

Abstract
Adenosine-secreting cellular brain implants constitute a promising therapeutic approach for the treatment of epilepsy. To engineer neural stem cells for therapeutic adenosine delivery, a reliable and fast analytical method is necessary to quantify cell-based adenosine release. Here we describe the development, optimization and validation of adenosine measurement using liquid chromatography – atmospheric pressure chemical ionization – tandem mass spectrometry (LC-APCI-MS/MS). LC-MS/MS in positive ion mode used selected reaction monitoring at m/z of 268.2/136.1 and 302.2/170.0 for adenosine and the internal standard, respectively. The bias was within 15% of the nominal value and evaluation of precision showed a relative standard deviation lower than 15% for all measured concentrations. The lower limit of quantification of adenosine was 15.6 ng/ml. Freeze and thaw stability and processed sample stability also fulfilled the acceptance criteria. Evaluation of the matrix effect showed that the method is not affected by relative matrix effects. The major advantages of this method are the absence of an extraction phase and the combination of the high selectivity and sensitivity characteristic for the LC-MS/MS technique, with a short run time of 4.5 min. These results demonstrate that this method is a useful tool to measure adenosine concentrations in culture medium released from stem cells in vitro.
Keywords: adenosine, liquid chromatography, tandem mass spectrometry, stem cells
1. Introduction
Adenosine is a purine ribonucleoside that plays important roles in many physiological processes. It is an endogenous neuromodulator of the brain and plays important protective roles in pathological conditions such as epilepsy [1]. Several studies already demonstrated that adenosine augmentation is a powerful strategy to suppress epileptic seizures, but adenosine cannot be given systemically because of side effects such as decreased heart rate, blood pressure and body temperature [2]. Therefore the local intracranial delivery of adenosine might be a good alternative and the transplantation of neural stem cells engineered to release adenosine might be an ideal solution to achieve focal long-term delivery of adenosine [3]. However, to screen engineered cell lines for therapeutic adenosine release, and to validate adenosine release prior to transplantation, an efficient method is needed to quantify adenosine secretion in vitro.
Several methods for separation and determination of adenosine in biological samples have been developed, using high performance liquid chromatography (± mass spectrometry) [4–22], capillary electrophoresis [23], or using an enzyme-coupled bioluminescence assay [24]. Since high performance liquid chromatography (HPLC) with tandem mass spectrometric (MS/MS) detection is now considered the method of choice for quantitative determination in biological fluids [25], methods using HPLC without MS/MS [4, 7, 8, 10, 12–14, 16–20] are not ideal for quantification of adenosine secretion in vitro. Among the published methods using LC with MS/MS, some are preceeded by an extraction phase or use UV-detection, which has a high limit of quantitation and a low selectivity [5, 6, 9, 11]. Two studies used the combination of LC and tandem MS without an extraction phase: one for analysis of renal vein perfusates [15] and one for analysis of microdialysates from rat brain [22] (one method with LC-MS/MS was developed to measure adenosine in Cordyceps sinensis, a chinese fungus, but was only published in Chinese [21]). However, use of these methods (with use of a similar column, ESI source and solvent system) in our laboratory did not result in reproducible and reliable quantification of adenosine in cell culture medium. Therefore we developed a new rapid method for the quantification of adenosine in cell culture medium using LC-MS/MS without the need for an extraction phase. In this paper we present the development, optimization and validation of an improved analytical method for the determination of adenosine using liquid chromatography – atmospheric pressure chemical ionization – tandem mass spectrometry (LC-APCI-MS/MS).
2. Materials and methods
2.1 Samples and standards
Neural stem cells were cultured under serum-free conditions in growth medium consisting of NS-A medium (StemCell Technologies SARL, Grenoble, France) with an additional 2 mM L-glutamine (Cambrex, Verviers, Belgium), 3 mM D-glucose (Sigma, Bornem, Belgium), 2% B27 (Invitrogen, Merelbeke, Belgium), 1% N2 supplement (Invitrogen, Merelbeke, Belgium) and 100 U/ml penicillin (Cambrex), 100 U/ml streptomycin (Cambrex) 20 ng/ml of human recombinant epidermal growth factor (EGF, Sigma) and 20 ng/ml of recombinant human basic fibroblast growth factor (bFGF, R&D, Minneapolis, MN, USA). Before samples were collected for adenosine quantification, growth medium was replaced by medium without growth factors and with addition of 50µM erythro-9-(2-hydroxy-3-nonyl)adenine hydrochloride (EHNA hydrochloride, Sigma) to prevent degradation of released adenosine. This will further be referred to as ‘medium’.
