

Abstract
Meningiomas are common tumors, representing 15-25% of all central nervous system tumors. NF2 gene inactivation on chromosome 22 has been shown as an early event in tumorigenesis; however, few factors underlying tumor growth and progression have been identified. Chromosomal abnormalities of 14q32 are often associated with meningioma pathogenesis and progression; therefore it has been proposed that an as yet unidentified tumor suppressor is present at this locus. MEG3 is an imprinted gene located at 14q32 that encodes a non-coding RNA with an anti-proliferative function. We found that MEG3 mRNA is highly expressed in normal arachnoidal cells. However, MEG3 is not expressed in the majority of human meningiomas or the human meningioma cell lines IOMM-Lee and CH157-MN. There is a strong association between loss of MEG3 expression and tumor grade. Allelic loss at the MEG3 locus is also observed in meningiomas, with increasing prevalence in higher grade tumors. In addition, there is an increase in CpG methylation within the promoter and the imprinting control region of MEG3 gene in meningiomas. Functionally, MEG3 suppresses DNA synthesis in both IOMM-Lee and CH157-MN cells by approximately 60% in BrdU incorporation assays. Colony-forming efficiency assays show that MEG3 inhibits colony formation in CH157-MN cells by approximately 80%. Furthermore, MEG3 stimulates p53-mediated transactivation in these cell lines. Therefore, these data are consistent with the hypothesis that MEG3, which encodes a non-coding RNA, may be a tumor suppressor gene at chromosome 14q32 involved in meningioma progression via a novel mechanism.
Keywords: MEG3 non-coding RNA, gene expression, anti-proliferation, meningiomas, pathogenesis and progression
Introduction
Meningiomas arise from the arachnoidal cells of the leptomeninges covering the brain and spinal cord, and account for 15-25% of all central nervous system tumors (1). Most meningiomas are slow growing and considered benign (WHO grade I). However, a subset of grade I meningiomas can recur, leading to compression of critical anatomic structures and clinically significant impairment of neurological function. Less than 20% of cases are classified as WHO grade II (atypical meningioma) or WHO grade III (anaplastic/malignant meningioma), and these exhibit more aggressive clinical behavior and have a higher risk of recurrence with increased morbidity and mortality (1).
Cytogenetic studies have revealed several chromosomal abnormalities in meningiomas, with losses of 22q, 1p, and 14q being most common. The inactivation of the NF2 gene at 22q12 has been identified as an early event in meningioma pathogenesis, but not associated with tumor progression (2). In contrast, abnormalities of chromosome 14, including 14q32, have been reported more frequently in higher-grade (WHO Grade II and III) as well as recurrent meningiomas (3-6). Therefore, it has been suggested that gene inactivation in this particular region is associated with progression of meningiomas from lower to higher grade, and may also be associated with tumor recurrence. However, relevant genes of interest in this region have not been discovered.
MEG3 is an imprinted gene with maternal expression which encodes a non-coding RNA. We have shown that MEG3 RNA expression is lost in the majority of clinically non-functioning human pituitary tumors and other cancer cell lines, and it suppresses cancer cell growth, stimulates p53-mediated transcriptional activation, and selectively activates p53 target genes (7, 8). MEG3 is highly expressed in the normal human brain (7).
Because MEG3 is located at 14q32, a region where chromosomal abnormalities are associated with meningioma progression, we hypothesized that MEG3 may represent a novel meningioma suppressor gene in this region. In this study we report the progressive loss of MEG3 expression in human meningiomas and inhibition of meningioma cell proliferation by MEG3.
Materials and Methods
Samples
Human meningioma samples were obtained from surgery and snap frozen at −80 °C. Matched whole blood samples were collected from each patient. Tumors were classified and graded according to the WHO grading system (1). Normal human brain and meningeal samples were obtained from the Harvard Brain Tissue Resource Center (Belmont, MA) and the Pathology Service at Massachusetts General Hospital. This study is approved by the Partners Healthcare Institutional Review Board.
In situ hybridization
Samples from normal human arachnoid tissue (including arachnoidal granulations) and human meningiomas were fixed in 4% paraformaldehyde for 3–4 h, rinsed with PBS, sectioned (5 μm) by a cryostat, and stored at −80 °C. In situ hybridization was performed as previously described (7), using MEG3 sense or anti-sense probes.
RNA extraction and RT-PCR
Total RNA was extracted from 46 human meningiomas (16 Grade I, 18 Grade II, and 12 Grade III) and the human meningioma cell lines IOMM-Lee and CH157-NM (obtained from Dr. DH Gutmann, Washington University School of Medicine, St. Louis, MO; we did not test these cell lines), using TRIzol Reagent (Invitrogen, Carlsbad, CA). Normal meningeal RNA samples were purchased from BioChain (Hayward, CA) and Analytical Biological Services (Wilmington, DE), or extracted from normal meningeal samples (see Samples, above). RT-PCR was performed as previously described (9), using MEG3-specific primers as well as GAPDH-specific primers as a control (sequences available upon request). RT reactions performed in the absence of reverse transcriptase were used as negative controls. Quantitative RT-PCR using TaqMan probes (Applied Biosystems, Foster City, CA) was performed as previously described (10).
Genomic DNA preparation
Tumor DNA was extracted from 27 snap-frozen meningioma samples using the DNeasy Tissue Kit (Qiagen, Valencia, CA). In addition, DNA samples from 27 corresponding peripheral blood leukocytes from the same patients were isolated using Puregene DNA extraction kit (Gentra Systems, Minneapolis, MN). Following DNA extraction, samples were amplified with the Whole Genome Amplification Kit (Sigma-Aldrich, St. Louis, MO). Normal meningeal genomic DNA samples were either purchased from BioChain (Hayward, CA) and Analytical Biological Services (Wilmington, DE) or extracted from normal meningeal samples.
