

Abstract
Under normal physiological conditions, the use of oxygen by cells of aerobic organisms generates potentially deleterious reactive oxygen metabolites. A chronic state of oxidative stress exists in cells because of an imbalance between prooxidants and antioxidants. The amount of oxidative damage increases as an organism ages and is postulated to be a major causal factor of senescence. Support for this hypothesis includes the following observations: (i) Overexpression of antioxidative enzymes retards the age-related accrual of oxidative damage and extends the maximum life-span of transgenic Drosophila melanogaster. (ii) Variations in longevity among different species inversely correlate with the rates of mitochondrial generation of the superoxide anion radical and hydrogen peroxide, (iii) Restriction of caloric intake lowers steady-state levels of oxidative stress and damage, retards age-associated changes, and extends the maximum life-span in mammals.
A common feature of the life cycle of virtually all multicellular organisms is the progressive decline in the efficiency of various physiological processes once the reproductive phase of life is over. A variety of strategies and models have been used to understand the nature of the mechanisms underlying the phenomenon of senescence. Frequently the purported explanations or hypotheses deal with the manifestations of aging, which are unlikely to be self-initiating, rather than with a more fundamental underlying cause accounting for the plethora of changes associated with senescence. To elucidate the mechanisms of aging, any causal hypothesis should explain the following three conditions: (i) why organisms undergo progressive and irreversible physiological decline in the latter part of life, (ii) why the life expectancy or rate of aging varies within and among species, and (iii) why experimental regimens such as caloric restriction delay the onset of a variety of age-associated physiological and pathological changes and extend the average and maximum life-span of animals. A mechanistic understanding of the effects of caloric restriction is important because of the efficacy of this regimen in the prolongation of the maximum life-span of mammals and because of its implications for human health.
A hypothesis ascribing one cause for aging postulates that the senescence-associated loss of functional capacity is due to the accumulation of molecular oxidative damage (1–4). This hypothesis is based on the fact that oxygen is potentially a toxic substance, and its use by aerobes, although necessary for their immediate survival, also may be hazardous to their long-term existence. The phenomenon of oxygen toxicity, sometimes referred to as the “oxygen paradox,” is inherent in the atomic structure of oxygen. Molecular oxygen is a biradical that upon single electron additions sequentially generates the partially reduced molecules , H2O2, and ·OH, which by further reactions can generate an array of additional reactive oxygen metabolites (ROMs) and cause extensive oxidative damage to biological macromolecules (5–11). This damage manifests as the peroxidation of membrane polyunsaturated fatty acid chains, modification of DNA (including base alterations, single-strand breaks, sister chromatid exchanges, and DNA-protein cross-links), and carbonylation and loss of sulfhydryls in proteins, among other changes. Carbonyl modifications of proteins occur in certain amino acid residues present near transition metal–binding sites and have been shown convincingly to cause enzymatic inactivation and enhance the likelihood of proteolysis (2).
There are several indications that the oxidant challenge to aerobic cells is not trivial. It is estimated that ~2 to 3% of the oxygen consumed by aerobic cells is diverted to the generation of and H2O2 (8). A typical cell in the rat may undergo 100,000 ROM attacks on DNA per day (3), and under steady-state conditions, ~10% of protein molecules may exhibit carbonyl modifications (2). The presence of the products of ROM interactions with macromolecules under steady-state conditions has led to the concept that antioxidative defenses are not fully efficient, that cells are chronically under oxidative stress, and that aging is a consequence of oxidative damage.
The Oxidative Stress Hypothesis of Aging
The basic tenet of the oxidative stress hypothesis is that senescence-related loss of function is due to the progressive and irreversible accrual of molecular oxidative damage (1–4). The predictions of this hypothesis, in the context of the above-stated three minimal conditions that need to he met for the validation of any causal hypothesis of aging, would thus be as follows: (i) The level of molecular oxidative damage increases during aging, (ii) Relatively longer life expectancy within and among species is associated with a correspondingly lower accrual of oxidative damage, (iii) Prolongation of life-span by regimens such as caloric restriction in mammals is associated with the amelioration of oxidative damage.