After medium replacement, samples (200 µl) were taken at different time points and stored at −20°C until processing. Adenosine and the internal standard (IS) 2-chloroadenosine were obtained from Sigma. Stock solutions of adenosine, IS (both prepared in pure water) and medium were stored at 4°C. For analysis of the samples, 20 µl IS (1000 ng/ml) was added to 40 µl of sample and shortly centrifuged. No extraction was performed. The calibration curve samples consisted of 40 µl medium with different adenosine concentrations (2000 ng/ml, 1000 ng/ml, 500 ng/ml, 250 ng/ml, 125 ng/ml, 62.5 ng/ml, 31.3 ng/ml, 15.6 ng/ml, 7.8 ng/ml and 0 ng/ml) and addition of 20 µl IS.
2.2 LC-MS/MS equipment
Liquid chromatography separations were performed using an Agilent LC 1100 HPLC system (Agilent Technologies, Santa Clara, US) equipped with a quaternary pump, a column oven and a 100 well-plate autosampler. The LC was coupled to an API 2000 Triple Quadrupole system (Applied Biosystems/MDS Sciex, Foster City, US) equipped with an APCI interface. Analyst Software 1.4.2 (Applied Biosystems) was used to control the instruments. A reversed phase column - the XBridge C8 column 4.6 mm × 75 mm, 3.5 µm (Waters, Zellik, Belgium) - was used for chromatographic separation. The temperature of the column oven was set at 40°C. The solvent system consisted of 2mM ammonium acetate in water (A) and 2mM ammonium acetate in methanol (B). An isocratic elution with 65% A and 35% B (v/v) was used. The flow rate was set to 500µL/min and an injection volume of 5µl was used. The total run time was 4.5 minutes.
2.3 Method validation
Validation was based on the guidelines of the US Food and Drug Administration and recent reviews [26–28].
Selectivity was tested by injection of 10 blank samples to check for interfering signals. Medium (without addition of adenosine) was used as blank sample. To obtain a regression model for the calibration curve, a dilution series of 10 concentrations of adenosine in medium with addition of IS was made. Per concentration, 5 replicates were analyzed and linearity was evaluated.
For evaluation of accuracy (bias) and precision different duplicates of 5 quality control (QC) samples at low (15.6 and 31.3 ng/ml), mean (125 and 500 ng/ml) and high (1000 ng/ml) adenosine concentrations (in medium with addition of IS) relative to the calibration range, were analyzed in 6 different days. The lower limit of quantification (LLOQ) was evaluated based on the detection results of adenosine in the accuracy and precision experiments.
Samples were stored at −20°C before analysis. Therefore evaluation of the freeze/thaw stability was necessary. Six replicates of QC samples with low (31.3 ng/ml), mean (250 ng/ml) and high (1000 ng/ml) adenosine concentrations (in medium with addition of IS) relative to the calibration range were analyzed before (control) and after three freeze/thaw cycles (treatment). To evaluate the processed sample stability, six replicates of QC samples of 10 concentrations of adenosine in medium with addition of IS were analyzed and then frozen at −20°C. Three months later, corresponding to the delay of analysis of the real samples, thawed samples were analyzed again. Results of the two analyses were compared.
Although electrospray ionization (ESI) has been reported to be much more prone to matrix effects, they may also occur with APCI [29]. Both the relative and absolute matrix effect were evaluated. To evaluate possible matrix effects, post-column infusion was performed by continuous injection of adenosine (1000 ng/ml) together with the mobile phase (elution consisting of 65% 2mM ammonium acetate in water and 35% 2mM ammonium acetate in methanol) or medium. This was repeated several times and the mean was calculated and plotted in a curve. Additionally the relative matrix effect was evaluated as described by Matuszewski [30]: calibration lines were prepared in 5 different lots of samples from medium taken from cultured stem cells and in 5 different lots of medium that had not been used in culture. Slopes from the standard lines were determined. It is recommended that the coefficient of variation (CV) of these slopes does not exceed 3–4% in order to conclude that the method is not affected by relative matrix effects. To determine the absolute matrix effect, six replicates of standard lines (1000, 500, 125, 31.3 and 15.6 ng/ml adenosine with addition of IS) in two different sets of samples were made. One consisting of a standard prepared in the mobile phase (set 1) and one set prepared in medium (set 2). The matrix effect was calculated as a percentage with the formula B/A × 100 (with A the mean peak areas for standards in the mobile phase and B the mean peak areas for standards in medium samples) [25].
2.4 Data analysis
Calibration curves with up to ten concentration points (see above) were acquired in every analytical run. Adenosine concentrations and peak area ratios were calculated using the Analyst Software 1.4.2. Further statistical analysis was performed using SPSS 15.0. P<0.05 was assessed to indicate a significant difference.
