

Abstract
Adeno-associated virus (AAV) serotype 8 appears to be the strongest of the natural serotypes reported to date for gene transfer in liver and muscle. In this study, we evaluated AAV8 in the brain by several methods, including biophotonic imaging of green fluorescent protein (GFP). In the adult rat hippocampus, levels of GFP expressed were clearly greater with AAV8 than with AAV2 or AAV5 by Western blot and biophotonic imaging and slightly but significantly greater than AAV1 by Western blot. In the substantia nigra, the GFP expression conferred by AAV8 was toxic to dopamine neurons, although toxicity could be avoided with dose titration. At the low dose at which there was no GFP toxicity from the GFP vector, another AAV8 vector for a disease-related (P301L) form of the microtubule-associated protein tau caused a 78% loss of dopamine neurons and significant amphetamine-stimulated rotational behavior. The AAV8 tau vector-induced cell loss was greater than that from AAV2 or AAV5 tau vectors, demonstrating that the increased gene transfer was functional. While the toxicity observed with GFP expression warrants great caution, the efficient AAV8 is promising for animal models of neurodegenerative diseases and potentially as well for gene therapy of brain diseases.
Keywords: adeno-associated virus, gene therapy, gene transfer, green fluorescent protein, hippocampus, neurodegeneration, neurodegenerative disease, neurotoxicity, substantia nigra, tau
INTRODUCTION
Improving the efficiency of viral vector gene transfer in the brain remains an important goal for gene therapy and other types of functional studies to: (1) achieve physiological levels of transgene product, (2) achieve widespread transgene expression away from the injection site, (3) transduce a broader range of cell types, (4) separate effective doses/volumes from those with side effects or toxicity, and (5) achieve high levels of transgene expression even when high-titer preparations are not available. For these reasons, we evaluated the adeno-associated virus (AAV) serotype 8 vector in the rat brain, which was recently shown to be more efficient than other AAV serotypes in liver [1] and muscle [2]. Gene therapy clinical trials are either under way or being pursued for diseases of the brain such as Alzheimer disease (AD), Parkinson disease (PD), Canavan disease, and Batten disease using the well-studied AAV2 serotype [3]. More efficient AAV serotypes may enhance therapeutic efficacy for these and other diseases of the brain.
Green fluorescent protein (GFP) is a useful reporter transgene that is well tolerated by neurons in the brain [4-6]. While GFP toxicity in various cell lines and primary neuronal cultures has been reported, to our knowledge, there are no reports demonstrating neuronal loss in the brain after singly expressed, untagged, cytoplasmic GFP gene transfer [7-9]. We therefore tested if AAV8 produces greater levels of GFP relative to serotypes AAV1, AAV2, and AAV5 and if elevated GFP expression is neurotoxic in a dose-dependent manner.
Viral vectors are also important for developing animal models of neurodegenerative diseases to study the disease process and explore novel therapies. The ability to control the onset of expression in adult or even aged animals may overcome the potential for adaptation/compensation during development in transgenic mice. The ability to target expression to brain regions that are most associated with specific diseases is another advantage. We previously expressed a form of α-synuclein associated with familial PD that resulted in significant degeneration of dopamine neurons in the substantia nigra (SN) of the rat [10]. More recently, we expressed a disease-related form of the microtubule-associated protein tau in the rat SN [11] because several neurodegenerative diseases involve neurofibrillary pathology (tau pathology) in the SN in humans, such as AD [12], a familial form of PD [13], frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) [14], progressive supranuclear palsy (PSP) [15], and corticobasal degeneration (CBD) [16,17]. Along with the tau pathology, there is degeneration of the SN in FTDP-17 [14], PSP [15], and CBD [16]. In a rat model for these diseases, AAV2 tau gene transfer resulted in significant degeneration of SN dopamine neurons, along with neurofibrillary tangle formation and rotational behavior, while AAV2 α-synuclein gene transfer was not effective in producing consistent motor deficits even though intraneuronal fibrils of α-synuclein had formed in the SN [11]. In an effort to induce a more intense model of SN degeneration, we evaluated AAV8 tau gene transfer to the rat SN at two vector doses.