Oxidative stress and damage increases during aging
Because of the relatively low steady-state concentrations of ROMs, together with their transient nature and the proximity of antioxidative defenses, it currently is not feasible to measure the in vivo rates of ROM generation. Therefore, tests that assay the concentrations of a variety of products of ROM reactions with macromolecules are used to ascertain the direction of the shift in the redox state of tissues. In both mammalian and insect tissues, the ratios of the redox couples, such as reduced glutathione:oxidized glutathione, NADPH:NADP+, and NADH:NAD+, tend to shift toward more prooxidant values during aging (12, 13). An age-associated elevation of the in vivo level of oxidative stress is indicated by an exponential increase in the exhalation of alkanes such as ethane and n-pentane, products of ROM-induced peroxidation of membrane lipids (13, 14). There is an age-related two-to threefold exponential increase in the concentration of oxidatively damaged proteins, which is indicated by a loss of protein –SH groups, protein carbonylation, and a loss of catalytic activity of enzymes (such as glutamine synthase and alcohol dehydrogenase) that are particularly susceptible to oxidative damage (2, 4). The concentration of 8-hydroxydeoxyguanosine, an indicator of DNA oxidative damage, also increases with age in various tissues of mammals and in insects (3, 15). The main point emerging from such studies is that molecular oxidative damage during aging is ubiquitous, substantial, and, like mortality rates, increases exponentially with age. If oxidative damage is indeed a fundamental causal factor in aging, as hypothesized, it follows that the progression of senescence is also exponential.
Age-associated increase in oxidative stress: Possible causes
The etiology of an age-associated increase in the amount of oxidative stress can be linked to three different factors: (i) an increase in the rate of generation of ROMs, (ii) a decline in antioxidative defenses, and (iii) a decline in the efficiency of repair or removal of damaged molecules. Rates of and H2O2 generation by mitochondria, which are the main source of production (16), increase with age in several different organs of mammals and in insects (17). One mechanism contributing to this increase is ROMs, generated by mitochondria, inflicting damage on the inner mitochondrial membrane, which by positive feedback induces a further increase in ROM generation. Mitochondria of houseflies exposed to different periods of hyperoxia exhibit a linear increase in the amounts of protein and DNA oxidative damage and an enhanced rate of H2O2 generation, when measured under normoxic conditions (4). In vitro exposure of mitochondria to ROM-generating systems, as well as to cross-linking agents such as glutaraldehyde, results in increased rates of H2O2 release. Another mechanism contributing to the increased production of and H2O2 may be the age-related imbalance among different mitochondrial oxidoreductases; for example, the relative decrease in cytochrome c oxidase activity that occurs during aging would tend to increase the autoxidizability of the upstream respiratory components, thereby elevating the rate of and H2O2 generation (18). It has also been suggested that the age-associated increase in mitochondrial DNA deletions, observed in several species, is induced by ROMs and results in abnormalities in the components of the electron transport chain (3). Notwithstanding, the functional significance of such deletions in aging has yet to be convincingly demonstrated.
No consistent pattern of age-associated changes in the activities of individual antioxidative enzymes or concentrations of low molecular weight substances has been detected (19, 20). However, the susceptibility of tissue homogenates and of live animals to experimentally induced oxidative stress increases with age (21), suggesting either a net decline in the antioxidative defenses or an increased availability of loosely bound transition metal ions, which can catalyze the scission of H2O2 into the highly reactive hydroxyl radical (·), believed to be the main agent of oxidative damage (or both). Information about age-associated changes in the efficiency of processes that repair or remove oxidatively damaged molecules is quite fragmentary. A decrease in the activity of neutral and alkaline proteases, which preferentially degrade oxidized proteins, has been found only in certain tissues during aging (2, 22). Overall, it seems that oxidative stress and damage increases during aging because of an increased rate of ROM generation and an increased susceptibility of tissues to oxidative damage.
Experimental tests of the oxidative stress hypothesis
Although the amount of accumulated oxidative damage during the normal adult life-span is extensive and rather substantial (increasing about two- to threefold), no claims of causality can be made on this basis without the demonstration that an experimentally induced attenuation in oxidative stress does indeed retard the rate of aging and prolong the life-span. A recent approach to reduce oxidative stress and damage involved an experimental elevation in enzymatic antioxidative defenses so that the possibility of ·OH generation was minimized (23, 24). The effects of simultaneous overexpression of Cu,Zn–superoxide dismutase (SOD) and catalase, which act in tandem to remove and H2O2 respectively, were determined in transgenic Drosophila melanogaster (Fig. 1). As compared with the diploid controls, transgenic flies carrying three copies of each of these genes exhibited (i) an up to one-third extension of the average and maximum life-span, (ii) a retardation in the age-related accumulation of oxidative damage to DNA and protein, (iii) less DNA oxidative damage in response to x-ray exposure of live flies, (iv) an attenuation in the age-associated increase in the rate of mitochondrial H2O2 generation, (v) an increase in the speed of walking, and (vi) a 30% increase in the metabolic potential (total amount of oxygen consumed during adult life per unit body weight). Together, these results indicate that an experimentally induced decrease in oxidative stress or damage retards age-associated deteriorative changes at the organelle and organismic levels and extends the chronological as well as the metabolic life of the flies.
Fig. 1.
