1.110-118.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Poole TL, McReynolds JL, Edrington TS, Byrd JA, Callaway TR, Nisbet DJ. J. Antimicrob. Chemother. 2006;58:359. doi: 10.1093/jac/dkl249. [DOI] [PubMed] [Google Scholar]
- 180.Kean EL, Wei Z. Glycoconjugate J. 1998;15:405. doi: 10.1023/a:1006982003957. [DOI] [PubMed] [Google Scholar]
- 181.Waals P. v.d. Tijdschrift voor Diergeneeskunde. 1973;98:853. [PubMed] [Google Scholar]
- 182.Lantzsch G, Binder H, Heerklotz H, Welzel P, Klose G. Langmuir. 1998;14:4095. [Google Scholar]
- 183.Graves-Woodward K, Pratt RF. Biochemistry. 1999;38:10533. doi: 10.1021/bi982309p. [DOI] [PubMed] [Google Scholar]
- 184.Marzian S, Happel M, Wagner U, Muller D, Welzel P, Fehlhaber H-W, Stark A, Schultz H-J, Markus A, Limbert M, van Heijenoort Y, van Heijenoort J. Tetrahedron. 1994;50:5299. [Google Scholar]
- 185.Kosmol R, Hennig L, Welzel P, Findeisen M, Muller D, Markus A, van Heijenoort J. J. Prakt. Chem. 1997;339:340. [Google Scholar]
- 186.McPherson DC, Popham DL. J. Bacteriol. 2003;185:1423. doi: 10.1128/JB.185.4.1423-1431.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Arbeloa, Segal H, Hugonnet J-E, Josseaume N, Dubost L, Brouard J-P, Gutmann L, Mengin-Lecreulx D, Arthur M. J. Bacteriol. 2004;186:1221. doi: 10.1128/JB.186.5.1221-1228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Popham DL, Young KD. Curr. Opin. Microbiol. 2003;6:594. doi: 10.1016/j.mib.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 189.Goffin C, Ghuysen JM. Microbiol. Mol. Biol. Rev. 1998;62:1079. doi: 10.1128/mmbr.62.4.1079-1093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hoskins J, Mastushima P, Mullen DL, Tang J, Zhao G, Meier TI, Nicas TI, Jaskunas SR. J. Bacteriol. 1999;181:6552. doi: 10.1128/jb.181.20.6552-6555.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wang TS, Manning SA, Walker S, Kahne D. J. Am. Chem. Soc. 2008;130:14068. doi: 10.1021/ja806016y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Perlstein DL, Zhang Y, Wang T-S, Kahne DE, Walker S. J. Am. Chem. Soc. 2007;129:12674. doi: 10.1021/ja075965y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yuan Y, Barrett D, Zhang Y, Kahne D, Sliz P, Walker S. Proc. Natl. Acad. Sci. USA. 2007;104:5348. doi: 10.1073/pnas.0701160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Perlstein DL, Wang T-SA, Doud EH, Kahne DE, Walker S. J. Am. Chem. Soc. 2010;132:48. doi: 10.1021/ja909325m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Di Berardino, Dijkstra A, Stuber D, Keck W, Gubler M. FEBS Lett. 1996;392:184. doi: 10.1016/0014-5793(96)00809-5. [DOI] [PubMed] [Google Scholar]
- 196.Park W, Matsuhashi M. J. Bacteriol. 1984;157:538. doi: 10.1128/jb.157.2.538-544.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Branstrom A, Midha S, Goldman RC. FEMS Microbiol. Lett. 2000;191:187. doi: 10.1111/j.1574-6968.2000.tb09338.x. [DOI] [PubMed] [Google Scholar]
- 198.Lovering L, de Cactro L, Strynadka NCJ. J. Mol. Biol. 2008;383:167. doi: 10.1016/j.jmb.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 199.Mani N, Sanchet P, Jiang Z-D, McNaney C, DeCenzo M, Knight B, Stankis M, Kuranda M, Rothstein DM. J. Antibiot. 1998;51:471. doi: 10.7164/antibiotics.51.471. [DOI] [PubMed] [Google Scholar]
- 200.Butaye P, Devriese LA, Haesebrouck F. Clin. Microbiol. Rev. 2003;16:175. doi: 10.1128/CMR.16.2.175-188.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Komatsuzawa H, Suzuki J, Sugai M, Miyake Y, Suginaka H. J. Anitmicrob. Chemother. 1994;33:1155. doi: 10.1093/jac/33.6.1155. [DOI] [PubMed] [Google Scholar]
- 202.Aarestrup FM, Bager F, Jensen NE, Madsen M, Meyling A, Wegener HK. APMIS. 1998;106:606. doi: 10.1111/j.1699-0463.1998.tb01391.x. [DOI] [PubMed] [Google Scholar]
- 203.Nishi H, Komatsuzawa H, Yamada S, Fujiwara T, Ohara M, Ohta K, Sugiyama M, Ishikawa T, Sugai M. Microbiol. Immunol. 2003;47:927. doi: 10.1111/j.1348-0421.2003.tb03466.x. [DOI] [PubMed] [Google Scholar]
- 204.Nishi H, Komatsuzawa H, Fujiwara T, McCallum N, Sugai M. Antimicrob. Agents Chemother. 2004;48:4800. doi: 10.1128/AAC.48.12.4800-4807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ruiz, Falcone B, Kahne D, Silhavy TJ. Cell. 2005;121:307. doi: 10.1016/j.cell.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 206.Edwards JE, McEwan NR, Wallace RJ. J. Appl. Microbiol. 2008;104:1617. doi: 10.1111/j.1365-2672.2007.03689.x. [DOI] [PubMed] [Google Scholar]
- 207.Anderson DI, Hughes D. Nat. Rev. Microbiol. 2010;8:260. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- 208.Metten K-H, Hobert K, Marzian S, Hackler UE, Heinz U, Welzel P, Aretz W, Bottger D, Hedtmann U, Seibert G, Markus A, Limbert M, van Heijenoort Y, van Heijenoort J. Tetrahedron. 1992;48:8401. [Google Scholar]
- 209.Bentley SD, Chater KF, Cerdeño-Tárraga A-M, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang C-H, Kieser T, Larke L, Murphy L, Oliver K, O’Niel S, Rabbinowitsch E, Rajandream M-A, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. Nature. 2002;417:141. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 210.Vollmer W, Holtje J-V. Antimicrob. Agents Chemother. 2000;44:1181. doi: 10.1128/aac.44.5.1181-1185.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ramachandran V, Chandrakala B, Kumar VP, Usha V, Solapure SM, de Sousa SM. Antimicrob. Agents Chemother. 2006;50:1425. doi: 10.1128/AAC.50.4.1425-1432.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hutchings MI, Hong H-J, Buttner MJ. Mol. Microbiol. 2006;59:923. doi: 10.1111/j.1365-2958.2005.04953.x. [DOI] [PubMed] [Google Scholar]
- 213.De Pascale G, Grigoriadou C, Losi D, Donadio S. J. Appl. Microbiol. 2007;103:133. doi: 10.1111/j.1365-2672.2006.03231.x. [DOI] [PubMed] [Google Scholar]
- 214.Cani PD, Delzenne NM. Curr. Pharm. Des. 2009;15:1546. doi: 10.2174/138161209788168164. [DOI] [PubMed] [Google Scholar]