RESULTS
AAV8 in Vitro
We tested the AAV8 vector for GFP gene transfer on human embryonic kidney 293 cells and neuron-enriched primary cultures. AAV8 did not efficiently transduce 293 cells relative to AAV2. We also tested AAV5 and AAV1 for the pattern of GFP expression: AAV2 >> AAV1 >> AAV8 > AAV5. For the neuronal cultures, AAV8 was efficient at transducing neurons (Figs. 1A and 1B), as we observed with AAV2 in this study and previously [5]. GFP epifluorescence peaked by 5 days and remained stable for at least another 5 days, the longest interval tested with either AAV2 or AAV8. Counterstaining with the neuronal marker NeuN determined that almost all of the neurons were transduced by AAV8 and counterstaining with the astroglial marker GFAP (glial acidic fibrillary protein) determined that astrocytes also expressed GFP (Fig. 1C). Despite highly neuroselective transduction in the brain, AAV2 also transduces cultured astrocytes [18]. Although AAV8 tended to transduce more cultured astrocytes than AAV2, we did not quantify this, and tests of GFP levels in the mixed cultures by Western blots at 10 days after infection did not show differences between AAV2 and AAV8 with equal concentrations tested for each vector and equal protein loading on the gels. AAV5 did not result in GFP expression (as detected by epifluorescence) in the mixed neuron/glia cultures at the concentrations tested (up 1 × 1011 vector genomes per 1 × 105 cells) and we did not test AAV1.
FIG. 1.

AAV8 in cultured cells and AAV1, AAV2, AAV5, and AAV8 in rat hippocampus. (A) GFP epifluorescence in mixed neuron/astroglia culture 5 days after adding AAV8 GFP (1 × 1011 vector genomes per 1 × 105 plated cells). (B) Colocalization of GFP (green) and the neuronal marker NeuN (red) demonstrating efficient neuronal gene transfer. (C) Colocalization of GFP (green) and the astroglial marker GFAP (red) in a microscopic field containing mostly astrocytes. (D) Green fluorescence from AAV1 GFP vector. (E) AAV2 GFP. (F) AAV5 GFP. (G) AAV8 GFP. Equal dose (2 × 1010 vector genomes), equal expression interval (4 weeks), and equal exposure time for image capture in D–G (1 s) demonstrate relatively robust GFP signal with AAV8. Sections shown in D–G are proximal to the injection track, are the peak sections for each subject shown, and represent the overall trend in relative brightness observed in 10 animals/serotype group. (H–K) Same order of serotypes as D–G with DAPI counterstain and with various exposure times. Bars: A, 160 μm; B, 100 μm; D, 610 μm; H, 305 μm. B and C, D–G, and H–K same magnifications.
In Vivo Hippocampus: Efficient Gene Transfer With AAV8
GFP epifluorescence from AAV8 vectors was consistently brighter relative to AAV1, AAV2, and AAV5 compared under equal conditions (Figs. 1D–1G) in 10 animals/serotype group. The AAV8 GFP vector produced strong GFP epifluorescence in hippocampus over larger portions than observed with AAV2 vectors in this study (Fig. 1) and previously [5]. AAV1 and AAV5 also led to more widespread expression relative to AAV2 in the hippocampus (Figs. 1 and 2) as previously reported [19].
FIG. 2.

GFP immunostaining of serial brain sections 4 weeks after injecting AAV vectors into the hippocampus demonstrating widespread expression with AAV1, AAV5, and AAV8 relative to AAV2. (A) AAV1 GFP. (B) AAV2 GFP. (C) AAV5 GFP. (D) AAV8 GFP. Each vector was injected at a dose of 7 × 109 vector genomes. Every sixth section was processed over a 2.8-mm front-to-back extent of the brain. The sequence for the 15 sections shown in A–D is indicated to the left of the sections in A. Bar: 6.2 mm. A–D same magnification.