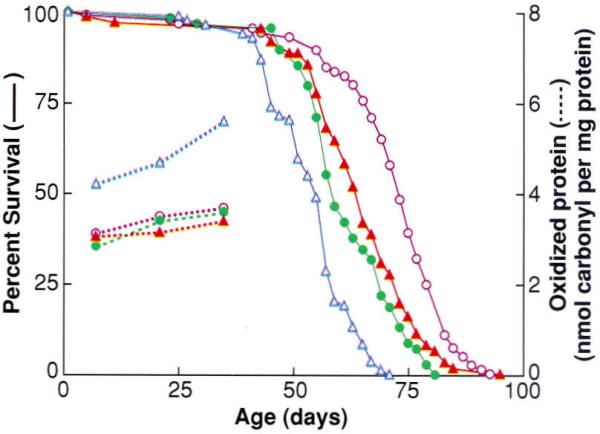
Effect of overexpression of antioxidative enzymes on life-span and protein oxidative damage in D. melanogaster. Survival curves (solid) and protein carbonyl content (dashed curves) at different ages for a control group (blue triangles) and three different lines (remaining symbols) of transgenic D. melanogaster overexpressing both Cu.Zn-superoxide dismutase and catalase. [Adapted from (23)]
Relation between intra- and interspecies variations in life-spans and oxidative stress
Individuals of the same chronological age vary in the degree of senescence-associated functional impairment and therefore differ in “physiological age.” Although information on the relation between oxidative stress and physiological age within a mammalian cohort is meager, an inverse relation has been demonstrated in insects. In the housefly, an indicator of impending death is the inability to fly, which was used to isolate two subpopulations differing by ~30% in their average and maximum life-span (MLS) (4, 25). As compared with the age-matched, short-lived flies, those with the longer life expectancy exhibited a lower rate of mitochondrial and H2O2, generation, a lower level of protein oxidative damage, a lesser amount of DNA oxidative damage, higher activities of SOD and catalase, and an elevation in the concentration of glutathione, a versatile intracellular reductant.
Variations in MLS among different species are often associated with differences in the metabolic rates (rate of oxygen consumption), metabolic potential (estimated as the total amount of energy consumed per gram of body weight during the life-span), and the level of oxidative stress. Various phylogenetic groups have different metabolic potentials; for example, ~25 kcal in dipteran flies, ~200 kcal in nonprimate mammals, ~800 kcal in humans, and ~1000 to 1500 kcal in birds (26). Rubner (27) pointed out that although the MLS of nonprimate mammals, such as the horse, cow, cat, dog, and guinea pig, varied by about fivefold, the metabolic potential was relatively similar, ~200 ± 25 kcal/g, indicating an inverse relation between metabolic rate and MLS. This relation was later encapsulated by Pearl (28) as the “rate of living” theory, which states that the “rate of energy expenditure would determine the length of life.”
A comparison among a group of nonprimate mammalian species indicated that the rates of mitochondrial and H2O2 generation in heart, kidney, and liver were directly correlated to the metabolic rate and inversely related to the species' MLS, providing a biochemical link between the concepts of rate of living and oxidative stress (4, 29) (Fig. 2A). Although the activities of individual antioxidative enzymes showed no uniform relation with MLS (4, 26) (Fig. 2, B and C), tissue homogenates from the relatively longer lived species were less susceptible to acute oxidative stress induced by exposure to x-rays (21). Another interspecies comparison involving five species of dipteran flies that varied about twofold in average life-span also indicated that longevity was inversely related to the rates of mitochondrial and H2O2 generation as well as the steady-state concentration of protein carbonyls (30).
Fig. 2.
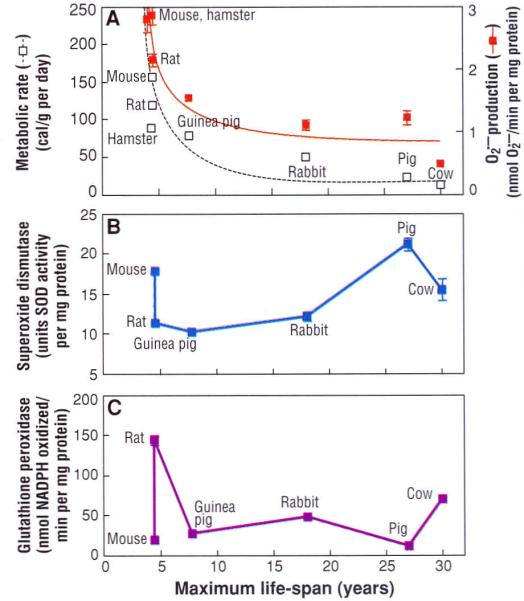
Comparison of maximum life-span (MLS) with (A) metabolic rate and rate of generation in submitochondrial particles, (B) total superoxide dismutase activity, and (C) glutathione peroxidase activity, in the heart of different mammalian species. [Adapted from (4) and (29)] MLS shows an inverse relation to metabolic rate (reported as calories consumed per gram of body weight per day) and generation, but no relation to antioxidative enzyme activity.
