

Abstract
In this study we investigated corresponding precursor and active forms of a p53 small molecule inhibitor for effect on temozolomide (TMZ) anti-tumor activity against glioblastoma (GBM), using both in vitro and in vivo experimental approaches. Results from in vitro cell viability analysis showed that the cytotoxic activity of TMZ was substantially increased when GBMs with wild-type p53 were co-treated with the active form of p53 inhibitor, and this heightened cytotoxic response was accompanied by increased PARP cleavage as well as elevated cellular phospho-H2AX. Analysis of the same series of GBMs, as intracranial xenografts in athymic mice, and administering corresponding p53 inhibitor precursor, that is converted to the active compound in vivo, yielded results consistent with the in vitro analyses: i.e., TMZ + p53 inhibitor precursor co-treatment, of three distinct wild-type p53 GBM xenografts, resulted in significant enhancement of TMZ anti-tumor effect relative to treatment with TMZ alone, as indicated by serial bioluminescence monitoring as well as survival analysis (p < 0.001 for co-treatment survival benefit in each case). Mice receiving intracranial injection with p53 null GBM showed similar survival benefit from TMZ treatment regardless of the presence or absence of p53 inhibitor precursor. In total, our results indicate that the p53 active and precursor inhibitor pair enhance TMZ cytotoxicity in vitro and in vivo, respectively, and do so in a p53-dependent manner.
Keywords: glioblastoma, p53, temozolomide, xenograft
Introduction
Attempts and approaches at manipulating p53 activity in treating human cancer have been numerous and diverse. For example, viral-mediated introduction and expression of wild-type TP53 in p53-defective tumor cells has been extensively investigated for more than a decade, including through clinical trial activity (1). Alternative approaches for increasing tumor cell wild-type p53 activity include the use of small molecules that promote p53 transcription, and the use of compounds that inhibit p53’s interaction with mdm2 (2).
Perhaps because of it being counter to conventional thinking about the role of tumor suppressor genes in cancer etiology, as well as being counterintuitive regarding the way in which tumor suppressor genes are viewed in relation to the treatment of cancer, there has been relatively little research directed towards the development of anti-tumor therapeutic strategies that include a p53 inhibitory component. Indeed, as a monotherapy, such a treatment approach could promote increased tumor cell proliferation and decreased tumor cell apoptosis. However, the potential consequences of attempted cell cycling by tumor cells with damaged DNA, resulting from genotoxic therapy with concurrent inhibition of p53, are interesting to consider. In fact, results from several studies, involving in vitro investigation of tumor cell lines, support enhanced cytotoxic chemotherapeutic response in association with p53 inhibition (3–6). Furthermore, with respect to GBM, the p53 small molecule inhibitor pifithrin-α, which was identified nearly a decade ago in association with a chemical library screen (7), has been shown to enhance in vitro cytotoxic effect of temozolomide (TMZ), a DNA alkylator, as well as the cytotoxic effect of chloroethylating nitrosoureas such as carmustine (8, 9).
In addition to reasons described above, in vivo investigation of p53 small molecule inhibitors, as part of a cancer treatment strategy, has been hindered due to limitations imposed by physical properties of the pifithrin-α reference compound (10). Recently, however, derivatives of the reference compound were described with respect to their potential in vivo use (11). In the current study we have tested one of these compounds, using an intracranial GBM xenograft therapy-response model, and present results indicating its enhancement of TMZ anti-tumor activity in vivo, and in a manner that is dependent on tumor cell p53 status.