3. Results
3.1 Optimization of MS parameters
Mass spectrometer analysis was set up in selected reaction monitoring (SRM) in positive polarity. Based on the component dependent parameters (table 1), the SRM transition of m/z 268.2/136.1 and 302.2/170.0 were selected respectively for adenosine and IS. Figure 1 shows the MS/MS spectra of both adenosine and the IS. Optimization of the parameters of the APCI interface was done by flow injection analysis. The obtained optimal parameters are shown in table 2.
Table 1.
SRM and component dependent parameters: Declustering Potential (DP), Focusing Potential (FP), Entrance Potential (EP), Collision cell Entrance Potential (CEP), Collision Energy (CE), Collision cell eXit Potential (CXP). IS = internal standard.
| Analyte | Q1 mass (amu) |
Q3 mass (amu) |
Dwell time (msec) |
DP | FP | EP | CEP | CE | CXP |
|---|---|---|---|---|---|---|---|---|---|
| IS | 302.2 | 170.0 | 150 | 30 | 370 | 7 | 16 | 25 | 6 |
| IS | 302.2 | 134.0 | 150 | 30 | 370 | 7 | 16 | 53 | 4 |
| Adenosine | 268.2 | 136.1 | 150 | 30 | 340 | 6 | 18 | 23 | 4 |
| Adenosine | 268.2 | 119 | 150 | 25 | 340 | 6 | 18 | 61 | 4 |
Figure 1.
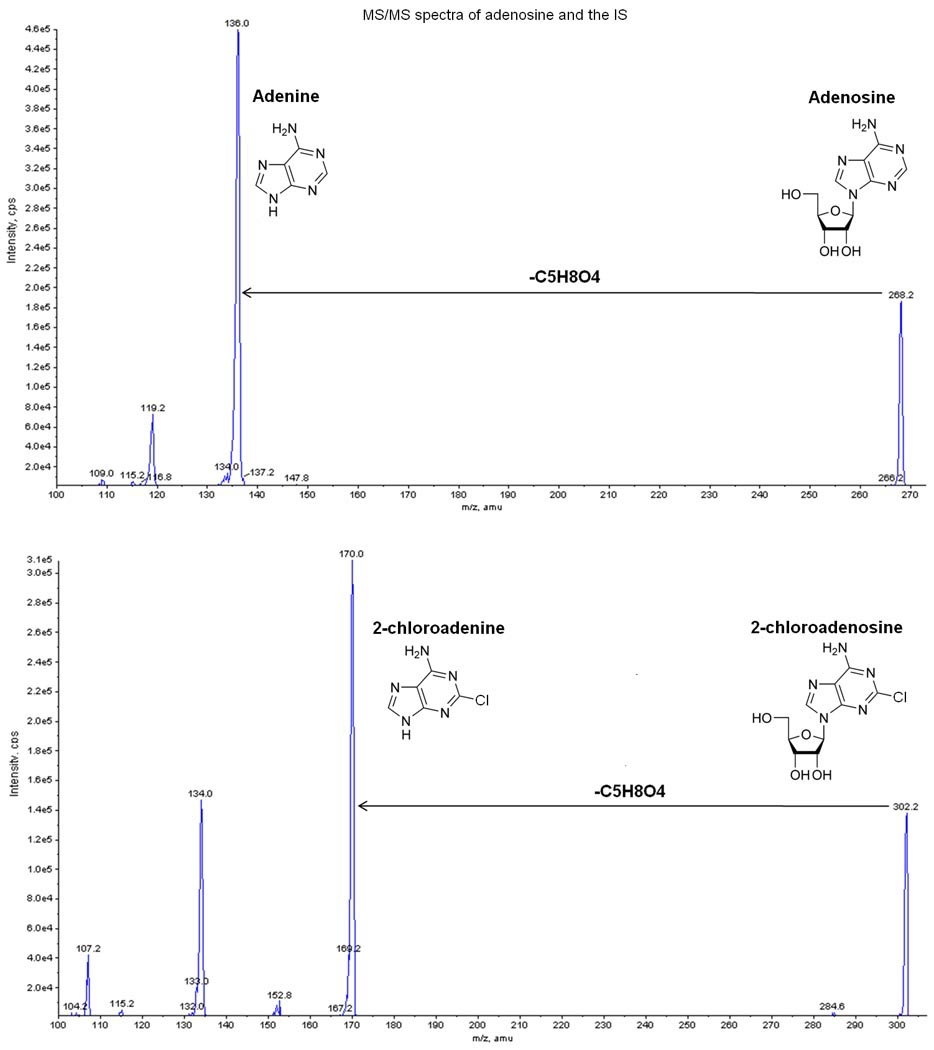
MS/MS spectra of both adenosine and the internal standard (IS: 2-chloroadenosine). m/z 268→136 and m/z 302→170 were used to monitor respectively adenosine and the IS.