Certain species have similar metabolic rates but different MLS and metabolic potentials. The white-footed mouse (Peromyscus leucopus; MLS ~8 years) and the house mouse (Mus musculus; MLS ~4 years) exhibit about a twofold difference in MLS and metabolic potential but have similar metabolic rates. The rates of mitochondrial and H2O2 generation in heart and brain were, respectively, 40 and 80% lower in Peromyscus (31), whereas catalase and glutathione peroxidase activities were about twice as high in Peromyscus than in Mus, and the concentration of protein carbonyls was 80% greater in the brain of Mus. A similar comparison between pigeon (MLS ~30 years) and rat (MLS ~4 years), which have similar body mass and metabolic rates, indicated that rates of mitochondrial and H2O2 generation in brain and heart were lower, whereas activities of SOD and glutathione peroxidase were higher in the pigeon (32, 33). Altogether, results of such comparative studies indicate that metabolic potential and MLS are inversely related to oxidative stress.
Caloric Restriction and Experimental Extension of MLS
Maximum rather than average life-span is believed to provide a more meaningful indicator of the underlying rate of aging because the latter may be prolonged entirely because of an optimization of the maintenance conditions rather than a slowdown of the rate of aging. Three regimens are known to extend the MLS of animals: (i) lowered ambient temperature in poikilotherms (cold-blooded animals) and hibernating mammals and (ii) a decrease in physical activity in poikilotherms, both of which decrease metabolic rate, and (iii) caloric restriction (4, 34). The MLS of adult houseflies can be extended ~2.5-fold by a 10°C reduction in ambient temperature or the abolition of flying, which requires a rate of oxygen consumption 50- to 100-fold that of the nonflying state (4). Increases in the MLS of flies are accompanied by decreases in the rates of mitochondrial and H2O2 generation as well as by decreases in the amounts of protein and DNA oxidative damage. Hibernating mammals such as the Turkish hamster also live longer under hibernation (35), indicating the life-lengthening effects of hypometabolic states in mammals as well.
The caloric restriction model
Although it has been known for about 60 years that a decrease in caloric intake by laboratory rats and mice, performed without malnutrition, extends the MLS, this phenomenon remained an underexplored curiosity until the mid-1970s (34). Notwithstanding, caloric restriction (CR) is now being increasingly used as a model regimen for understanding the basic mechanisms of aging, primarily because it causes an unambiguous, robust, and reproducible extension of MLS and delays many, although not all, age-associated biochemical, physiological, and behavioral changes. Life-span extension by CR has also been reported in fish, spiders, Daphnia (water-flea), and other nonrodent species, indicating a broad relation between energy intake and aging.
Although presently no definitive information exists on whether or not CR similarly retards age-related deleterious changes and extends longevity in humans, three studies on nonhuman primates (primarily rhesus monkeys) are in progress (36). The available results suggest that CR can he safely carried out in primates and that certain physiological effects of CR, which occur in rodents (for example, decreased blood glucose and insulin concentrations, improved insulin sensitivity, and lowering of body temperature), are also observed in rhesus monkeys (37). Whether MLS extension is achieved in monkeys subjected to CR should become known in the future.
It has been demonstrated that a reduction in caloric intake per se, rather than of a particular nutrient, is required to increase MLS (34). A notable feature of the relation between caloric intake and life-span is that food consumption above an optimal level (that is, the amount below which CR has a life-threatening effect on the animal) progressively shortens longevity (Fig. 3) (38). The usual practice of ad libitum feeding of laboratory rodents promotes obesity and an earlier appearance of age-associated pathologies. Another complication associated with ad libitum feeding is the wide variation in caloric intake among individuals of a cohort. It has been suggested that laboratory rodents for gerontological or toxicological studies should be fed a controlled caloric intake (39).
Fig. 3.