Copy number analysis for chromosomal loss
Quantitative real time PCR was used to quantify gene copy number. The starting relative copy number DNA at each locus in a tumor sample was given by the formula 2-ΔΔCT, where ΔΔCT = CT(tumor – reference) minus CT(normal – reference)(11). RNase P, a housekeeping gene, was used as the reference gene. This gene has only one copy per haploid cell and was amplified in parallel with experimental samples for normalizing the results in order to allow relative quantification analysis. The normal DNA extracted from peripheral blood leukocytes from the same patient was designated as 1.0 by this equation, and all other samples were calculated in relation to this value. In order for the ΔΔCT to be valid, the efficiencies of the reference and target should be approximately equal. A calibration curve was constructed using serial dilutions of template DNA (198,000pg/μl to 19.8pg/μl) and the plot of log input amount versus ΔCT (target probe – reference RNase P) had a slope < 0.1 for each primer probe. PCR amplification efficiencies (E) were determined according to the equation: E = 10(−1/slope). The efficiency for each primer probe was: 1.96 for RNase P, 2.00 for DLK1, 1.97 for D14S119; r = −1.00. The quantitative real time PCR was performed using a 25 μl working master mix containing: 50ng of the template DNA in 1xTaqMan universal Master Mix (Applied Biosystems, Foster City, CA), 200 nM final concentration of the primers, and the probe (FAM labeled, Applied Biosystems, Foster City, CA). The reaction was run in a SmartCycler II (Cephid, Sunnyvale, CA, thermal cycler), using the following cycling parameters: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C (denature) for 15 s with 60°C for 1 min (annealing extension). The sequence of the genomic probes and primers that mapped to region 14q32.1 to 14q32.3 (D14S831, D14S1006 for DLK1, WI-16835 for MEG3, and D14S119) were obtained from the genome databases. Sequences of primers and TaqMan probes are available upon request. Single copy loss was considered to be present in tumors in which the highest value of the standard deviation was below one (12).
Methylation analysis of genomic DNA
Genomic DNA from 6 Grade I, 8 Grade II, 4 Grade III human meningiomas, or from 2 normal human meningeal samples was treated with sodium bisulfite using the MethylDetector Bisulfite Modification Kit (Active Motif, Carlsbad, CA). PCR amplification of treated DNA at MEG3 promoter (R1) and enhancer (R4), and imprinting control (IG-DMR) region, and the cloning of PCR products were performed as previously described (9, 10). Ten to twenty clones from each PCR product were examined by sequencing. The percentage of methylation at each particular CpG site among these 10 to 20 clones was recorded; then the percentage of methylation at each CpG site with the genomic region was averaged. Therefore the data represent overall percentage of methylated CpG sites within a particular genomic region. All data are expressed as the mean ± standard deviation (SD) for descriptive statistics and ± standard error of the mean (SEM) for comparing groups. Repeated measures of ANOVA were used to analyze data where appropriate. A p<0.05 was considered significant.
Expression vectors
For the BrdU incorporation assay, MEG3 and DLK1 cDNA were cloned into a pCMS-d2EGFP vector, which expresses both destabilized green fluorescent protein (d2EGFP) and MEG3 or DLK1 cDNA. For the colony formation assay and transient transfections and luciferase assays, MEG3, DLK1, and GADD45γ cDNA were cloned into a pCI-neo vector (Promega). Other plasmids used in luciferase assays include p53-Luc (Startagene, La Jolla, CA) and pCMVβ (BD Clontech, Palo Alto, CA).
Cell culture, transfection, and luciferase assay
Human meningioma derived cell lines IOMM-Lee and CH157-NM were cultured in DMEM supplemented with 10% heat-inactivated FBS and penicillin/streptomycin. Cells were transfected with Mirus TransIT-LT1 reagent (Mirus Corp, Madison, WI) as previously described (13). For luciferase assays, cells in 12-well plates were transfected with plasmid DNAs containing 50 ng p53-Luc, 0.2 μg pCMVβ, and 50 ng pCI-neo-MEG3 as indicated. Cells were lysed and luciferase activities were measured as previously described (13). The luciferase activity was normalized against the β-galactosidase activity from the same well. Each experiment was repeated at least four times. Statistical analysis was performed using a t-test.
Growth suppression assays
Growth suppression of meningioma cell lines IOMM-Lee and CH157-NM by MEG3 was measured by BrdU incorporation assay and colony formation assay, as previously described (8, 14). Each experiment was repeated at least three times. Statistical analysis was performed using a t-test.
5-aza-2′-deoxycytidine treatment
CH157-MN cells were seeded in 100 mm cell culture dishes ad cultured in medium containing 5 μM 5-aza-2′-deoxycytidine (Sigma-Aldrich) or vehicle for 5 days. The culture medium was changed and fresh agent added daily. RNA extraction and RT-PCR for MEG3 and GAPDH RNA was performed as previously described (9).
Western blot
Cells were lysed with radioimmune precipitation assay buffer to obtain total protein and Western blotting was performed as previously described (13). The blot was probed with antibody DO-1 (Santa Cruz Biotechnology, Santa Cruz, CA) to detect p53 protein.
Results
MEG3 expression in normal human arachnoidal cells, meningiomas, and meningioma cell lines
We first examined MEG3 expression in normal human meningeal cells, meningiomas, and meningioma cell lines. MEG3 mRNA was readily detected by RT-PCR in all 9 normal human meningeal samples (see Fig 1A, Lanes N, for representative samples). However, MEG3 mRNA was present only in 3 of 9 Grade I (Fig 1A, top left, Lanes 3, 4, and 5) and 1 of 11 Grade II (Fig 1A, top right, Lane 6) meningiomas. None of the 7 Grade III meningiomas examined expressed MEG3 mRNA (Fig 1A, bottom left). The difference in MEG3 expression between normal and combined tumor samples was significant (normal vs all tumors: p<0.0001; normal vs Grade I tumors: p=0.0294; normal vs Grade II tumors: p<0.0001; normal vs Grade III tumors: p<0.0001; Grade I vs combined Grade II/III: p=0.0297) using Fisher’s Exact 2-Tail Test.