Materials and Methods
In vitro experiments
GBM xenografts used in this study have been previously described (12,13), as has the modification of xenografts for bioluminescence imaging (13). Culturing of xenograft cells were as non-adherent neurospheres in neurobasal media (Invitrogen, San Diego, CA), while U87 cells (American Type Culture Collection) were propagated as monolayer cultures in DMEM supplemented with 10% fetal calf serum. Temozolomide (TMZ: obtained as Temodar from Schering-Plough, Kenilworth, NJ) and active form p53 inhibitor (cyclic pifithrin-α p-nitro, Calbiochem, San Diego, CA) were dissolved in dimethyl-sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO) as 20 and 5 mM stock solutions, respectively. For bioluminescence viability analysis, cells were treated with DMSO, TMZ (added to concentrations of 50 or 100 μM), p53 inhibitor (concentration of 10 μM), or a combination of TMZ and p53 inhibitor, with chemical agents added to media 1x/day for 3 consecutive days. Cell culture specimens were examined for bioluminescence signal using a Xenogen imaging system (Caliper Life Sciences, Alameda, CA), following the addition of 25 μl of 20 mg/ml sodium luciferin (Gold Biotechnology, St. Louis, MO) in phosphate buffered saline (PBS, Invitrogen).
Flow cytometry cell cycle analysis
U87 cells were treated 1x/day for 3 days with DMSO only, 10 μM p53 inhibitor, 50 or 100 μM TMZ, or 50 or 100 μM TMZ + 10 μM p53-inhibitor. At 1, 4, and 7 days following final treatment the cells were harvested, washed with PBS, and fixed with cold 70% ethanol. Cells were stained with propidium iodide and examined by flow-cytometry (BD LSR II, Becton-Dickinson, Franklin Lakes, NJ), with results analyzed using FlowJo software (Ashland, OR).
Immunoblot analysis
Primary antibodies used for immunoblot analysis (previously described: see reference 13) were for detection of PARP (Cell Signaling Tech, Danvers, MA), phospho-histone H2AX (Cell Signaling Tech), p53 (Sigma-Aldrich), p21 (Santa Cruz Biotech, Santa Cruz, CA), MGMT (R&D Systems, Minneapolis, MN), or beta-tubulin (Millipore, Danvers, MA). Secondary antibodies used were either anti-mouse or anti-rabbit (Cell Signaling Tech), or anti-goat (Santa Cruz Biotech).
Methylation-specific PCR
Anyalysis of MGMT promoter methylation was as described by Esteller et al (14). PCR products were resolved in 4.5% agarose gels (NuSieve 3:1, Lonza, Inc., Allendale, NJ), and were subsequently stained using ethidium bromide.
In vivo experiments
Procedures used for intracranial tumor therapy-response experiments, including monitoring of tumor growth and response to therapy by bioluminescence imaging, have been previously described (13). Treatment groups for the experiments reported here were as follows: three days intraperitoneal DMSO and oral suspension vehicle (OraPlus, Paddock Laboratories, Minneapolis, MN) by gavage (control group); three days 10 mg/kg TMZ in oral suspension vehicle for GBM 12, GBM 14 and U87, and 50 mg/kg for GBM 26; three days 0.25 mg p53 inhibitor precursor (pifithrin-α p-nitro, Calbiochem) in DMSO by intraperitoneal injection; or 3 days combination TMZ and p53 inhibitor as indicated for their administration as single agents.
Results
GBM xenograft explant cultures were incubated with TMZ +/− cyclic pifithrin-α p-nitro (Supplementary Figure 1: see reference 11) to assess in vitro effects of this p53 inhibitor on tumor cell TMZ cytotoxic response. Results for p53 wild-type (p53wt) GBM 26 showed substantial decreases in viable cell number resulting from TMZ + p53 inhibitor co-treatments, in relation to cells treated with TMZ alone (Figures 1A, 1B). Importantly, cyclic pifithrin-α p-nitro alone showed no anti-tumor effect. In contrast to the results for GBM 26, GBM 12 cells, which express no endogenous p53 (p53null: see reference 12), showed similar cytotoxic response to TMZ irrespective of the presence or absence of the p53 inhibitor (Figure 1C). As shown by others (11), cyclic pifithrin-α p-nitro inhibits DNA damage-associated induction of the negative cell cycle regulator p21, but only in p53wt cells (Figure 1D). Also, and consistent with the p53 small molecule inhibitor treatments, pre-treating cells with p53 siRNA enhanced TMZ cytotoxic response of GBM 26 cells, relative to pretreating cells with non-specific siRNA, whereas GBM 12 cells responded similarly to TMZ whether pre-incubated with p53 or non-specific siRNA (Supplementary Figure 2).