Table 2.
optimized parameters for the APCI interface
| Parameter | Value |
|---|---|
| Collision Activated Dissociated gas | 2 psi |
| Gas 1 (nebulizer gas) | 60 psi |
| Gas 2 (turbo gas) | 15 psi |
| Curtain gas | 50 psi |
| Temperature | 500 °C |
| Interface heater | ON |
| Nebulizer Current | 2 µA |
3.2 Interference with adenosine measurement
Analysis of the first calibration curves resulted in very low adenosine peaks with even no detectable adenosine at a concentration of 500 ng/ml. The different compounds of the medium were analyzed and revealed an interference of the NS-A medium with adenosine. In vivo, adenosine is metabolized via 2 major enzymes: adenosine kinase and adenosine deaminase [31]. The different compounds of the medium were evaluated to search for presence of the enzymes. After addition of the adenosine deaminase inhibitor EHNA to the NS-A medium, correct measurements were obtained. Since the stem cells were cultured in NS-A medium, sample collection and analysis was also performed using this medium. To prevent adenosine breakdown during sample collection and analysis, EHNA was added to the medium at the start of the sample collection. It has been demonstrated that addition of EHNA to cultured neurons or astrocytes does not give acute toxicity to the cells after EHNA addition [32]. To avoid interference with the analysis EHNA was also added to the calibration curves.
Figure 2 represents a chromatogram of an analysis of a sample derived from an adenosine releasing cell clone.
Figure 2.
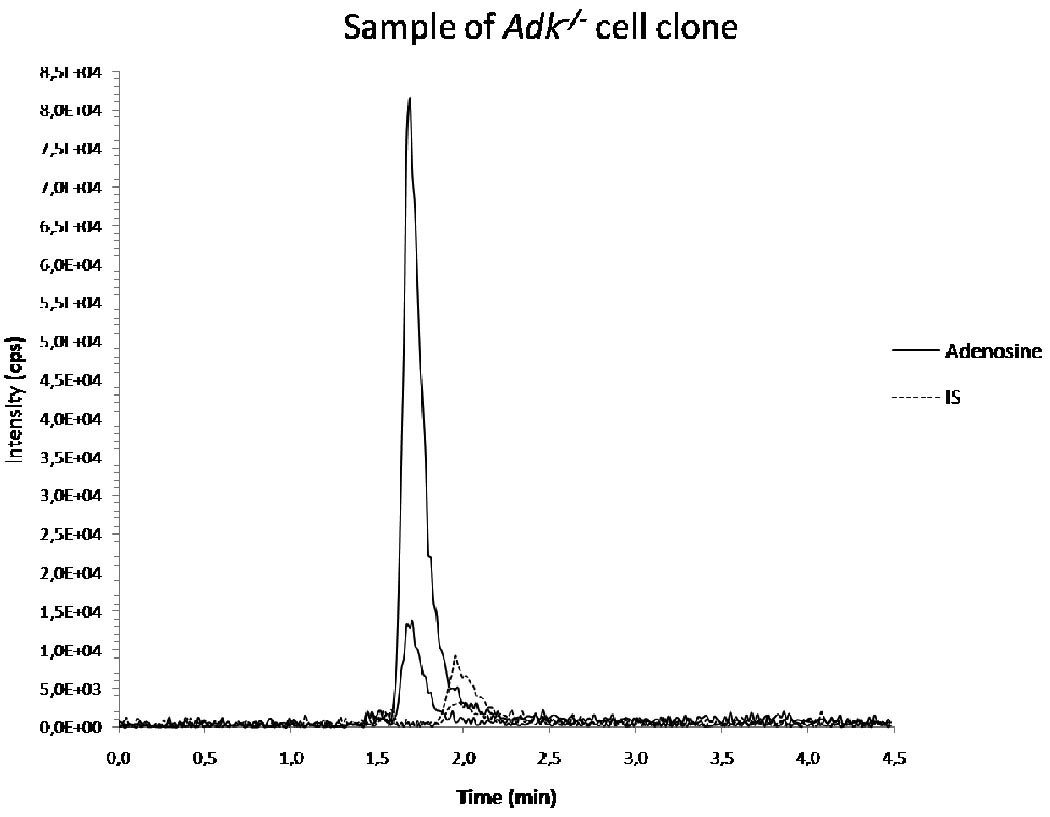
chromatogram of analysis of a sample of Adk−/− cells collected after 24 hours. IS = internal standard.