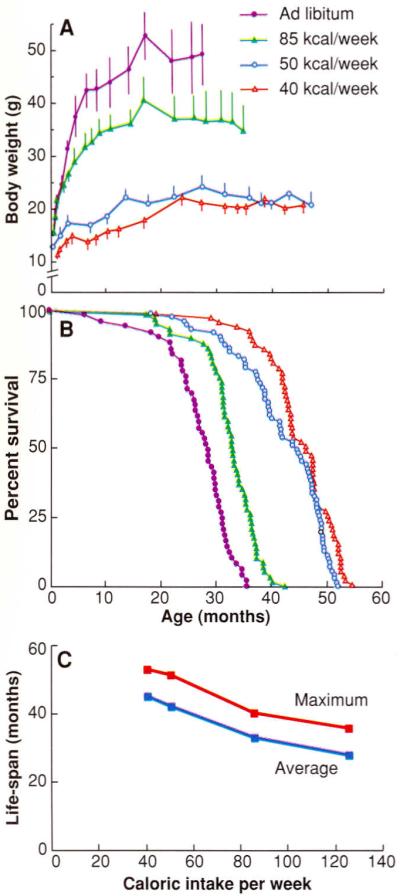
Effect of caloric intake on (A) body weight, (B) percent survival, and (C) life-span in female C3B10F1, mice. Symbols in (A) apply to (B) as well. [Adapted from (38)]
Besides reducing mortality, CR elicits a broad array of physiological alterations (34). About 80 to 90% of the ~300 age-sensitive changes examined in rodents fed a CR diet, including those in behavior and learning, immune responses, gene expression, enzyme activities, hormonal action, glucose intolerance, DNA repair capacities, and rates of protein synthesis, exhibited a “delayed aging” profile. Certain effects of CR in rats can be relatively swift; for example, the level of corticosterone in blood increases after only 1 week (40), and the concentration of blood glucose drops ~20% after 5 days while insulin levels decrease >50% in 3 weeks (41). It thus appears that CR can rapidly affect the physiological state.
The mechanism by which CR prolongs MLS has been variously postulated to be retardation of growth, reduction of body fat, delaying neuroendocrine or immunologic changes, increase in DNA repair capacities, altered gene expression, enhanced apoptosis, reduction of body tempetature and depression of metabolic rate, and amelioration of oxidative stress or damage, among others (34, 42). Such purported mechanisms vary in the degree to which each may satisfactorily explain the entire spectrum of CR-induced alterations. Some of these postulates, however, are no longer valid; for example, CR started at 12 months of age increases the MLS of mice (43), which argues against the growth retardation hypothesis. Reduction in body fat, an invariable consequence of CR, does not seem to be related to MLS because of the lack of a clear relation between body fat and longevity of ad libitum–fed rats and, in fact, a positive correlation among those on CR (44).
Whether the mechanism by which CR extends MLS involves a decrease in the rate of metabolism is controversial. However, this issue is of considerable relevance here because of the direct relation between the rate of oxygen consumption and mitochondrial rate of and H2O2 generation. Applying the general principles of Pearl's rate of living theory, Sacher suggested that CR may extend MLS by reducing the metabolic rate (45). However, this view was challenged because the rate of oxygen consumption per unit lean body weight in male Fischer 344 rats was unaffected by CR imposed at 6 weeks of age (34). Nonetheless, in another study (46), the metabolic rate in Fischer rats was found to decrease in response to CR started at 6 months of age. Mice subjected to CR and housed at room temperature (~21°C) exhibit a daily phase of up to a 13°C drop in body temperature (47), whereas rats on CR show a reduction ~2°C (48). A recognized consequence of a decrease in body temperature is the reduction in the rate of oxygen consumption (49). Furthermore, the concentration of triiodothyronine, which elevates metabolic rate, is markedly reduced in rodents on CR (34). It thus seems possible that MLS extension by CR in rodents is associated with a hypometabolic state.
Caloric restriction and attenuation of oxidative stress
If the hypothesis that oxidative stress is a major causal factor in the aging process is indeed valid, then one can predict that CR would lower the steady-state level of oxidative stress, retard the age-associated accrual of oxidative damage, and increase metabolic potential. Experimental results tend to support these predictions. Rodents subjected to CR show attenuation of age-associated increases in rates of mitochondrial and H2O2 generation (50), slower accrual of oxidative damage (50, 51), decreased alkane production (52), and delayed loss of membrane fluidity (53). Activities of individual antioxidative enzymes in different tissues do not follow a consistent pattern during aging or in response to CR (19, 20, 50, 54); however, CR decreases the in vitro susceptibility of tissues to acute oxidative stress (55). Metabolic potential, estimated on the basis of total caloric intake per gram of body weight during the life-span, is elevated by CR. For example, increases of 46 and 25%, respectively, occurred in male Fischer rats (56) and female C3B10F, mice (38).
The highest degree of oxidative damage as well as its attenuation by CR occur in tissues such as brain, heart, and skeletal muscle, which are composed primarily of long-lived, postmitotic cells. These tissues are also the targets of several age-related degenerative disorders in which oxidative stress has been implicated (5, 54). The protein carbonyl content in the brain of 15-month-old ad libitum–fed mice can be reduced by 5 weeks of CR, and that of 15-month-old CR-fed mice, increased to the ad libitum level by 5 weeks of free feeding (57). The effects of CR on oxidative stress and damage in the brain thus appear to be rapidly inducible and reversible.