Figure 1.
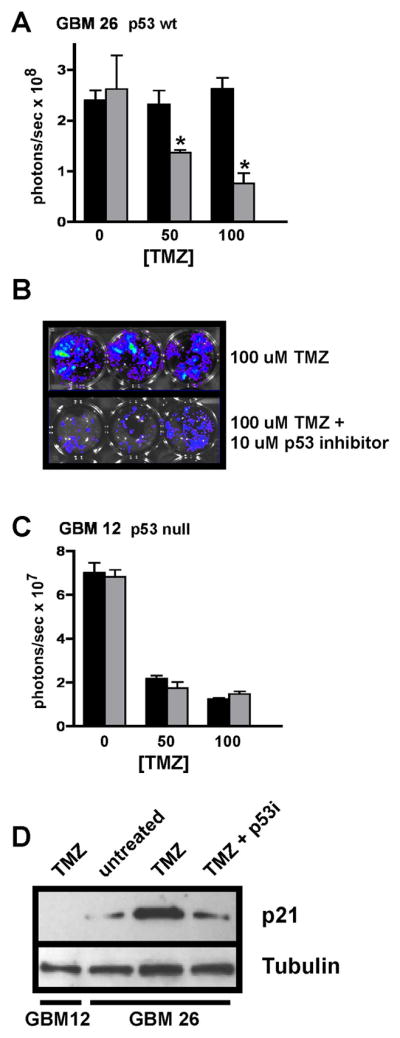
In vitro cell viability analysis of GBM xenograft explant cultures subjected to TMZ +/− p53 inhibitor treatments. A) GBM 26 cells were treated with TMZ only (black) at the indicated concentrations, or with TMZ + 10 μM cyclic pifithrin-α p-nitro (gray). Treatments were administered 1x/day for three consecutive days (each treatment group consisting of triplicate samples), and 4 days following final treatment the luminescence of each treatment group (Figure 1B) was determined. Average values for each treatment group are shown, with standard error of mean indicated. Asterisks denote comparisons for which there is a significant difference (p < 0.05) between TMZ only vs. TMZ + p53 inhibitor treatments. C) The same experimental design as described for GBM 26 was used for assessing p53null GBM 12 cell response to treatments, with results presented as indicated for panel A. D) Immunoblot results for p21 expression in cells treated with TMZ +/− p53 inhibitor (p53i). TMZ (100 μM) induces p21 expression in cells with wild-type p53 (GBM 26), and this induction is suppressed through concurrent treatment with p53 inhibitor (incubations as described above for panels A and C). In contrast to the results for GBM 26, p21 is not detected in p53null GBM 12 cells, irrespective of treatment (shown) or lack of treatment with TMZ. Replicate samples for this analysis yielded similar results.
To further evaluate cyclic pifithrin-α p-nitro for in vitro activity, as well as to contrast results from the use of this compound with those previously reported for the pifithrin-α reference compound (Supplementary Figure 1C; see reference 8), U87 cells were incubated with TMZ +/− cyclic pifithrin-α p-nitro and examined by flow cytometric cell cycle analysis. Similar to results reported for the reference compound (8), higher sub-G1 cell fractions were observed in samples treated with TMZ + cyclic pifithrin-α p-nitro, relative to TMZ treatment alone. Sub-G1 ratios for U87 cells subjected to co-treatment vs. treatment with 100 μM TMZ only were 1.97, 2.25, and 1.54, at days 1, 4, and 7 following final treatment, respectively (Figures 2A, 2B). Consistent with the GBM xenograft explant culture cytotoxic response results described above (Figure 1), results from U87 flow cytometry analysis showed little difference in cell cycle distributions between U87 treated with cyclic pifithrin-α p-nitro vs. mock-treated control cells (Figure 2A).
Figure 2.