Figure 1.

MEG3 RNA is expressed in normal human meninges but not in the majority of human meningiomas. A, RT-PCR readily detected MEG3 RNA in normal human meningeal (Lanes N), but only in 4 of 9 typical meningiomas (Top panel, left: Lane 2, 3, 4, and 5) and 1 of 11 atypical meningiomas (Top Panel, right: Lane 6). None of 7 anaplastic meningiomas (Bottom panel, left) or tumor cell lines (Bottom panel, right: Lane 1, IOMM-Lee; Lane 2, CH157-MN; Lane 3, a pituitary tumor derived cell line PDFS) expressed MEG3 RNA as examined by RT-PCR. M: molecular weight marker. B, in situ hybridization shows that MEG3 RNA is present in the arachnoidal cells of human meningeal samples (left panel), but no MEG3 RNA was detected in one typical meningioma sample (Tumor No. 5 in Fig 1A). Nuclei were stained by hematoxylin in the tumor slide.
No MEG3 mRNA was detected in the human meningioma derived cell lines IOMM-Lee and CH157-MN (Fig 1A, bottom right, Lane 1 and 2). Using in situ hybridization, we observed that MEG3 mRNA was abundantly present in the arachnoidal cells (Fig 1B, left). In contrast, no MEG3 mRNA was detected by in situ hybridization in several Grade I meningiomas, including Tumor No. 5 (Fig 1B, right); this tumor showed positive MEG3 mRNA expression by RT-PCR (Fig 1A, top left, Lane 5), suggesting that even if MEG3 mRNA is expressed in some tumors, its expression levels are low compared to that in the normal samples.
Quantitative RT-PCR was performed to assess the relative MEG3 expression levels in meningiomas compared to that in the normal human meningeal samples. In addition to the 27 samples used for the regular RT-PCR shown in Figure 1, 19 additional meningioma samples were included (7 Grade I, 7 Grade II, and 5 Grade III). The relative MEG3 RNA expression level in each tumor was compared with the average level of MEG3 RNA determined from 6 normal human meningeal samples (Table 1). Among 16 Grade I tumors, quantitative RT-PCR detected MEG3 RNA in 9 tumors, ranging from only 0.23% to 7.8% of the average MEG3 RNA level in the normal tissues. In the 18 Grade II tumors, MEG3 RNA was detectable at low levels in 6 tumor samples, ranging from 0.13% to 0.39% of the average MEG3 RNA level in the normal tissues. Only one Grade II tumor expressed a level of MEG3 RNA comparable to normal tissue. In the 12 Grade III tumors, MEG3 RNA was detected in only one sample, at a level of approximately 1% of that in the normal tissue (Table 1). Overall, MEG3 is expressed in normal arachnoidal cells but is expressed at low levels in some grade I meningiomas and is absent in the majority of Grade II and almost all Grade Ill meningiomas.
Table 1.
MEG3 RNA Expression, Gene Copy Loss, and Methylation in Promoter (R1), Enhancer(R4), and Imprinting Control Region (IG-DMR) in Meningiomas
| Samples | MEG3 Level* |
Loss of MEG3 Allele |
R1 Methylation |
Mean ± SD | R4 Methylation |
Mean ± SD | IG-DMR Methylation |
Mean ± SD |
|---|---|---|---|---|---|---|---|---|
| Normal | ||||||||
| NM1** | 100% | --*** | 7% | 6.0±1.41 | 26% | 17.06±12.72 | 42% | 50.06.0±1.0 |
| NM2 | 100% | -- | 5% | 8% | 59% | |||
| Grade I | ||||||||
| I-1 | UD | No | 6% | 15.4±27.0 | 71% | 69.2±17.4 | 48% | 56.4±9.9 |
| I-2 | 0.97% | No | 90% | 87% | 59% | |||
| I-3 | 2.75% | No | 22% | 87% | 74% | |||
| I-4 | 3.17% | No | 6% | 43% | 56% | |||
| I-5 | 7.80% | No | 3% | 49% | 56% | |||
| I-6 | UD | No | 3% | 85% | 41% | |||
| I-7 | 0.42% | No | 1% | 55% | 56% | |||
| I-8 | UD | No | 1% | 89% | 70% | |||
| I-9 | UD | No | 7% | 57% | 48% | |||
| I-10 | UD | -- | -- | -- | -- | |||
| I-11 | 2.75% | -- | -- | -- | -- | |||
| I-12 | 0.23% | -- | -- | -- | -- | |||
| I-13 | 4.80% | -- | -- | -- | -- | |||
| I-14 | 3.13% | -- | -- | -- | -- | |||
| I-15 | UD | -- | -- | -- | -- | |||
| I-16 | UD | -- | -- | -- | -- | |||
| Grade II | ||||||||
| II-1 | UD | Yes | 15% | 14.4±5.6 | 57% | 43.6±13.1 | 61.0±4.8 | |
| II-2 | UD | Yes | -- | -- | 62% | |||
| II-3 | 0.33% | No | 13% | 48% | -- | |||
| II-4 | UD | No | 11% | 52% | 71% | |||
| II-5 | UD | No | -- | 59% | ||||
| II-6 | 161% | No | 21% | 42% | ||||
| II-7 | 0.39% | No | 22% | 34% | 53% | |||
| II-8 | 0.35% | No | 11% | 55% | 60% | |||
| II-9 | UD | No | 18% | 47% | 58% | |||
| II-10 | UD | No | 4% | 14% | 64% | |||
| II-11 | UD | Yes | -- | -- | 61% | |||
| II-12 | 0.13% | -- | -- | -- | -- | |||
| I-I13 | UD | -- | -- | -- | -- | |||
| II-14 | UD | -- | -- | -- | -- | |||
| II-15 | 0.16% | -- | -- | -- | -- | |||
| II-16 | UD | -- | -- | -- | -- | |||
| II-17 | 0.22% | -- | -- | -- | -- | |||
| II-18 | UD | -- | -- | -- | -- | |||
| Grade III | ||||||||
| III-1 | UD | Yes | -- | 27.0±21.26 | -- | 58.3±19.3 | -- | 68.8±5.1 |
| III-2 | UD | Yes | -- | -- | -- | |||
| III-3 | UD | Yes | 58% | 87% | 67% | |||
| III-4 | UD | No | 22% | 45% | 76% | |||
| III-5 | UD | No | 10% | 37% | 73% | |||
| III-6 | UD | Yes | -- | -- | -- | |||
| III-7 | UD | No | 18% | 64% | 63% | |||
| III-8 | UD | -- | -- | -- | -- | |||
| III-9 | 1.09% | -- | -- | -- | -- | |||
| III-10 | UD | -- | -- | -- | -- | |||
| III-11 | UD | -- | -- | -- | -- | |||
| III-12 | UD | -- | -- | -- | -- |
Shown as the percentage of the average level in the normal tissue. The average level in the normal tissue, obtained from 6 normal meningeal samples, is set as 100%. UD: undetectable.