We compared the spread of gene transfer among AAV1, AAV2, AAV5, and AAV8 vectors 4 weeks after hippocampal injection of equal particle doses (Fig. 2). We estimated volumes of hippocampus containing immunostained GFP cell bodies and processes on the injected side by the Cavalieri method from 1.8 to 4.6 mm posterior to bregma. The transduced volumes from 4 animals per vector group were (mean mm3 ± SEM) AAV1, 11.28 ± 0.96; AAV2, 5.02 ± 1.07; AAV5, 10.47 ± 0.80; AAV8, 11.04 ± 1.39. We compared the transduced volumes by ANOVA/Bonferroni multiple comparison test. AAV1, AAV5, and AAV8 all were significantly different from AAV2 (P < 0.05), although there were no differences among AAV1, AAV5, and AAV8. The volume of the entire hippocampus on the injected side within the sampled region was consistent among the 16 animals analyzed, 14.57 ± 0.23 mm3, and for AAV1, AAV5, and AAV8, 72–77% of the injected hippocampus contained GFP-expressing cell bodies and processes at this vector dose.
Transgene expression in neuron perikarya in regions that innervate and are distal to the injection site is a well-known phenomenon with AAV vectors [5,19-22]. After hippocampal injections of AAV8 GFP, we observed GFP-expressing neurons in medial septum/diagonal band, entorhinal cortex, and contralateral uninjected hippocampus, particularly hilus and CA3/CA4 (ipsilateral entorhinal cortex and contralateral hippocampus shown in Supplemental Fig. 1). We also observed examples of cells in these regions after hippocampal injections of AAV1, AAV2, or AAV5 (data not shown). We did not attempt to count the distal cells to compare serotypes in this regard.
Cell bodies expressing GFP after injections of AAV8 had neuronal morphology. GFP colocalized with the neuronal marker NeuN (Figs. 3A–3C) but not the astroglial marker GFAP (not shown) as seen with AAV2. In addition to the pyramidal and granule cells, a subset of GABAergic neurons (parvalbumin immunoreactive) was efficiently transduced in the stratum oriens, pyramidal and granule cell layers, and hilus with AAV8 (Figs. 3F–3H).
FIG. 3.

Neuronal and GABAergic expression of AAV8 in hippocampus. (A) GFP expression in hippocampus 4 weeks after injecting 2 × 1010 vector genomes. (B) NeuN counterstain, same section as A. (C) Merged image shows that the majority of pyramidal and granule neurons on this section was transduced. (D) Higher magnification of injected side showing colocalization of GFP and NeuN. (E) Uninjected contralateral hippocampus with NeuN and DAPI counterstains. GFP was strongly expressed in contralateral afferents in CA1 and inner molecular layer of dentate gyrus. (F) GFP epifluorescence on injected side. (G) Immunolabeling for parvalbumin, a marker of GABAergic neurons, same field as F. (H) Merger of F and G shows examples of cells expressing both markers. Bars: A, 305 μm; D, 77 μm; F, 194 μm. A–C, D and E, and F–H same magnifications.