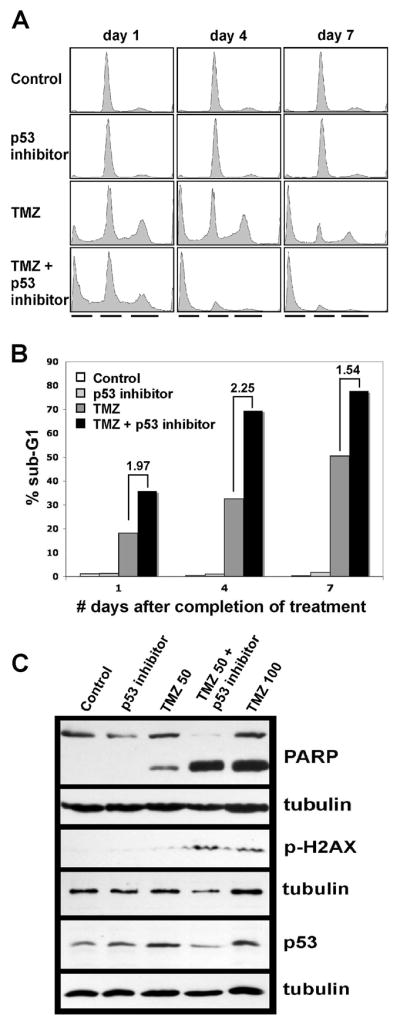
Flow cytometry and immunoblot analysis of U87 cells subjected to TMZ +/− cyclic pifithrin-α p-nitro inhibitor treatments. A) Cell treatments were as follows: control = DMSO only; p53 inhibitor only = 10 μM cyclic pifithrin-α p-nitro administered daily for 3 consecutive days; TMZ only = 50 or 100 μM TMZ administered daily for 3 consecutive days; 50 or 100 μM TMZ + 10 μM p53 inhibitor administered daily for 3 consecutive days. Cells were harvested from individual plates on the days indicated (values represent days following final treatment administration) and prepared for flow cytometry analysis. Associated flow cytometry profiles show similar amounts of and predominant G1 phase cell fractions in control and p53 inhibitor only samples at each time point, whereas time-dependent increases in proportion of sub-G1 cell fractions are evident in the TMZ only and the TMZ + p53 inhibitor series (black lines beneath each column of profiles indicate, from left to right, the positions of the sub-G1, G1, and G2/M cell fractions). Results shown are for 100 uM TMZ treatments. B) Bar graph showing sub-G1 ratios for TMZ + p53 inhibitor vs. TMZ only treatments, and indicating that the proportion of sub-G1 cells is largest, at each time point, in cultures subjected to combined TMZ + p53 inhibitor treatment. C) Immunoblot results for U87 cells subjected to control and p53 inhibitor only treatments as described above, and to combination treatments with 50 or 100 uM TMZ +/− p53 inhibitor. Results are for cells 4 days after final treatment administration, and show presence or absence of cleaved PARP, presence or absence of phospho-H2AX, and relative amounts of p53 protein; each tubulin result is presented beneath the corresponding result obtained from the same filter, and address total protein loading variation. Note the lack of effect of TMZ on p53 expression (middle and far right lanes of p53 immunoblot panel), which is consistently observed in tumors expressing p53.
Immunoblot analysis of U87 cells incubated with TMZ +/− cyclic pifithrin-α p-nitro showed increased PARP cleavage resulting from combined treatment, in comparison with cells treated with the same concentration of TMZ alone (Figure 2C), thereby suggesting increased caspase activity in association with the enhanced cytotoxic response to TMZ + p53 inhibitor. Heightened apoptotic response to combination treatment was also indicated by increased phospho-H2AX in cells incubated with TMZ + p53 inhibitor, relative to cells incubated with the same concentration of TMZ alone. Levels of p53 protein were similar between culture treatments (Figure 2C), consistent with the inhibitory activity of pifithrin-α compounds involving suppression of existing p53 activity, rather than suppression of p53 expression.