In conclusion, several lines of evidence suggest that the accrual of oxidative damage is a common feature of senescence, transcending phylogenetic boundaries. Characteristic variations observed in the rates of mitochondrial and H2O2 generation among different species and tissues suggest that the rates of ROM production are genetically determined. Similarly, species-specific variations in the enzymatic antioxidative defenses together with the MLS extension by the overexpression of Cu, Zn–superoxide dismutase and catalase in transgenic Drosophila implicate specific genes in the control of oxidative stress and aging. The extension of MLS by experimental regimens such as CR in mammals and hypometabolic states in poikilotherms, which decrease the rates of ROM generation, point toward the involvement of environmental-genetic interactions in the governance of longevity. The recent finding that the life-span extension of Clk mutants of Caenorhabditis elegans, is associated with a hypometabolic phenotype is consistent with this concept (58). Although the present discussion has focused on the damaging effects of ROMs, it has been extensively documented that ROMs also modulate gene expression (59), which further broadens their possible role in the aging process.
Acknowledgments
We thank R. Mockett for assistance in the preparation of the manuscript. Research of the authors is supported by grants from the National Institute on Aging, NIH (R.S.S. and R.W.), and the American Cancer Society (R.W.). This is publication number 96-14 from the Madison Geriatric Research, Education, and Clinical Center. We apologize to our colleagues whose relevant work could not be cited because of constraints of space.
REFERENCES AND NOTES
- 1.Harman D. J. Gerontol. 1956;11:298. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 2.Stadtman ER. Science. 1992;257:1220. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 3.Ames BN, Shigenaga MK, Hagen TM. Proc. Natl. Acad Sci. U.S.A. 1993;90:7915. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]; Richter C. In: Current Topics in Bioenergetics. Lee CP, editor. vol. 17. Academic Press; San Diego, CA: 1994. pp. 1–19. [Google Scholar]
- 4.Sohal RS, Orr WC. In: Molecular Aspects of Aging. Esser K, Martin GM, editors. Wiley; New York: 1995. pp. 109–127. [Google Scholar]
- 5.Davies KJA. Biochem. Soc. Symp. 1995;61:1231. doi: 10.1042/bss0610001. [DOI] [PubMed] [Google Scholar]
- 6.Fridovich I. Science. 1978;201:875. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- 7.Pryor WA. Photochem. Photobiol. 1978;28:787. doi: 10.1111/j.1751-1097.1978.tb07020.x. [DOI] [PubMed] [Google Scholar]
- 8.Chance B, Sies H, Boveris A. Physiol. Rev. 1979;59:527. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 9.Halliwell B, Gutteridge JMC. Methods Enzy-mol. 1990;186:1. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- 10.Feig DI, Reid TM, Loeb LA. Cancer Res. 1994;54(suppl.):1890S. [PubMed] [Google Scholar]
- 11.McCord JM. Proc. Soc. Exp. Biol. Med. 1995;209:112. doi: 10.3181/00379727-209-43885c. [DOI] [PubMed] [Google Scholar]
- 12.Noy N, Schwartz H, Gafni A. Mech. Ageing Dev. 1985;29:63. doi: 10.1016/0047-6374(85)90047-8. [DOI] [PubMed] [Google Scholar]
- 13.Sohal RS. In: Advances in Myochemistry. Benzi G, editor. vol. 2. John Libbey; Paris: 1989. pp. 21–34. [Google Scholar]
- 14.Sagai M, Ichinose T. Life Sci. 1980;27:731. doi: 10.1016/0024-3205(80)90326-4. [DOI] [PubMed] [Google Scholar]