NM: normal meningeal sample
--: not examined
Copy number loss at the MEG3 locus in meningiomas
We next performed copy number analysis to determine whether there is MEG3 gene loss in meningiomas. Four markers were analyzed: D14S831 located at 14q32.1; D14S1006 located at 14q32.2, within the DLK1 gene; WI-16835 located at 14q32.2/3, within the MEG3 gene; and D14S119 at 14q32.3. As summarized in Table 1, copy number loss between 14q32.1 and 14q32.3, including the MEG3 gene locus, was found in 3 of 10 Grade II and 4 of 7 Grade III meningiomas. No copy number loss at this region was found in any Grade I meningioma. For those tumors with copy number loss at 14q32, we also analyzed another marker located at 14q12. No copy number loss was detected at 14q12 in any tumors (data not shown). Therefore, there is specific loss at 14q32, containing the MEG3 gene, in these higher grade meningiomas. Notably, none of the tumors with MEG3 gene copy number loss express MEG3 RNA (as determined by RT-PCR).
Genomic DNA methylation in the promoter, enhancer, and imprinting control region of MEG3 gene
The status of CpG methylation in the promoter (R1), enhancer (R4), and IG-DMR region of the MEG3 gene was examined in 6 Grade I, 8 Grade II, 4 Grade III human meningiomas, and 2 normal human meningeal samples. These functional regions have been described in our previous publications (9, 10). In two normal human meningeal samples, the percent of methylated CpGs in the promoter R1 region was very low (6.0±1.41, mean±SD). There is an increase in the degree of CpG methylation in this region in tumors (15.4±27 for Grade I, p=0.1769, compared to that in normal tissue; 14.4±5.6 for Grade II, p=0.0037; and 27.0±18.4 for Grade III, p=0.0712) (Table 1).
For the R4 region with enhancer activity, approximately 17% of CpG dinucleotides are methylated in the normal human meningeal samples (17±12.72). The percentage of CpG methylation in the tumors is 69.2±17.4 for Grade I (p=0.0187, compared to that in normal tissue), 43.63±13.1 for Grade II (p=0.073), and 58.3±19.3 for Grade III (p=0.02553) (Table 1).
For the imprinting controlling IG-DMR, methylation was found in approximately 50% of the CpG dinucleotides in the normal meningeal samples (50±12%). There was a statistically significant increase in methylation in the tumors (56.4±9.9% for Grade I, 61.0±4.8% for Grade II, and 69.8±5.1% for Grade III). The degree of methylation significantly correlated with tumor grade (one way ANOVA test, p=0.038) (Table 1).
There is no statistically significant correlation between the extent of CpG methylation in each individual region and MEG3 RNA expression. Clearly, mechanisms other that DNA hypermethylation also contribute to MEG3 gene silencing in meningiomas. However, in samples without MEG3 RNA expression, the percentages of methylation are significantly higher at CpG positions 1, 10, and 17 in the enhancer region (R4) and positions 2, 3, and 5 in the IG-DMR region compared to those in the samples with MEG3 RNA expression. Therefore, these are potential hot-spots of methylation which may be linked to transcriptional silencing of MEG3.
To explore the functional role of DNA methylation in the silencing of MEG3 transcription in meningioma cells, we treated the human meningioma cell line CH157-MN cells with 5-aza-2′-deoxycytidine, a demethylating agent. As shown in Figure 2, treatment of 5-aza-2′-deoxycytidine resulted in MEG3 RNA expression.
Figure 2.
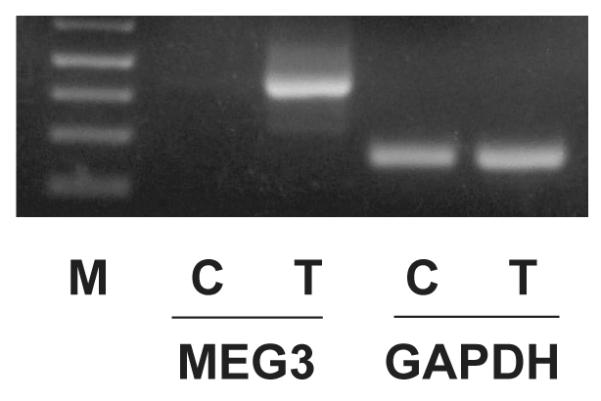
5-aza-2′-deoxycytidine treatment results in MEG3 RNA expression in CH157-MN cells as examined by RT-PCR. M: molecular marker; C: vehicle treatment control; T: 5-aza-2′-deoxycytidine treatment.