Previous study of pifithrin-α p-nitro derivatives has revealed that 2-aminothiazole salt cyclization (Figures 1A, 1B) is required for the formation of active p53 inhibitor, and that this cyclization occurs spontaneously in vivo, albeit over a period of several hours (11). Because active form p53 inhibitors have relatively short half-lives in vivo, it has been suggested that the kinetics of inactive, open form precursor conversion to corresponding cyclized, active form compound can be used to advantage for achieving prolonged cell exposure to active state inhibitors in vivo, and thereby achieve prolonged p53 inhibitory effect (11). In accord with this line of reasoning, we investigated the open form precursor of cyclic pifithrin-α p-nitro (Supplementary Figure 1A) for effect on TMZ in vivo anti-tumor activity, using a previously-described intracranial GBM xenograft therapy-response model (13). Mice injected with p53wt tumor cells (GBM 14, GBM 26, and U87), or with cells from p53null GBM 12, were monitored for luminescence increases indicative of log phase growth of injected cells, at which time TMZ (gavage) and p53 inhibitor precursor (intraperitoneal injection) were administered singularly or concurrently 1x/day for three consecutive days (treatment periods indicated by vertical gray bars, Figures 3A, 3B). For mice injected with p53wt GBM, survival was significantly extended by combined TMZ + p53 inhibitor treatment, relative to TMZ treatment alone, and this effect was observed for three p53wt tumors (p < 0.001 for each: Figure 3A, Table 1). Survival benefits were consistent with corresponding results from bioluminescence monitoring, which revealed prolonged decrease of p53wt intracranial tumor luminescence in mice receiving TMZ + p53 inhibitor (Figure 3B). It is worth noting that p53 inhibitor precursor enhancement of TMZ cytotoxicity was observed irrespective of p53wt GBM sensitivity to TMZ alone, as TMZ resistant GBM 26, with unmethylated MGMT promoter and highly expressed MGMT protein, as well as GBM 14 and U87, in which MGMT promoter methylation is accompanied by a lack of detectable MGMT protein (Figures 3C, 3D), all showed significantly increased response to combination treatment. Conversely, mice with p53null GBM 12 intracranial tumor did not experience significant survival benefit from combined TMZ + p53 inhibitor precursor treatment, in comparison to mice receiving treatment with TMZ only (Figure 3A, Table 1).
Figure 3.

Intracranial xenograft therapy-response to TMZ +/− p53 inhibitor precursor treatments. Groups of 32 mice received intracranial tumor cell injection (300,000 cells/mouse) using luciferase-modified GBM, and at times when log-phase growth was indicated by BLI, each series of mice was randomized into four treatment groups with 8 mice/group, and treatments were initiated. Treatment groups were as follows: control (intraperitoneal injection of DMSO + gavage with TMZ suspension vehicle, 1x/day for 3 days: broken black line); p53 inhibitor precursor only (0.25 mg in 50 ul DMSO administered by intraperitoneal injection 1x/day for 3 days: broken gray line); TMZ only (10 mg/kg administered in oral suspension vehicle by gavage, 1x/day for 3 days for GBMs 12, 14, and U87, and 50 mg/kg 1x/day for 3 days for GBM 26: solid black line); or combination TMZ + p53 inhibitor (solid gray line), as indicated for each agent’s use alone. Treatment periods are indicated by a vertical gray bar in each of the graphs in panels A and B. A) Survival plots for each experiment show that combined TMZ + p53 inhibitor precursor treatment significantly extends symptom-free survival relative to TMZ alone in mice with p53wt tumor (GBM 26, GBM 14, and U87), but not in mice with p53null GBM 12. P-values indicated at the top of each graph are for TMZ alone vs. TMZ + p53 inhibitor precursor comparisons. B) Corresponding bioluminescence imaging curves, with plots beginning at day of treatment initiation, and showing either lack of effect or suppression of luminescence resulting from TMZ +/− p53 inhibitor precursor treatments (mean luminescence values plotted for each treatment group, with standard error of mean indicated). For the plots involving GBM 26, GBM 14, and U87, the number of days between log phase tumor growth for TMZ only and TMZ + p53 inhibitor precursor treatment groups is indicated, and the order of these differences (largest to smallest) is consistent with the order for corresponding length of survival benefit (see Table 1). To the far right are luminescence intensity overlay images. For mice with intracranial GBM 14, GBM 26, or U87, mice with lowest and highest intracranial tumor luminescence in TMZ only and TMZ + p53 inhibitor treatment groups, respectively, are shown, with images captured at days indicated by the arrowheads along the TMZ + p53 inhibitor plots in panel A. For GBM 12, which does not show additional response to combination treatment beyond that associated with TMZ alone, image overlays are shown for mice that define the median tumor luminescence from each group at the day indicated by the arrowhead over the corresponding graph in panel A. C) MGMT promoter methylation analysis showing lack of methylation in GBM 26, which has corresponding detectable protein expression (panel D), and displays resistance to TMZ alone (panel A). Results for tumors GBM 12, GBM 14, and U87 show presence of MGMT promoter ethylation, and corresponding lack of detectable protein by immunoblot.