We quantified levels of GFP with biophotonic imaging for serotype comparisons and for a time course of AAV8-mediated expression (Fig. 4). Vector doses for all the biophotonic imaging studies were 2 × 1010 vector genomes. Survival intervals after hippocampal injections were 4 weeks for the serotype comparisons and 1, 2, and 4 weeks for the AAV8 time course. We imaged intact phosphate-buffered saline (PBS)-perfused brains with the GFP filter set and measured the photon emission at a constant exposure time. The results from three animals/group at 4 weeks were as follows (mean photons/s ± SEM): AAV1, 2.60 ± 0.38 × 1010; AAV2, 6.03 ± 1.06 × 109; AAV5, 3.80 ± 0.86 × 109; AAV8, 2.37 ± 0.64 × 1010. ANOVA/Tukey’s multiple comparison test determined that AAV1 was significantly different from AAV2 and AAV5 (both P < 0.05), AAV8 was significantly different from AAV2 and AAV5 (both P < 0.05), and there were no differences between either AAV2 and AAV5 or AAV1 and AAV8. The results from three animals/group with AAV8 at 1 and 2 weeks were as follows: 1 week, 2.37 ± 0.55 × 109 photons/s; 2 weeks, 2.46 ± 0.59 × 1010 photons/s. ANOVA/Tukey’s multiple comparison test determined a significant difference between 1 and 2 weeks (P < 0.05) and no difference between 2 and 4 weeks.
FIG. 4.

Biophotonic imaging of GFP in brains that were injected with AAV vectors into hippocampus. (A) Serotype comparisons at equal dose (2 × 1010 vector genomes) and expression interval (4 weeks). Three brains per vector group are shown (in columns) as indicated. The technique yields quantitative data, described under Results. (B) 1-, 2-, and 4-week time course of AAV8 GFP expression at the same vector dose as in A. Three brains per interval shown (in columns) as indicated.
We also evaluated GFP expression levels by Western blot at 4 weeks (Fig. 5). We estimated the relative intensities of the GFP bands with the Scion (Frederick, MD, USA) imaging program. We compared AAV2, AAV5, and AAV8 at a dose of 7 × 109 vector genomes (Fig. 5A). We performed three separate analyses with the AAV5 and AAV8 samples diluted 3- to 5-fold to match the intensity of the AAV2 bands. The average increase in expression of AAV5 relative to AAV2 with three or four animals/group was 13-fold. By the same analyses, AAV8 was 26-fold stronger than AAV2. In a separate study, we compared AAV1 and AAV8 at a dose of 2 × 1010 vector genomes with three animals/group (Fig. 5B). While the density of GFP bands appeared similar for AAV1 and AAV8, image analysis detected a significant 42% greater signal for AAV8 relative to AAV1 (P < 0.02, t test, N = 3/group).
FIG. 5.
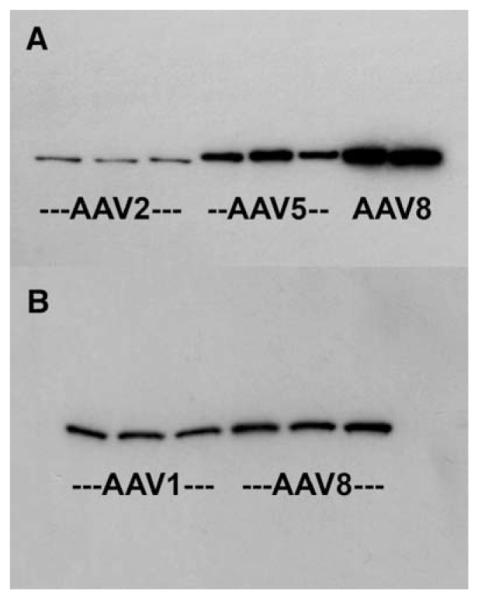
GFP levels evaluated by Western blot. (A) GFP immunoblot showing 31-kDa GFP bands from dissected hippocampi 4 weeks after injection of 7 × 109 vector genomes of AAV. On this blot, three AAV2 brains (60 μg protein loaded), three AAV5 brains (12 μg protein loaded), and two AAV8 brains (12 μg protein loaded) were assayed. (B) GFP immunoblot comparing AAV1 and AAV8 at a dose of 2 × 1010 vector genomes after 4 weeks (12 μg protein loaded in all lanes). There was a small but significantly greater expression of GFP from AAV8 relative to AAV1 (see Results).