Suppression of meningioma cell growth by MEG3 cDNA
To investigate the functional relevance of MEG3 in human meningiomas, we tested its ability to suppress in vitro cell growth of meningioma cell lines IOMM-Lee and CH157-MN. Transfection of a MEG3 expression vector into IOMM-Lee and CH157-MN cells resulted in suppression of BrdU incorporation by approximately 60% (Fig 3A). However, when the transfection was performed with a similar expression vector in which the CMV promoter sequence controlling MEG3 expression was deleted, no suppression of BrdU incorporation was observed, indicating that expression of MEG3 RNA in the transfected cells is required for suppression of DNA synthesis. Transfection of a DLK1 expression vector showed no suppression of BrdU incorporation (Fig 3A). In colony-forming efficiency assays, MEG3 suppressed colony formation in CH157-MN cells by approximately 80%, similar to GADD45γ, a known growth suppressor (15). Again, DLK1 failed to suppress colony formation in CH157-MN cells (Fig 3B, 3C).
Figure 3.

MEG3 suppresses human meningioma cell proliferation. A, Suppression of DNA synthesis by MEG3-expression as measured by BrdU incorporation assay in IOMM-Lee cells. B, Suppression of DNA synthesis by MEG3-expressing as measured by BrdU incorporation assay in CH157-MN cells. Ctr: transfection with an empty expression vector as a control; MEG3: transfection with MEG3-expressing vector; dp: transfection with the same MEG3 expression vector but without CMV promoter; DLK: transfection with a DLK1-expressing vector. No inhibition of DNA synthesis was observed when cells were transfected with a MEG3 containing vector with the CMV promoter deleted (dp), or a DLK-expressing vector (DLK). Data are represented as mean ± SD for BrdU-labeling index from at least three independent experiments. C: In colony formation assay, transfection of MEG3- or GADD45γ-expressing vector into CH157-MN cells resulted in a significant decrease in colony number compared to the empty expression vector (Ctr), while transfection of DLK-expressing vector does not affect colony formation. D: The percentage reduction in colony numbers in each transfected cell culture plate. Data are represented as mean ± SD for BrdU-labeling index from at least three independent experiments. *: p<0.001.
Stimulation of p53-mediated transactivation by MEG3 in meningioma cells
To begin to understand the molecular mechanism by which MEG3 suppresses meningioma cell growth, we examined whether MEG3 can affect the function of p53, one of the most important tumor suppressors which functions a sequence-specific transcription factor. Both meningioma cell lines IOMM-Lee and CH157-MN express p53 protein (Fig 4A). When a p53-responsive reporter plasmid was transfected into these cells, luciferase activity was detected in the cell lysate. When a MEG3 expression vector was co-transfected with this p53-responsive reporter, reporter activity was increased by approximately 4 fold (Fig 4B, 4C). Therefore, MEG3 is able to stimulate p53-mediated transactivation in IOMM-Lee and CH157-MN cells.
Figure 4.

A: Both CH157-NM (Lane 1) and IOMM-Lee (Lane 2) cell line express p53 protein as examined by Western blot. B and C: MEG3 stimulates p53-mediated transactivation in IOMM-Lee (B) and CH157-NM (C) cells. The relative luciferase activity from cells without MEG3-expressing vector co-transfection is designated as 1. Data are represented as mean ± SD from at least three independent experiments. *: p<0.001.
Discussion
It has long been suggested that chromosome 14q32 contains a tumor suppressor gene involved in meningioma pathogenesis and progression (3-6, 16-18). However, the potential 14q32 tumor suppressor has not yet been discovered. Our data indicate that MEG3 may be an excellent candidate for this tumor suppressor, because 1) the MEG3 gene is located at chromosome 14q32; 2) MEG3 RNA is highly expressed in normal arachnoidal cells, the likely cell of origin for meningiomas, but not expressed in the majority of meningiomas; 3) loss of MEG3 RNA expression as well as loss of MEG3 gene copy number is more common in higher grade meningiomas and there is an overall increase in CpG methylation in tumors associated with tumor grade; and 4) MEG3 RNA expression in human meningioma cell lines strongly suppresses tumor cell growth and activates p53-mediated transactivation.
Early cytogenetic studies revealed monosomy of chromosome 22 in up to 70% of meningiomas, and subsequent studies have identified loss of heterozygosity (LOH) at polymorphic markers on 22q in 40% to 70% of meningiomas (19-22). At 22q12.2, a key gene of interest, NF2, has been identified to be associated with meningioma pathogenesis (23, 24), which encodes a tumor suppressor known as merlin or schwannomin, a member of the protein 4.1 superfamily, functioning to link cell surface signaling to intracellular pathways (25). Because loss of merlin expression is observed in meningiomas regardless of tumor grade, NF2 inactivation is an early event in meningioma pathogenesis and is not associated with tumor progression (2). In contrast, loss of MEG3 expression and loss of MEG3 gene copy is more common in higher grade meningiomas, suggesting that loss of MEG3 function may not only be associated with tumor pathogenesis but also with progression. Of the two human meningioma cell lines used in our functional studies, IOMM-Lee is merlin-positive, but CH157-NM is merlin-negative. The fact that MEG3 suppresses in vitro proliferation of both cell lines indicates that the function of MEG3 is independent of merlin. Consistent with our data, previous studies with large tumor numbers have shown that there is no or minimal correlation of 14q and 22q loss in human meningiomas (16, 26-28).