Table 1.
Summary of TMZ +/− p53 Inhibitor Xenograft Studies
| Tumor |
Control |
p53i only |
TMZ only |
TMZ + p53i |
p-value |
|---|---|---|---|---|---|
| GBM 14 | 28.1 | 27.6 | 76.8 | >142 (>85.0%) | < 0.001 |
| 29.5 | 28.5 | 73.5 | 145 (97.3%) | ||
| GBM 26 | 48.3 | 48.6 | 51.1 | 82.9 (62.2%) | < 0.001 |
| 48.0 | 49.0 | 54.0 | 85.0 (57.4%) | ||
| U87 | 29.9 | 29.6 | 36.9 | 53.7 (45.5%) | < 0.001 |
| 30.0 | 30.0 | 37.0 | 51.0 (37.8%) | ||
| GBM 12 | 15.8 | 16.3 | 50.9 | 52.0 (2.2%) | = 0.178 |
| 16.0 | 16.0 | 50.5 | 54.0 (6.9%) |
Mean (black) and median (gray) values in days for duration of symptom-free survival of mice receiving intracranial injection of indicated tumor cells and receiving indicated treatments. Values in parentheses represent mean or median percent survival difference in comparing TMZ only vs. TMZ +p53 inhibitor (p53i). P-values for TMZ only vs. TMZ + p53i survival comparisons are indicated to the right.
Discussion
Research for examining potential clinical applications of p53 small molecule inhibitors has often involved experimental paradigms that address the protection of normal cells, rather than determining effects on tumor cell response to therapy that includes p53 inhibition. Specifically, there is considerable interest regarding whether p53 small molecule inhibitors can be used to spare normal cells from p53-mediated apoptosis that occurs from oxidative stress during stroke, renal injury, or cardiac arrest (15), as well as from the genotoxic effects of cancer treatments (7). Small molecule inhibition of p53 in tumor cells, for enhancing the anti-cancer effects of therapy, has not attracted much attention, even though there is ample, related evidence in support of this concept. This support, however, primarily stems from the use of viral gene product expression (4) and RNA interference (3, 6) to suppress tumor cell p53 function, and the perspective of the associated studies has been to investigate the role of p53 as a determinant of tumor cell response to cytotoxic therapy, rather than to pursue p53 as a target for augmenting the effects of therapy.
In the current study we have contrasted p53wt vs. p53null GBM xenograft response to cytotoxic therapy that includes the use of a small molecule inhibitor of p53. Significantly extended survival benefit from combination therapy was evident among series of mice with intracranial p53wt GBM, whereas mice with intracranial p53null GBM did not experience survival benefit from combination therapy. The issue regarding the response of p53 missense mutants to p53 inhibitor treatment is complex due to the diversity of p53 mutations in human cancer, the distinct transcriptional effects of individual p53 mutations, and the transcription-independent cellular activities of mutant p53 (16–18). Current studies are in progress to address the variability of p53 missense mutant GBM response to p53 inhibitor augmented cytotoxic therapy.