In Vivo Substantia Nigra: GFP Neurotoxicity with AAV8
Similar to hippocampus, AAV8 GFP fluorescence in the SN was generally brighter than that of AAV2 or AAV5 and neuroselective in terms of NeuN and GFAP counterstaining: GFP colocalized with the neuronal but not the astroglial marker. AAV8 efficiently labeled both SN pars compacta dopamine neurons (labeled for tyrosine hydroxylase (TH)) and GABAergic neurons (labeled for parvalbumin) in the SN pars reticulata as did AAV2 [11]. GFP-expressing cells were found in striatum after injections of AAV2, AAV5, or AAV8 in SN. GFAP, OX-42 (microglial marker), and DAPI counterstaining were slightly elevated along the needle track only, although the pattern was identical with that of injections of the vector diluent, so the AAV8 did not induce astrogliosis or microgliosis.
AAV8 GFP expression did, however, cause neuronal loss in the SN. Injections with AAV8 GFP at a dose of 5 × 1010 vector genomes resulted in unprecedented bright fluorescence as in hippocampus, although we observed considerably fewer GFP-positive cells in the SN pars compacta than with similar doses of AAV2 or AAV5 GFP. We therefore tested four doses of the AAV8 GFP vector and found more GFP-positive cells in the pars compacta at the lower doses, consistent with the hypothesis that toxic levels of GFP were achieved at the high dose. TH staining showed loss of labeling after AAV8 GFP gene transfer relative to the contralateral uninjected side (1 × 1010 particle dose shown in Fig. 6). While the results shown in Fig. 6 suggest a loss of TH phenotype rather than loss of cells at this dose, i.e., pars compacta GFP-expressing cells were negative for TH (arrows in Fig. 6), there was clear loss of both TH- and GFP-expressing cells at the fivefold higher dose. Quantification of TH neurons by stereology showed a 41% loss relative to uninjected SN in the highest dose group (Table 1A), and at the lowest dose, there was no dopaminergic cell loss (Table 1A; Fig. 7). We found a significant dose response for AAV8 GFP vector dose and the number of TH neurons estimated by stereology (ANOVA, F3,13 = 5.25, P < 0.02). To determine whether the TH cell loss was attributable to GFP toxicity rather than AAV8 infection, we compared equal doses of the AAV8 GFP with AAV8 Empty vector, the same construct without the GFP DNA. We were limited to the dose that we could test because of the titer of the AAV8 Empty vector preparation, 2 × 1012 vector genomes/ml, which resulted in an injection of 8 × 109 vector genomes to the animal. Comparing the AAV8 GFP and the AAV8 Empty vectors at this dose to uninjected SN by Dunnett’s multiple comparison test against a single control group resulted in significantly fewer cells with the AAV8 GFP (P < 0.05) but not the AAV8 Empty vector (Table 1A), supporting that the cell loss was due to GFP toxicity rather than AAV8 infection.
FIG. 6.

AAV8 GFP gene transfer induces partial loss of substantia nigra dopamine neurons. (A) Tyrosine hydroxylase immunofluorescence of uninjected substantia nigra, contralateral to B. (B) Tyrosine hydroxylase immunofluorescence 4 weeks after injection of 1 × 1010 vector genomes of AAV8 GFP. (C) Same section as B showing GFP epifluorescence. Arrows point to cells in pars compacta expressing GFP but not TH, showing that the AAV8 GFP vector at this dose resulted in loss of endogenous gene expression, not cell loss. Bar: 60 μm. A–C, same magnification.