MEG3 was identified as the human counterpart of a mouse imprinted gene Gtl2 (29), identified by gene trapping in an attempt to isolate genes involved in early development (30). Gtl2 is closely linked to another imprinted gene Dlk1 (31, 32), a paternally expressed gene whose function may be involved in the control of growth and differentiation (33-37). Studies have intensively focused on the genomic characterization and imprinting control of the Dlk1 and Gtl2/Meg3 locus (38-41). However, the physiological function of MEG3 remained unknown until we reported its anti-proliferative activity in human cancer cells (7) and showed loss of MEG3 expression and promoter hypermethylation in pituitary adenomas (9). Subsequently, a number of reports have shown loss of MEG3 expression and promoter hypermethylation in several types of human tumors, including pituitary adenomas, neuroblastomas, pheochromocytomas, Wilms tumors, and other carcinomas, underscoring its potential tumor suppressive function (9, 42, 43).
In our study, DLK1 served as an important control. DLK1, also located at 14q32 and closely linked to MEG3, is an imprinted gene but with paternal expression. DLK1 encodes a protein that contains an extracellular domain with 6 EGF-like repeats, a transmembrane domain and a short cytoplasmic tail. DLK1 regulates the differentiation of different cell lineages, including preadipocytes, skeletal stem cells, thymocytes, and adrenal gland cells (34-37). Up-regulation of DLK1 has recently been reported in some tumors (44, 45). However, our data show that only MEG3 suppresses meningioma cell growth, while DLK1 has no such effect on these cells. These data are consistent with our hypothesis that MEG3 is a specific candidate tumor suppressor at 14q32.
Mutations in the TP53 tumor suppressor gene have been identified in more than 50% of human tumors, and more than 90% of cancers contain defects in the p53 pathway (46). However, the involvement of p53 in meningiomas remains elusive. In general, high levels of p53 protein expression (2, 47) and occasional TP53 mutations have been found in high grade meningiomas (48). The p53 protein is regulated by MDM2, which inhibits p53 function and promotes its degradation. This p53/MDM2 interaction is inhibited by p14ARF. In the absence of a TP53 gene mutation, loss of MDM2 protein expression and a high percentage of p14ARF gene methylation have been reported in high grade meningiomas (49), consistent with our previous observations that MEG3 expression leads to p53 protein accumulation and MDM2 down-regulation (8). Here we report that MEG3 enhances p53-mediated transcription in meningiomas. Therefore, it is possible that in normal arachnoidal cells, p53 and MEG3 function together to keep cell proliferation under control. In this conceptual schema, loss of MEG3 expression would lead to impairment of p53 function, resulting in uncontrolled cell proliferation and subsequent tumor development, even though the meningioma cells could respond by expressing more p53 protein to reverse this impairment. Future studies to investigate MEG3 and p53 expression in these tumors would be important to support this potential mechanism.
It has yet to be determined how MEG3 interacts with p53. Lacking a solid open reading frame (ORF) and lacking a Kozak consensus sequence in any of its short ORFs, it was suggested that MEG3 functions as a non-coding RNA (50). Recently, using untranslatable MEG3 cDNA mutants, we have provided the first experimental evidence for its non-coding RNA nature (8). As shown in this study, a MEG3 expression vector without a promoter fails to suppress DNA synthesis in both IOMM-Lee and CH157 cells, indicating that transcription of MEG3 RNA is necessary for its growth suppressive function. Further investigation of the molecular interaction between MEG3 and p53 may reveal a novel mechanism for control of cell proliferation and meningioma pathogenesis involving non-coding RNAs.
In conclusion, our data strongly suggest MEG3 as a candidate tumor suppressor gene at 14q32 associated with the pathogenesis and progression of human meningiomas. As an imprinted gene encoding a non-coding RNA, it appears to suppress tumor development via entirely novel mechanisms. Further investigation of MEG3 could therefore provide important information regarding the pathogenesis of human meningiomas; reveal novel mechanisms to broaden our knowledge of the involvement of non-coding RNAs in human tumor biology; and eventually point to new therapeutic strategies for these tumors.
Acknowledgments
We thank Dr. DH Gutmann (Washington University School of Medicine, St. Louis, MO) for kindly providing human meningioma cell lines IOMM-Lee and CH157-MN. This work was supported in part by National Institute of Health R01-DK-40947 (A. Klibanski) and The Guthart Family Foundation.
References
- 1.Louis OHDN, Wiestler OD, Cavenee WK. World Health Organization Classification of Tumours of the Central Nervous System. IARC Press; Lyon: 2007. [Google Scholar]
- 2.Lusis E, Gutmann DH. Meningioma: an update. Curr Opin Neurol. 2004;17(6):687–92. doi: 10.1097/00019052-200412000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Simon M, von Deimling A, Larson JJ, et al. Allelic losses on chromosomes 14, 10, and 1 in atypical and malignant meningiomas: a genetic model of meningioma progression. Cancer Res. 1995;55(20):4696–701. [PubMed] [Google Scholar]