3.3 Validation of the method
Selectivity and calibration model
Injection of ten blank samples of medium to evaluate the selectivity, showed no interfering signals. Secondly, five replicates of a dilution series of 10 adenosine concentrations in medium were analyzed to create a calibration curve. The relation of the analyte (i.e. adenosine) and the corresponding response was evaluated. The best fitting calibration curve was obtained by linear regression with 1/x weighting factor (r = 0.9989). The linearity was confirmed by statistical analysis (p<0.05). Only in the low concentration range (7.8 and 15.6 ng/ml adenosine) outliers with a bias of more than 20% were found (1/5 samples with 15.6 ng/ml adenosine concentration; 3/5 samples with 7.8 ng/ml adenosine). In the other concentration groups all calibration points had a bias lower than 15% (range 0 – 13.4%). The bias of the mean per concentration level was lower than 15% in all concentration groups after exclusion of the outliers.
Accuracy (bias) and precision
Accuracy (bias) and precision were analyzed on 5 QC samples in duplicate on 6 different days. The accuracy must be within ±15% of the nominal value; for evaluation of precision the relative standard deviation (RSD) must be lower than 15% (or 20% near the LLOQ). The bias was calculated as the percentage deviation of the observed mean value from the respective reference value (table 3). The results were within 15% of the nominal value at all measured concentrations. The within-day and between-day precision were evaluated using one-way analysis of variance. The results, expressed as percentage RSD for the 5 different concentrations, are shown in table 3. The RSD was lower than 15% for all measured concentrations.
Table 3.
results of inaccuracy (bias) and precision measurements. Inaccuracy (bias) is expressed in %; precision experiments in % relative standard deviation (RSD). Bias was within 15% of the nominal value at all concentration levels. For the precision experiments, the RSD results were lower than 15% at all concentration levels.
| Adenosine(ng/ml) | 1000 | 500 | 125 | 31.3 | 15.6 |
|---|---|---|---|---|---|
| Inaccuracy (bias)(%) | 1.35 | 2.65 | 0.66 | 2.04 | 0.19 |
| Within-day precision (%RSD) | 3.13 | 4,47 | 6.82 | 9.60 | 5.93 |
| Between–day precision(%RSD) | 2.49 | 4.18 | 0* | 0* | 5.15 |
If the ‘mean square between groups’ < ‘mean square within groups’, the RSD is set ‘0’. The mean squares are obtained from the analysis of variance-table [27].
Precision and accuracy of QC samples with 15.6 ng/ml adenosine were within the acceptance criteria. Measurements of the lower concentration at 7.8 ng/ml resulted more often in outliers (see also calibration model). Therefore we set our LLOQ of adenosine with this method at 15.6 ng/ml.
Stability
To evaluate freeze and thaw stability, the mean of the replicate QC samples after three freeze/thaw cycles (treatment) was compared to the mean before treatment. The mean measured concentration of the samples with 1000 ng adenosine/ml was 98.2% of the mean before treatment. Of the samples with 250 ng/ml and 31.3 ng/ml this was respectively 100.1% and 100.2%. Since this is within 90–110% of the control samples, it fulfills the acceptance criteria.
Processed sample stability was evaluated by use of parametric tests. No significant difference was found between the first analysis and the analysis after 3 months freezing at −20°C (p>0.05).
Matrix effect
The matrix effect and the possibility of ionization suppression or enhancement were evaluated by comparing the results of the different sets as described in the methods. Figure 3 represents the result of continuous injection of adenosine (1000 ng/ml) and mobile phase or medium. There is a decrease in intensity of adenosine in both the mobile phase and medium, meaning this is not due to matrix effect at that time point (1.4 to 1.6 minutes). At 1.7 minutes, when the adenosine peak is expected in our method, the adenosine signal measured in medium is higher than the adenosine signal in mobile phase, indicating ionization enhancement. This time point is marked in figure 3. The IS is expected around 2 minutes. At that time, a drop in adenosine signal is seen in the graph with medium injection, but not with the mobile phase. This is suggestive for ionization suppression for the IS.
Figure 3.