- 15.Agarwal S, Sohal RS. Proc. Aatl. Acad. Sci. U.S.A. 1994;91:12332. doi: 10.1073/pnas.91.25.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turrens JF, McCord JM. In: Free Radicals, Lipoproteins, and Membrane Lipids. Paulet AC, Douste-Blazy L, Paoletti R, editors. Plenum; New York: 1990. pp. 203–212. [Google Scholar]
- 17.Sohal RS, Brunk UT. Mutat. Res. 1992;275:295. doi: 10.1016/0921-8734(92)90033-l. [DOI] [PubMed] [Google Scholar]
- 18.Sohal RS. Free Radical Biol. Med. 1993;14:583. doi: 10.1016/0891-5849(93)90139-l. [DOI] [PubMed] [Google Scholar]
- 19.Matsuo M, Gomi F, Dooley MM. Mech. Ageing Dev. 1992;64:273. doi: 10.1016/0047-6374(92)90084-q. [DOI] [PubMed] [Google Scholar]
- 20.Rikans LE, Moore DR, Snowden CD. Biochim. Biophys. Ada. 1991;1074:195. doi: 10.1016/0304-4165(91)90061-k. [DOI] [PubMed] [Google Scholar]
- 21.Agarwal S, Sohal RS. Exp. Gerontol. 1996;31:387. doi: 10.1016/0531-5565(95)02039-x. [DOI] [PubMed] [Google Scholar]
- 22.Starke-Reed PE, Oliver CN. Arch. Biochem. Biophys. 1989;275:559. doi: 10.1016/0003-9861(89)90402-5. [DOI] [PubMed] [Google Scholar]; Agarwal S, Sohal RS. ibid. 1994;309:24. doi: 10.1006/abbi.1994.1078. [DOI] [PubMed] [Google Scholar]
- 23.Orr WC, Sohal RS. Science. 1994;263:1128. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- 24.Sohal RS, Agarwal S, Orr WC. J. Biol. Chem. 1995;270:15671. doi: 10.1074/jbc.270.26.15671. [DOI] [PubMed] [Google Scholar]
- 25.Sohal RS, Toy PL, Allen RG. Mech. Ageing Dev. 1986;36:71. doi: 10.1016/0047-6374(86)90140-5. [DOI] [PubMed] [Google Scholar]
- 26.Sohal RS. Aging Clin. Exp. Res. 1993;5:3. [Google Scholar]
- 27.Rubner M. Das Problem der Lebensdauer und Seine Beziehungen zum Wachstrum und Ernäbrung. Oldenbourg; Munich: 1908. [Google Scholar]
- 28.Pearl R. The Rate of Living. Knopf; New York: 1928. [Google Scholar]
- 29.Ku H-H, Brunk UT, Sohal RS. Free Radical Biol. Med. 1993;15:621. doi: 10.1016/0891-5849(93)90165-q. [DOI] [PubMed] [Google Scholar]
- 30.Sohal RS, Sohal BH, Orr WC. ibid. 1995;19:499. doi: 10.1016/0891-5849(95)00037-x. [DOI] [PubMed] [Google Scholar]
- 31.Sohal RS, Ku H-H, Agarwal S. Biochem. Biophys. Res. Commun. 1993;196:7. doi: 10.1006/bbrc.1993.2208. [DOI] [PubMed] [Google Scholar]
- 32.Ku H-H, Sohal RS. Mech. Ageing Dev. 1993;72:67. doi: 10.1016/0047-6374(93)90132-b. [DOI] [PubMed] [Google Scholar]
- 33.Barja G, Cadenas S, Rojas C, Perez-Campo R, Lopes-Torres M. Free Radical Res. 1994;21:317. doi: 10.3109/10715769409056584. [DOI] [PubMed] [Google Scholar]
- 34.Weindruch R, Walford RL. The Retardation of Aging and Disease by Dietary Restriction. Thomas; Springfield, IL: 1988. [Google Scholar]; Masoro EJ, Shimokawa I, Yu BP. Ann. N. Y. Acad. Sci. 1991;621337 doi: 10.1111/j.1749-6632.1991.tb16990.x. [DOI] [PubMed] [Google Scholar]; Yu BP, editor. Modulation of Aging Processes by Dietary Restriction. CRC Press; Boca Raton, FL: 1994. [Google Scholar]
- 35.Lyman CP, O'Brien RC, Greene GC, Papafrangos ED. Science. 1981;212:668. doi: 10.1126/science.7221552. [DOI] [PubMed] [Google Scholar]
- 36.Ingram DK, et al. J. Gerontol. 1990;45:B148. doi: 10.1093/geronj/45.5.b148. [DOI] [PubMed] [Google Scholar]; Kemnitze JW, et al. ibid. 1993;48:B17. [Google Scholar]; Hansen BC, Ortmeyer HK, Bodkin NL. Obesity Res. 1995;3(suppl. 2):S199. doi: 10.1002/j.1550-8528.1995.tb00464.x. [DOI] [PubMed] [Google Scholar]