- 4.Weber RG, Bostrom J, Wolter M, et al. Analysis of genomic alterations in benign, atypical, and anaplastic meningiomas: toward a genetic model of meningioma progression. Proc Natl Acad Sci U S A. 1997;94(26):14719–24. doi: 10.1073/pnas.94.26.14719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menon AG, Rutter JL, von Sattel JP, et al. Frequent loss of chromosome 14 in atypical and malignant meningioma: identification of a putative ‘tumor progression’ locus. Oncogene. 1997;14(5):611–6. doi: 10.1038/sj.onc.1200853. [DOI] [PubMed] [Google Scholar]
- 6.Cai DX, Banerjee R, Scheithauer BW, Lohse CM, Kleinschmidt-Demasters BK, Perry A. Chromosome 1p and 14q FISH analysis in clinicopathologic subsets of meningioma: diagnostic and prognostic implications. J Neuropathol Exp Neurol. 2001;60(6):628–36. doi: 10.1093/jnen/60.6.628. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Zhou Y, Mehta KR, et al. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J Clin Endocrinol Metab. 2003;88(11):5119–26. doi: 10.1210/jc.2003-030222. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Y, Zhong Y, Wang Y, et al. Activation of p53 by MEG3 non-coding RNA. J Biol Chem. 2007;282(34):24731–42. doi: 10.1074/jbc.M702029200. [DOI] [PubMed] [Google Scholar]
- 9.Zhao J, Dahle D, Zhou Y, Zhang X, Klibanski A. Hypermethylation of the promoter region is associated with the loss of MEG3 gene expression in human pituitary tumors. J Clin Endocrinol Metab. 2005;90(4):2179–86. doi: 10.1210/jc.2004-1848. [DOI] [PubMed] [Google Scholar]
- 10.Gejman R, Batista DL, Zhong Y, et al. Selective loss of MEG3 expression and intergenic differentially methylated region hypermethylation in the MEG3/DLK1 locus in human clinically nonfunctioning pituitary adenomas. J Clin Endocrinol Metab. 2008;93(10):4119–25. doi: 10.1210/jc.2007-2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 12.Senchenko V, Liu J, Braga E, et al. Deletion mapping using quantitative real-time PCR identifies two distinct 3p21.3 regions affected in most cervical carcinomas. Oncogene. 2003;22(19):2984–92. doi: 10.1038/sj.onc.1206429. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Y, Mehta KR, Choi AP, Scolavino S, Zhang X. DNA damage-induced inhibition of securin expression is mediated by p53. J Biol Chem. 2003;278(1):462–70. doi: 10.1074/jbc.M203793200. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, Sun H, Danila DC, et al. Loss of expression of GADD45 gamma, a growth inhibitory gene, in human pituitary adenomas: implications for tumorigenesis. J Clin Endocrinol Metab. 2002;87(3):1262–7. doi: 10.1210/jcem.87.3.8315. [DOI] [PubMed] [Google Scholar]
- 15.Takekawa M, Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95(4):521–30. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- 16.Leone PE, Bello MJ, de Campos JM, et al. NF2 gene mutations and allelic status of 1p, 14q and 22q in sporadic meningiomas. Oncogene. 1999;18(13):2231–9. doi: 10.1038/sj.onc.1202531. [DOI] [PubMed] [Google Scholar]
- 17.Schneider BF, Shashi V, von Kap-herr C, Golden WL. Loss of chromosomes 22 and 14 in the malignant progression of meningiomas. A comparative study of fluorescence in situ hybridization (FISH) and standard cytogenetic analysis. Cancer Genet Cytogenet. 1995;85(2):101–4. doi: 10.1016/0165-4608(95)00154-9. [DOI] [PubMed] [Google Scholar]
- 18.Tse JY, Ng HK, Lau KM, Lo KW, Poon WS, Huang DP. Loss of heterozygosity of chromosome 14q in low- and high-grade meningiomas. Hum Pathol. 1997;28(7):779–85. doi: 10.1016/s0046-8177(97)90149-0. [DOI] [PubMed] [Google Scholar]
- 19.Zang KD. Cytological and cytogenetical studies on human meningioma. Cancer Genet Cytogenet. 1982;6(3):249–74. doi: 10.1016/0165-4608(82)90063-2. [DOI] [PubMed] [Google Scholar]
- 20.Seizinger BR, de la Monte S, Atkins L, Gusella JF, Martuza RL. Molecular genetic approach to human meningioma: loss of genes on chromosome 22. Proc Natl Acad Sci U S A. 1987;84(15):5419–23. doi: 10.1073/pnas.84.15.5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dumanski JP, Carlbom E, Collins VP, Nordenskjold M. Deletion mapping of a locus on human chromosome 22 involved in the oncogenesis of meningioma. Proc Natl Acad Sci U S A. 1987;84(24):9275–9. doi: 10.1073/pnas.84.24.9275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casalone R, Simi P, Granata P, et al. Correlation between cytogenetic and histopathological findings in 65 human meningiomas. Cancer Genet Cytogenet. 1990;45(2):237–43. doi: 10.1016/0165-4608(90)90088-r. [DOI] [PubMed] [Google Scholar]
- 23.Ikeda K, Saeki Y, Gonzalez-Agosti C, Ramesh V, Chiocca EA. Inhibition of NF2-negative and NF2-positive primary human meningioma cell proliferation by overexpression of merlin due to vector-mediated gene transfer. J Neurosurg. 1999;91(1):85–92. doi: 10.3171/jns.1999.91.1.0085. [DOI] [PubMed] [Google Scholar]
- 24.Kalamarides M, Niwa-Kawakita M, Leblois H, et al. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. 2002;16(9):1060–5. doi: 10.1101/gad.226302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curto M, McClatchey AI. Nf2/Merlin: a coordinator of receptor signalling and intercellular contact. British Journal of Cancer. 2008;98(2):256–62. doi: 10.1038/sj.bjc.6604002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pfisterer WK, Coons SW, Aboul-Enein F, Hendricks WP, Scheck AC, Preul MC. Implicating chromosomal aberrations with meningioma growth and recurrence: results from FISH and MIB-I analysis of grades I and II meningioma tissue. J Neurooncol. 2008;87(1):43–50. doi: 10.1007/s11060-007-9498-9. [DOI] [PubMed] [Google Scholar]