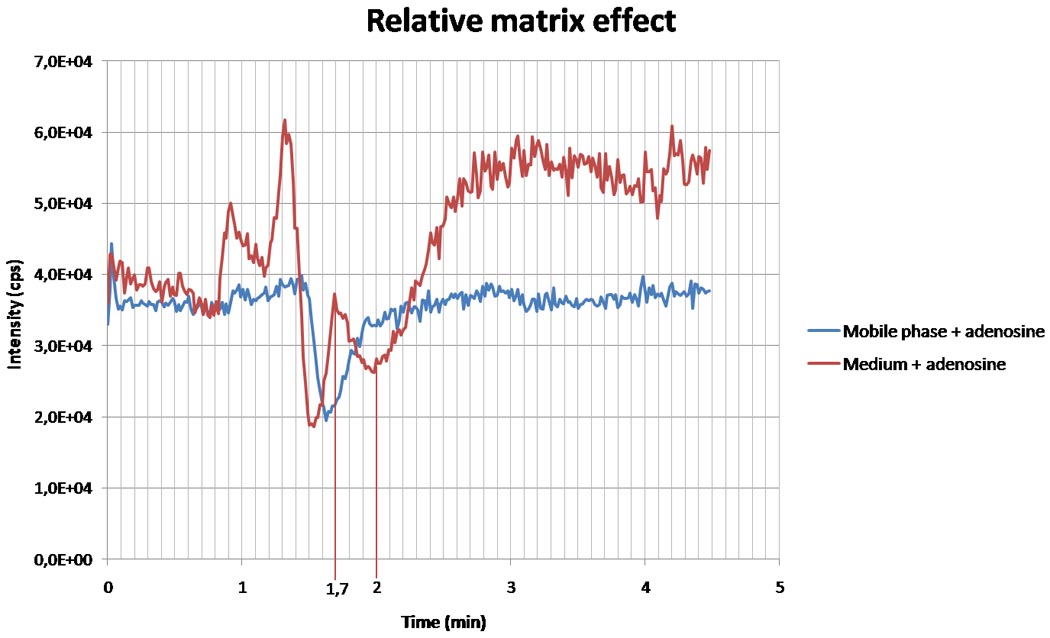
relative matrix effect measured via continuous injection of adenosine (1000 ng/ml) and mobile phase (blue curve) or medium (red curve). After 1.7 minutes, at the time of the expected adenosine peak, adenosine signal in medium is higher than the adenosine signal in mobile phase, indicating ionization enhancement. Around 2 minutes, when the peak of the internal standard is expected, a decrease in adenosine is found in the medium, but not in the mobile phase, suggestive for ionization suppression.
Calibration lines for testing the relative matrix effect were prepared in 5 different lots of different media (i.e. medium taken from cells and medium not been used in culture) as described in the methods. The CV of the slopes of the standard lines did not exceed 3–4%: 2.3% in the medium taken from cells and 2.6% in the medium not been used in culture. This means that the method is not affected by relative matrix effects [30].
The absolute matrix effect was also investigated, both for adenosine and the IS. Evaluation of the mean peak areas of adenosine in mobile phase compared to medium showed an ionization enhancement in medium ranging from 157% to 184% at different concentrations. For the IS the opposite was found: an ionization suppression in the medium compared to the mobile phase ranging from 38% to 43% (table 4).
Table 4.
evaluation of the absolute matrix effect (ME) of the mean peak area (mpa) of adenosine and the internal standard (IS). The matrix effect (ME) is expressed as the ratio of the mpa of set 2 (standard line in medium) to the mpa of set 1 (standard line in the mobile phase) multiplied by 100. A value of >100% indicates ionization enhancement, a value of <100% indicates ionization suppression.
| Concentration adenosine (ng/ml) |
mpa adenosine | ME adenosine (B1/A1%) |
mpa IS | ME IS (B2/A2%) |
||
|---|---|---|---|---|---|---|
| Set 1 (mobile phase) |
Set 2 (medium) |
Set 1 (mobile phase) |
Set 2 (medium) |
|||
| 15.6 | 4240 | 7140 | 168 | 150000 | 62900 | 42 |
| 31.2 | 4700 | 8660 | 184 | 145000 | 63000 | 43 |
| 125 | 33800 | 58300 | 172 | 151000 | 65200 | 43 |
| 500 | 143000 | 231000 | 162 | 154000 | 62700 | 41 |
| 1000 | 293000 | 461000 | 157 | 163000 | 62600 | 38 |
| A1 | B1 | A2 | B2 | |||
4. Discussion and conclusions
We developed and validated an analytical method for the determination of adenosine in cell culture supernatants using liquid chromatography – atmospheric pressure chemical ionization – tandem mass spectrometry (LC-APCI-MS/MS). LC-MS/MS has been widely used for the determination of drugs and their metabolites in biological samples because of its high sensitivity and specificity [25]. Chromatographic separation was performed with the XBridge C8 column and an isocratic elution of 65% 2mM ammonium acetate in water and 35% 2mM ammonium acetate in methanol (v/v). Analysis of the first samples showed interference of the NS-A medium with the adenosine determination. This problem was solved after addition of the adenosine deaminase inhibitor EHNA to the medium indicating that one or several on the medium components contained traces of the adenosine degrading enzyme adenosine deaminase. Consequently, in all subsequent experiments EHNA was added during sample preparation.