TABLE 1.
| (A) Tyrosine hydroxylase neurons after gene transfera | ||||
|---|---|---|---|---|
| Treatment | Dose | TH neurons | N | Comparison |
| Uninjected | — | 9458 ± 169 | 5 | |
| AAV8 Empty | 8 | 9502 ± 189 | 3 | Uninjected, NS |
| AAV8 GFP | 4 | 9471 ± 161 | 3 | |
| AAV8 GFP | 8 | 8031 ± 569 | 3 | Uninjected, P < 0.05 |
| AAV8 GFP | 10 | 6586 ± 829 | 5 | |
| AAV8 GFP | 50 | 5550 ± 516 | 3 | AAV8 GFP dose 4, P < 0.01 |
| AAV8 tau | 4 | 2112 ± 512 | 3 | AAV8 GFP equal dose, P < 0.001 |
| AAV8 tau | 10 | 2415 ± 637 | 3 | AAV8 GFP equal dose, P < 0.001 |
| AAV2 tau | 4 | 6526 ± 615 | 4 | AAV8 tau dose 4, P < 0.001 |
| AAV5 tau | 6 | 7491 ± 316 | 3 | AAV8 tau dose 4, P < 0.001 |
| (B) Rotational behavior after gene transferb | |||||
|---|---|---|---|---|---|
| Vector | Dose | Ipsilateral | Contralateral | N | P |
| Uninjected | — | 12.0 ± 5.5 | 6.7 ± 4.2 | 6 | NS |
| AAV8 GFP | 4 | 19.7 ± 5.2 | 12.3 ± 3.2 | 3 | NS |
| AAV8 GFP | 10 | 18.8 ± 6.6 | 7.6 ± 2.4 | 5 | NS |
| AAV5 tau | 6 | 6.7 ± 4.8 | 13.0 ± 12.5 | 3 | NS |
| AAV8 tau | 4 | 38.7 ± 8.1 | 2.7 ± 2.7 | 3 | <0.02 |
| AAV8 tau | 10 | 22.0 ± 3.2 | 3.0 ± 2.5 | 3 | <0.001 |
Number of substantia nigra pars compacta dopamine neurons (±SEM) estimated by stereology at the indicated dose (×109 vector genomes) 4 weeks after gene transfer (for the vector groups). Significant dose–response relationship for number of TH neurons found with the four doses of the AAV8 GFP vector by ANOVA, P < 0.02 (see Results). Other comparisons used Dunnet’s and Bonferroni’s multiple comparison tests (see Results).
Amphetamine-stimulated rotations (±SEM) 4 weeks after gene transfer (for the vector groups, dose × 109 vector genomes). Side-to-side differences were observed in the AAV8 tau but not the GFP or AAV5 tau vector groups (t test).
FIG. 7.

AAV8 tau gene transfer induces robust lesion of substantia nigra dopamine neurons. (A) GFP immunostaining 4 weeks after injection of 4 × 109 vector genomes of AAV8 GFP. (B) Adjacent section from A showing that this dose led to no change in TH staining on the injected side (right side of image) relative to the contralateral uninjected side. (C) Human tau immunostaining 4 weeks after injection of 6 × 109 vector genomes of AAV5 tau. (D) TH staining on adjacent section from C. (E) Human tau immunostaining 4 weeks after injection of 4 × 109 vector genomes of the AAV8 tau vector. A blank area in the pars compacta devoid of tau staining was always observed after these injections, unlike with AAV5 tau in C. (F) TH staining on an adjacent section from E showing dramatic loss of TH neurons on the side injected with tau, which was not observed with AAV5 in D. Bars: A, 200 μm; B, 320 μm. A, C, and E same magnification; B, D, and F same magnification.