- 37.Kemnitz JW, et al. Am. J. Physiol. 1994;266:E540. doi: 10.1152/ajpendo.1994.266.4.E540. [DOI] [PubMed] [Google Scholar]; Lane MA, et al. ibid. 1995;268:E941. [Google Scholar]; Bodkin NL, Ortmeyer HK, Hansen BC. J. Gerontol. 1995;50:B142. doi: 10.1093/gerona/50a.3.b142. [DOI] [PubMed] [Google Scholar]; Lane MA, et al. Proc. Natl. Acad. Sci. U.S.A. 1996;93:4159. [Google Scholar]
- 38.Weindruch R, Walford RL, Fligiel S, Guthrie D. J. Nutr. 1986;116:641. doi: 10.1093/jn/116.4.641. [DOI] [PubMed] [Google Scholar]
- 39.Abelson PH. Science. 1995;270:215. doi: 10.1126/science.270.5234.215. [DOI] [PubMed] [Google Scholar]; Hart RW, Neuman DA, Robinson RT, editors. Dietary Restriction: Implications for the Design and Interpretation of Toxicity and Carcinogenicity Studies. ILSI Press; Washington, DC: 1995. [Google Scholar]
- 40.Nelson JF. personal communication [Google Scholar]
- 41.Cartee GD, Dean DJ. Am. J. Physiol. 1994;266:E946. doi: 10.1152/ajpendo.1994.266.6.E946. [DOI] [PubMed] [Google Scholar]
- 42.Van Remmen H, Ward W, Sabia RF, Richardson A. In: Handbook of Physiology. Masoro EJ, editor. Oxford Univ. Press; New York: 1995. pp. 171–234. [Google Scholar]; Warner HR, Fernandes G, Wang E. J. Gerontol. 1995;50:B107. doi: 10.1093/gerona/50a.3.b107. [DOI] [PubMed] [Google Scholar]
- 43.Weindruch R, Walford RL. Science. 1982;215:1415. doi: 10.1126/science.7063854. [DOI] [PubMed] [Google Scholar]
- 44.Bertrand HA, Lynd FT, Masoro EJ, Yu BP. J. Gerontol. 1980;35:827. doi: 10.1093/geronj/35.6.827. [DOI] [PubMed] [Google Scholar]
- 45.Sacher GA. In: Handbook of the Biology of Aging. Finch CE, Hayflick L, editors. Van Nostrand Reinhold; New York: 1977. pp. 582–638. [Google Scholar]
- 46.Gonzales-Pacheco DM, Buss WC, Koehler KM, Woodside WF, Alpert SS. J. Nutr. 1993;123:90. doi: 10.1093/jn/123.1.90. [DOI] [PubMed] [Google Scholar]
- 47.Koizumi A, Tsukada M, Wada Y, Masuda H, Weindruch R. ibid. 1992;122:1446. doi: 10.1093/jn/122.7.1446. [DOI] [PubMed] [Google Scholar]
- 48.Duffy PH, Feuers R, Nakamura KD, Leakey J, Hart RW. Chronobiol. Int. 1990;7:113. doi: 10.3109/07420529009056963. [DOI] [PubMed] [Google Scholar]
- 49.Prosser CL. In: Comparative Animal Physiology. Prosser CL, editor. Saunders; Philadelphia: 1973. pp. 362–428. [Google Scholar]
- 50.Sohal RS, Ku H-H, Agarwal S, Forster MJ, Lai H. Mech. Ageing Dev. 1994;74:121. doi: 10.1016/0047-6374(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 51.Sohal RS, Agarwal S, Candas M, Forster MJ, Lai H. ibid. 1994;76:215. doi: 10.1016/0047-6374(94)91595-4. [DOI] [PubMed] [Google Scholar]
- 52.Matsuo M, Gomi F, Kuramoto K, Sagai M. J. Gerontol. 1993;48:B133. doi: 10.1093/geronj/48.4.b133. [DOI] [PubMed] [Google Scholar]
- 53.Chen JJ, Yu BP. Free Radical Biol. Med. 1994;17:411. doi: 10.1016/0891-5849(94)90167-8. [DOI] [PubMed] [Google Scholar]; Choi JH, Yu BP. ibid. 1995;18:133. doi: 10.1016/0891-5849(94)00106-t. [DOI] [PubMed] [Google Scholar]
- 54.Weindruch R, Warner HR, Starke-Reed PE. In: Free Radicals in Aging. Yu BP, editor. CRC Press; Boca Raton, FL: 1993. pp. 269–275. [Google Scholar]
- 55.Sohal RS, Dubey A. unpublished data [Google Scholar]
- 56.Masoro EJ, Yu BP, Bertrand HA. Proc. Natl. Acad. Sci. U.S.A. 1982;79:4239. doi: 10.1073/pnas.79.13.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dubey A, Forster MJ, Lai H, Sohal RS. Arch. Biochem. Biophys. doi: 10.1006/abbi.1995.0037. in press. [DOI] [PubMed] [Google Scholar]
- 58.Lakowski B, Hakimi S. Science. 1996;272:1010. doi: 10.1126/science.272.5264.1010. [DOI] [PubMed] [Google Scholar]
- 59.Janssen YM, Van Houten B, Borm PJ, Mossman BT. Lab. Invest. 1993;69:261. [PubMed] [Google Scholar]