There are numerous candidates to consider as mediators of p53 inhibitor enhanced cytotoxic response, with our results currently supporting more extensive investigation of p53 inhibitor mediated suppression of p21 (Figure 1D), as well as investigation of apoptotic response effectors that are inferred by our preliminary results (Figure 2; Supplementary Figure 3). There are, of course, additional aspects of this cytotoxic response to be investigated, one of which is indicated in a recent report showing that p53 inhibition can suppress TMZ associated induction of MGMT (19). In the case of the tumor cell sources used here, this could be a contributing factor associated with p53 inhibitor enhancement of TMZ cytotoxicity against GBM 26, for which MGMT is unmethylated and expressed, but is probably less likely for GBM 14 and U87 tumors, for which MGMT is presumably silenced through MGMT promoter methylation (Figure panels 3C and 3D).
In addition to investigating the molecular biology of pathways and proteins associated with enhanced TMZ cytotoxic effect through concurrent p53 inhibition, the identification of surrogate markers that can be used to assess p53 inhibitor activity in tumors is critically important. The ability to analyze tumors for temporal variations in the expression and/or activity of biomarkers that respond to p53 inhibition, in combination with corresponding analysis of tumor extracts for temporal variations in p53 inhibitor content (e.g., through use of HPLC), will provide key information regarding inhibitor pharmacodynamics for optimizing combination therapy dosing and sequencing.
Results presented here provide some basis for considering the subgroup of GBM that would benefit from therapy that includes p53 inhibition. As concerns this point, it is significant that p53 inhibition enhances TMZ cytotoxicity in p53wt tumors irrespective of their sensitivity to TMZ monotherapy. Consequently, this therapeutic approach could conceivably benefit the two-thirds of GBM patients whose tumors are of wild-type p53 status (20). Looking beyond the GBM-specific interests of this report, it seems plausible that the results of our study are generalizable to other types of cancer, as well as to other types of genotoxic therapy.
An additional observation associated with this study is that TMZ + p53 inhibitor treatment is without adverse effect, as evidenced by autopsy analysis of animal subjects that showed no abnormal cellular proliferation in tissue from mice receiving TMZ + p53 inhibitor treatment (e.g., gastrointestinal tract: supplementary Figure 4A). This, in combination with the long term survival of mice that received TMZ + p53 inhibitor (see GBM 14 results in Figure 3A), as well as the similarity in group body weight patterns for TMZ only and TMZ + p53 inhibitor treated mice (Supplementary Figure 4B), suggest a lack of acute as well as of long-term effects of this therapy. Nonetheless, the simultaneous administration of a DNA damaging agent with an inhibitor of DNA repair could promote undesirable systemic side effects, including the development of independent cancers in other organs. To address this concern, future research should perhaps include strategies for the local delivery of inhibitor to tumor, in order to minimize side effects resulting from combination therapy. Irrespective of the methods of delivery that are used, the long-term consequences of this therapeutic approach will need to be investigated in detail.