Microtubule-Associated Protein Tau Neurodegeneration Disease Model with AAV8
Based on the boost of GFP expression we observed in hippocampus, we tested an AAV8 vector for the P301L form of tau. This mutation is associated with neurodegeneration in humans [14,23] and resulted in a significant rat model of SN neurodegeneration in AAV2 context [11]. The AAV8 tau vector resulted in profound lesioning of the SN, while an AAV5 tau vector did not (Fig. 7; Table 1A). Two doses of the tau vector resulted in 74–78% loss of TH neurons relative to uninjected SN (Table 1A). At both doses, there was significantly more cell loss with AAV8 tau relative to the equal dose of AAV8 GFP (ANOVA and Bonferroni’s multiple comparison test, P < 0.001). In addition, the AAV8 tau (dose 4 × 109 vector genomes) caused significantly more cell loss than an equal dose of AAV2 tau (ANOVA/Bonferroni’s multiple comparison test, P < 0.001) or a higher dose of AAV5 tau (6 × 109 vector genomes; ANOVA/Bonferroni’s multiple comparison test, P < 0.001). We tested the rats for amphetamine-stimulated rotational behavior, which occurs when there is a pronounced side-to-side imbalance in nigrostriatal dopamine, as significant behavioral effects typically require 50% loss of SN dopamine neurons and 75–90% loss of striatal dopamine [24,25]. Consistent with the robust dopaminergic cell loss resulting from AAV8 tau, we found significant amphetamine-stimulated turning in this group, but not in the AAV5 tau group or GFP controls after unilateral injections to the SN (Table 1B). The enhanced transgene expression observed in hippocampus with AAV8 relative to AAV5 was therefore also functionally observed with tau vectors in the SN in terms of cell loss and behavior.
DISCUSSION
AAV8 produced strong transgene expression in rat neurons in culture and in the brain. We measured GFP expression levels by Western blot and biophotonic imaging and found greater levels with AAV8 relative to AAV2 or AAV5. The spread of expression in hippocampus with AAV8 was greater than with AAV2 but not AAV5. While AAV5 produced greater hippocampal volumes of GFP expression and greater levels of GFP on Westerns relative to AAV2, the biophotonic imaging resulted in a similar signal for AAV2 and AAV5. These data are consistent with the report by Paterna et al. [22], which showed that AAV5 produces greater spread while AAV2 produces greater intraneuronal levels of transgene expression, and demonstrate that the Western blots, the volume estimates, and the biophotonic imaging measure different properties of GFP expression. We conclude that features of AAV8 combine the widespread expression within the hippocampus of AAV5 with the high intraneuronal expression levels of AAV2. AAV1 is also an efficient vector for hippocampus, with a similar signal from biophotonic imaging and a similar spread relative to AAV8. While there was significantly more GFP on Westerns of dissected hippocampi with AAV8 than with AAV1 the difference was not pronounced, although GFP epifluorescence on sections was in all cases noticeably brighter with AAV8 than with AAV1. This may suggest a greater range of expression levels in specific cells with AAV8.
The improved neuronal gene transfer of AAV8 relative to AAV2 in the brain was not observed in primary neuronal cultures. Similar to AAV2 [18], AAV8 resulted in transduction of astrocytes in culture but not in the brain, while AAV5 did not produce detectable transduction in cultured neurons or glia. These results demonstrate that AAV gene transfer of primary neuronal cultures is a poor predictor of gene transfer in the brain.
The high levels of GFP derived from AAV8 were neurotoxic to dopamine neurons in the SN at higher but not lower vector doses. To our knowledge, this marks the first quantified example of untagged, singly expressed, cytoplasmic GFP toxicity in the brain and certainly indicates great caution should be used with high-efficiency vectors such as AAV8. While these results were unequivocal, it is unclear why there was no apparent neuronal loss in the hippocampus, as visualization of NeuN did not suggest any loss of pyramidal or granule neurons associated with the GFP. The results suggest that dopamine neurons are more susceptible to GFP toxicity, although there are several other potential explanations including more efficient gene expression in dopamine neurons or differential tolerance to infusion of volumes of several microliters that could render neurons vulnerable.
Despite the toxicity of the GFP reporter gene after AAV8 gene transfer in this study, tau was significantly more toxic to dopamine neurons. At two doses, AAV8 tau gene transfer induced a 74–78% loss of dopamine neurons relative to uninjected controls and significant rotational behavior. In contrast, similar doses of AAV8 GFP, AAV2 tau, or AAV5 tau did not cause as much cell loss. These results are consistent with what we observed in hippocampus with respect to transduction efficiency of AAV8 relative to AAV2 or AAV5.