- 27.Pfisterer WK, Hank NC, Preul MC, et al. Diagnostic and prognostic significance of genetic regional heterogeneity in meningiomas. Neuro Oncol. 2004;6(4):290–9. doi: 10.1215/S1152851704000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sayagues JM, Tabernero MD, Maillo A, et al. Incidence of numerical chromosome aberrations in meningioma tumors as revealed by fluorescence in situ hybridization using 10 chromosome-specific probes. Cytometry. 2002;50(3):153–9. doi: 10.1002/cyto.10075. [DOI] [PubMed] [Google Scholar]
- 29.Miyoshi N, Wagatsuma H, Wakana S, et al. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells. 2000;5(3):211–20. doi: 10.1046/j.1365-2443.2000.00320.x. [DOI] [PubMed] [Google Scholar]
- 30.Schuster-Gossler K, Simon-Chazottes D, Guenet JL, Zachgo J, Gossler A. Gtl2lacZ, an insertional mutation on mouse chromosome 12 with parental origin-dependent phenotype. Mamm Genome. 1996;7(1):20–4. doi: 10.1007/s003359900006. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt JV, Matteson PG, Jones BK, Guan XJ, Tilghman SM. The Dlk1 and Gtl2 genes are linked and reciprocally imprinted. Genes Dev. 2000;14(16):1997–2002. [PMC free article] [PubMed] [Google Scholar]
- 32.Takada S, Tevendale M, Baker J, et al. Delta-like and gtl2 are reciprocally expressed, differentially methylated linked imprinted genes on mouse chromosome 12. Curr Biol. 2000;10(18):1135–8. doi: 10.1016/s0960-9822(00)00704-1. [DOI] [PubMed] [Google Scholar]
- 33.Smas CM, Sul HS. Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell. 1993;73(4):725–34. doi: 10.1016/0092-8674(93)90252-l. [DOI] [PubMed] [Google Scholar]
- 34.Friedrichsen BN, Carlsson C, Moldrup A, et al. Expression, biosynthesis and release of preadipocyte factor-1/ delta-like protein/fetal antigen-1 in pancreatic beta-cells: possible physiological implications. J Endocrinol. 2003;176(2):257–66. doi: 10.1677/joe.0.1760257. [DOI] [PubMed] [Google Scholar]
- 35.Abdallah BM, Jensen CH, Gutierrez G, Leslie RG, Jensen TG, Kassem M. Regulation of human skeletal stem cells differentiation by Dlk1/Pref-1. J Bone Miner Res. 2004;19(5):841–52. doi: 10.1359/JBMR.040118. [DOI] [PubMed] [Google Scholar]
- 36.Laborda J. The role of the epidermal growth factor-like protein dlk in cell differentiation. Histol Histopathol. 2000;15(1):119–29. doi: 10.14670/HH-15.119. [DOI] [PubMed] [Google Scholar]
- 37.Lee K, Villena JA, Moon YS, et al. Inhibition of adipogenesis and development of glucose intolerance by soluble preadipocyte factor-1 (Pref-1) J Clin Invest. 2003;111(4):453–61. doi: 10.1172/JCI15924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charlier C, Segers K, Karim L, et al. The callipyge mutation enhances the expression of coregulated imprinted genes in cis without affecting their imprinting status. Nat Genet. 2001;27(4):367–9. doi: 10.1038/86856. [DOI] [PubMed] [Google Scholar]
- 39.Paulsen M, Takada S, Youngson NA, et al. Comparative sequence analysis of the imprinted Dlk1-Gtl2 locus in three mammalian species reveals highly conserved genomic elements and refines comparison with the Igf2-H19 region. Genome Res. 2001;11(12):2085–94. doi: 10.1101/gr.206901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takada S, Paulsen M, Tevendale M, et al. Epigenetic analysis of the Dlk1-Gtl2 imprinted domain on mouse chromosome 12: implications for imprinting control from comparison with Igf2-H19. Hum Mol Genet. 2002;11(1):77–86. doi: 10.1093/hmg/11.1.77. [DOI] [PubMed] [Google Scholar]
- 41.Wylie AA, Murphy SK, Orton TC, Jirtle RL. Novel imprinted DLK1/GTL2 domain on human chromosome 14 contains motifs that mimic those implicated in IGF2/H19 regulation. Genome Res. 2000;10(11):1711–8. doi: 10.1101/gr.161600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Astuti D, Latif F, Wagner K, et al. Epigenetic alteration at the DLK1-GTL2 imprinted domain in human neoplasia: analysis of neuroblastoma, phaeochromocytoma and Wilms‘ tumour. Br J Cancer. 2005;92(8):1574–80. doi: 10.1038/sj.bjc.6602478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pantoja C, de Los Rios L, Matheu A, Antequera F, Serrano M. Inactivation of imprinted genes induced by cellular stress and tumorigenesis. Cancer Res. 2005;65(1):26–33. [PubMed] [Google Scholar]
- 44.Yin D, Xie D, Sakajiri S, et al. DLK1: increased expression in gliomas and associated with oncogenic activities. Oncogene. 2006;25(13):1852–61. doi: 10.1038/sj.onc.1209219. [DOI] [PubMed] [Google Scholar]
- 45.Kawakami T, Chano T, Minami K, Okabe H, Okada Y, Okamoto K. Imprinted DLK1 is a putative tumor suppressor gene and inactivated by epimutation at the region upstream of GTL2 in human renal cell carcinoma. Hum Mol Genet. 2006;15(6):821–30. doi: 10.1093/hmg/ddl001. [DOI] [PubMed] [Google Scholar]
- 46.Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- 47.Collins VP. Brain tumours: classification and genes. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 2):ii2–11. doi: 10.1136/jnnp.2004.040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho H, Ha SY, Park SH, Park K, Chae YS. Role of p53 gene mutation in tumor aggressiveness of intracranial meningiomas. J Korean Med Sci. 1999;14(2):199–205. doi: 10.3346/jkms.1999.14.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amatya VJ, Takeshima Y, Inai K. Methylation of p14(ARF) gene in meningiomas and its correlation to the p53 expression and mutation. Mod Pathol. 2004;17(6):705–10. doi: 10.1038/modpathol.3800111. [DOI] [PubMed] [Google Scholar]
- 50.Schuster-Gossler K, Bilinski P, Sado T, Ferguson-Smith A, Gossler A. The mouse Gtl2 gene is differentially expressed during embryonic development, encodes multiple alternatively spliced transcripts, and may act as an RNA. Dev Dyn. 1998;212(2):214–28. doi: 10.1002/(SICI)1097-0177(199806)212:2<214::AID-AJA6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]