Supplementary Material
Acknowledgments
Supported by NIH Grants NS049720, CA097257, CA108961, CA127716, CA100011
References
- 1.Roth JA, Nguyen D, Lawrence DD, et al. Retrovirus-mediated wild-type p53 gene transfer to tumors of patients with lung cancer. Nat Med. 1996;2:985–91. doi: 10.1038/nm0996-985. [DOI] [PubMed] [Google Scholar]
- 2.Levesque AA, Eastman A. p53-based cancer therapies: Is defective p53 the Achilles heel of the tumor? Carcinogenesis. 2007;28:13–20. doi: 10.1093/carcin/bgl214. [DOI] [PubMed] [Google Scholar]
- 3.Johnson KR, Fan W. Reduced expression of p53 and p21WAF1/CIP1 sensitizes human breast cancer cells to paclitaxel and its combination with 5-fluorouracil. Anticancer Res. 2002;22:3197–204. [PubMed] [Google Scholar]
- 4.Wang Y, Zhu S, Cloughesy TF, Liau LM, Mischel PS. p53 disruption profoundly alters the response of human glioblastoma cells to DNA topoisomerase I inhibition. Oncogene. 2004;23:1283–90. doi: 10.1038/sj.onc.1207244. [DOI] [PubMed] [Google Scholar]
- 5.Kuo PC, Liu HF, Chao JI. Survivin and p53 modulate quercetin-induced cell growth inhibition and apoptosis in human lung carcinoma cells. J Biol Chem. 2004;279:55875–85. doi: 10.1074/jbc.M407985200. [DOI] [PubMed] [Google Scholar]
- 6.Bartz SR, Zhang Z, Burchard J, et al. Small interfering RNA screens reveal enhanced cisplatin cytotoxicity in tumor cells having both BRCA network and TP53 disruptions. Mol Cell Biol. 2006;26:9377–86. doi: 10.1128/MCB.01229-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Komarov PG, Komarova EA, Kondratov RV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–7. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 8.Xu GW, Mymryk JS, Cairncross JG. Pharmaceutical-mediated inactivation of p53 sensitizes U87MG glioma cells to BCNU and temozolomide. Int J Cancer. 2005;116:187–92. doi: 10.1002/ijc.21071. [DOI] [PubMed] [Google Scholar]
- 9.Batista LF, Roos WP, Christmann M, Menck CF, Kaina B. Differential sensitivity of malignant glioma cells to methylating and chloroethylating anticancer drugs: p53 determines the switch by regulating xpc, ddb2, and DNA double-strand breaks. Cancer Res. 2007;67:11886–95. doi: 10.1158/0008-5472.CAN-07-2964. [DOI] [PubMed] [Google Scholar]
- 10.Gary RK, Jensen DA. The p53 inhibitor pifithrin-alpha forms a sparingly soluble derivative via intramolecular cyclization under physiological conditions. Mol Pharm. 2005;2:462–74. doi: 10.1021/mp050055d. [DOI] [PubMed] [Google Scholar]
- 11.Pietrancosta N, Moumen A, Dono R, et al. Imino-tetrahydro-benzothiazole derivatives as p53 inhibitors: discovery of a highly potent in vivo inhibitor and its action mechanism. J Med Chem. 2006;49:3645–52. doi: 10.1021/jm060318n. [DOI] [PubMed] [Google Scholar]
- 12.Giannini C, Sarkaria JN, Saito A, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005;7:164–76. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarkaria JN, Yang L, Grogan PT, et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol Cancer Ther. 2007;6:1167–74. doi: 10.1158/1535-7163.MCT-06-0691. [DOI] [PubMed] [Google Scholar]
- 14.Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999;59:793–7. [PubMed] [Google Scholar]
- 15.Strosznajder RP, Jesko H, Banasik M, Tanaka S. Effects of p53 inhibitor on survival and death of cells subjected to oxidative stress. J Physiol Pharmacol. 2005;56 (Suppl 4):215–21. [PubMed] [Google Scholar]
- 16.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 17.Kakudo Y, Shibata H, Otsuka K, Kato S, Ishioka C. Lack of correlation between p53-dependent transcriptional activity and the ability to induce apoptosis among 179 mutant p53s. Cancer Res. 2005;65:2108–14. doi: 10.1158/0008-5472.CAN-04-2935. [DOI] [PubMed] [Google Scholar]
- 18.Fuster JJ, Sanz-González SM, Moll UM, Andrés V. Classic and novel roles of p53: prospects for anticancer therapy. Trends Mol Med. 2007;13:192–9. doi: 10.1016/j.molmed.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Blough MD, Zlatescu MC, Cairncross JG. O6-methylguanine-DNA methyltransferase regulation by p53 in astrocytic cells. Cancer Res. 2007;67:580–4. doi: 10.1158/0008-5472.CAN-06-2782. [DOI] [PubMed] [Google Scholar]
- 20.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–53. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.