Our results show that adenosine can be quantified within a broad concentration range from 15.6 ng/ml to 2000 ng/ml with good accuracy and precision while maintaining freeze and thaw stability and processed sample stability. Evaluation of matrix effect using post-column infusion suggested ionization enhancement for adenosine and ionization suppression for the IS. Assessment of the absolute matrix effect confirmed these results showing the presence of a matrix effect for adenosine and IS measurements in medium compared to mobile phase. An ionization enhancement (157–184%) of adenosine and ionization suppression (38–43%) of the IS are found. However testing the relative matrix effect according to the method of Matuszewski [30] showed that the method is not affected by relative matrix effects. All samples taken from the cultured cells contain medium and all calibration curves are made in the same medium with reproducible results, confirming that the absolute matrix effect does not interfere with the results of analysis of our samples.
Earlier techniques for quantification of adenosine have not been tested for cell culture medium. Our method developed here is reliable to measure adenosine concentrations released from cultured stem cells into medium and therefore gives an additional value. This new analytical method for the quantification of adenosine has several advantages: combination of the high selectivity and sensitivity characteristic for LC-MS/MS technique, with a short run time of 4.5 min, but without the need for an extraction phase. Our new detection method for adenosine may have therapeutic implications, since the local delivery of adenosine via stem cells is considered to be a therapeutic option for future treatments of several neurological disorders [33]. Prior to transplantation of stem cells, reliable determination of adenosine released in vitro is necessary and may be performed using this method.
ACKNOWLEDGEMENTS
Annelies Van Dycke is supported by a junior researcher (‘Aspirant’) grant from the Fund for Scientific Research-Flanders.
Detlev Boison is Director of the Epilepsy Program at Legacy Research and supported by grants NS061844, NS057475, NS058780, NS057538 and MH083973 from the National Institutes of Health (NIH).
Professor Paul Boon is a Senior Clinical Investigator of the Fund for Scientific Research-Flanders and is supported by grants from the Fund for Scientific Research-Flanders and Ghent University Research Fund and by the Clinical Epilepsy Grant from Ghent University Hospital.
Footnotes
DISCLOSURE OF CONFLICTS OF INTEREST
None of the authors has any conflict of interest to disclose
Reference List
- 1.Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Int. Rev. Neurobiol. 2005;63:191. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- 2.Dunwiddie TV. Adv. Neurol. 1999;79:1001. [PubMed] [Google Scholar]
- 3.Boison D. Epilepsy Res. 2009;85:131. doi: 10.1016/j.eplepsyres.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delabar U, Kloor D, Luippold G, Muhlbauer B. J. Chromatogr. B Biomed. Sci. Appl. 1999;724:231. doi: 10.1016/s0378-4347(98)00580-5. [DOI] [PubMed] [Google Scholar]
- 5.Dolezalova P, Krijt J, Chladek J, Nemcova D, Hoza J. Rheumatology. (Oxford) 2005;44:74. doi: 10.1093/rheumatology/keh401. [DOI] [PubMed] [Google Scholar]
- 6.Dudley E, Lemiere F, Van DW, Tuytten R, El-Sharkawi S, Brenton AG, Esmans EL, Newton RP. Rapid Commun. Mass Spectrom. 2004;18:2730. doi: 10.1002/rcm.1685. [DOI] [PubMed] [Google Scholar]
- 7.Gayden RH, Watts BA, III, Beach RE, Benedict CR. J. Chromatogr. 1991;536:265. doi: 10.1016/s0021-9673(01)89259-1. [DOI] [PubMed] [Google Scholar]
- 8.Giannattasio S, Gagliardi S, Samaja M, Marra E. Brain Res. Brain Res. Protoc. 2003;10:168. doi: 10.1016/s1385-299x(02)00215-5. [DOI] [PubMed] [Google Scholar]
- 9.Graham RL, McClean S, O'Kane EJ, Theakston D, Shaw C. Biochem. Biophys. Res. Commun. 2005;333:88. doi: 10.1016/j.bbrc.2005.05.077. [DOI] [PubMed] [Google Scholar]
- 10.Hagberg H, Andersson P, Lacarewicz J, Jacobson I, Butcher S, Sandberg M. J. Neurochem. 1987;49:227. doi: 10.1111/j.1471-4159.1987.tb03419.x. [DOI] [PubMed] [Google Scholar]
- 11.Huang LF, Guo FQ, Liang YZ, Li BY, Cheng BM. J. Pharm. Biomed. Anal. 2004;36:877. doi: 10.1016/j.jpba.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 12.Liebich HM, Di SC, Wixforth A, Schmid HR. J. Chromatogr. A. 1997;763:193. doi: 10.1016/s0021-9673(96)00757-1. [DOI] [PubMed] [Google Scholar]
- 13.Mei DA, Gross GJ, Nithipatikom K. Anal. Biochem. 1996;238:34. doi: 10.1006/abio.1996.0246. [DOI] [PubMed] [Google Scholar]
- 14.Pazzagli M, Corsi C, Latini S, Pedata F, Pepeu G. Eur. J. Pharmacol. 1994;254:277. doi: 10.1016/0014-2999(94)90465-0. [DOI] [PubMed] [Google Scholar]
- 15.Ren J, Mi Z, Jackson EK. J. Pharmacol. Exp. Ther. 2008;325:920. doi: 10.1124/jpet.108.137752. [DOI] [PubMed] [Google Scholar]
- 16.Scalia S, Simeoni S, Dalpiaz A, Villani S. J. Pharm. Biomed. Anal. 2001;24:1131. doi: 10.1016/s0731-7085(00)00572-0. [DOI] [PubMed] [Google Scholar]
- 17.Sottofattori E, Anzaldi M, Ottonello L. J. Pharm. Biomed. Anal. 2001;24:1143. doi: 10.1016/s0731-7085(00)00574-4. [DOI] [PubMed] [Google Scholar]
- 18.Svensson JO, Jonzon B. J. Chromatogr. 1990;529:437. doi: 10.1016/s0378-4347(00)83851-7. [DOI] [PubMed] [Google Scholar]
- 19.Wojcik WJ, Neff NH. J. Neurochem. 1982;39:280. doi: 10.1111/j.1471-4159.1982.tb04736.x. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto T, Moriwaki Y, Takahashi S, Fujita T, Tsutsumi Z, Yamakita J, Shimizu K, Shiota M, Ohta S, Higashino K. J. Chromatogr. B Biomed. Sci. Appl. 1998;719:55. doi: 10.1016/s0378-4347(98)00402-2. [DOI] [PubMed] [Google Scholar]
- 21.Yang Z, Chi SY, Zhang CH, Wu A. Zhongguo Zhong. Yao Za Zhi. 2007;32:2018. [PubMed] [Google Scholar]
- 22.Zhu Y, Wong PS, Zhou Q, Sotoyama H, Kissinger PT. J. Pharm. Biomed. Anal. 2001;26:967. doi: 10.1016/s0731-7085(01)00450-2. [DOI] [PubMed] [Google Scholar]
- 23.Zhao R, Xu G, Yue B, Liebich HM, Zhang Y. J. Chromatogr. A. 1998;828:489. doi: 10.1016/s0021-9673(98)00589-5. [DOI] [PubMed] [Google Scholar]
- 24.Boison D, Scheurer L, Tseng JL, Aebischer P, Mohler H. Exp. Neurol. 1999;160:164. doi: 10.1006/exnr.1999.7209. [DOI] [PubMed] [Google Scholar]
- 25.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Anal. Chem. 2003;75:3019. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 26.Peters FT, Drummer OH, Musshoff F. Forensic Sci. Int. 2007;165:216. doi: 10.1016/j.forsciint.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 27.Peters FT. In: Applications of Liquid Chromatography-Mass Spectrometry in Toxicology. Polettini A, editor. London: Pharmaceutical Press; 2006. p. 71. [Google Scholar]
- 28.US Department of Health and Human Services. 2001 doi: 10.3109/15360288.2015.1037530. www.fda.gov/cder/guidance/index.htm. [DOI] [PubMed]
- 29.Souverain S, Rudaz S, Veuthey JL. J. Chromatogr. A. 2004;1058:61. [PubMed] [Google Scholar]
- 30.Matuszewski BK. J Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2006;830:293. doi: 10.1016/j.jchromb.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Boison D. Neurodegener. Dis. 2007;4:28. doi: 10.1159/000100356. [DOI] [PubMed] [Google Scholar]
- 32.Parkinson FE, Xiong W. J. Neurochem. 2004;88:1305. doi: 10.1046/j.1471-4159.2003.02266.x. [DOI] [PubMed] [Google Scholar]
- 33.Boison D. Curr. Opin. Pharmacol. 2008;8:2. doi: 10.1016/j.coph.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]