Vector-based neurodegenerative disease models are an important complement to neurotoxin lesions and transgenic mice. Relative to neurotoxins, the vector approach may better mimic slow disease progression and, more importantly, specifically model tau pathophysiology. Relative to transgenic mice, the vector approach may be more targeted to specific brain regions and ages associated with specific neurodegenerative diseases. We are hopeful these advantages for a disease model will translate for drug development. While the observation of GFP reporter gene toxicity with AAV8 was alarming, it could be controlled with dose titration. At the lower doses, the AAV8 tau vector produced an enhanced model of SN degeneration, significant for studying diseases involving neurofibrillary pathology and loss of dopamine neurons such as FTDP-17, PSP, and CBD. This will be important for our goals of studying the disease process longitudinally within animals by various analyses of motor function and brain imaging. Because therapies are limited or nonexistent for these pernicious diseases and there are familial forms of these diseases, as is the case for AD and PD, gene therapy is worth considering. The enhanced efficiency of serotype 8 could be important for the ongoing efforts in brain gene therapy using AAV [3].
MATERIALS AND METHODS
DNA and AAV constructs
An expression cassette flanked with AAV2 terminal repeats was packaged into recombinant AAV2 or cross-packaged into recombinant AAV1, AAV5, or AAV8. The promoter/enhancer combination used to drive expression included the hybrid cytomegalovirus/chicken β-actin promoter and the 3′ enhancer woodchuck hepatitis virus posttranscriptional regulatory element as described previously [5]. Plasmids for the reporter control GFP and the P301L form of human tau including exons 2, 3, and 10 (four repeat microtubule-binding domains, 4R2N) were described previously [5,26]. An empty vector construct that had the same terminal repeats and promoter/enhancer as above but no transgene coding sequence was packaged.
The method for packaging plasmids in recombinant AAV was based on previously described methods [22,27]. Human embryonic kidney 293 cells were cotransfected with the AAV2 terminal repeat-containing plasmid and packaging plasmid(s) as follows: for AAV2, pDG [28]; for AAV5, pXYZ5 [27]; for AAV8, p5E18-VD2/8 and pAd-deltaF6 [29]. The cell lysate was applied to a discontinuous gradient of iodixanol (OptiPrep, Greiner Bio-One, Longwood, FL, USA) and centrifuged. The AAV was then removed and concentrated and washed using Millipore (Billerica, MA, USA) Biomax 100 Ultrafree-15 units. AAV vector stocks were titered for copies of vector genomes, by dot blotting against standard curves of known amounts of DNA using nonradioactive psoralen–biotin and BrightStar kits from Ambion (Austin, TX, USA). Vector preps used in the study ranged from 1.1 × 1012 to 1.2 × 1013 vector genomes/ml and equal dose comparisons were made by normalizing titers with the diluent, lactated Ringer’s solution (Baxter, Deerfield, IL, USA). Two or three batches of the vectors AAV1 GFP, AAV2 GFP, AAV8 GFP, and AAV8 tau were tested, yielding consistent results.
Animals and stereotaxic injections
Male Sprague–Dawley rats (3 months of age, from Harlan, Indianapolis, IN, USA) were anesthetized with a cocktail of 3 ml xylazine (20 mg/ml, from Butler, Columbus, OH, USA), 3 ml ketamine (100 mg/ml, from Fort Dodge Animal Health, Fort Dodge, IA, USA), and 1 ml acepromazine (10 mg/ml, from Boehringer Ingelheim, St. Joseph, MO, USA) administered intramuscularly at a dose of 1 ml/kg. Viral stocks were injected through a 27-gauge cannula connected via 26-gauge internal diameter polyethylene tubing to a 10-μl Hamilton syringe mounted to a microinjection pump (CMA/Microdialysis, North Chelmsford, MA, USA) at a rate of 0.2 μl/min. The stereotaxic injection coordinates for hippocampus were 3.6 mm bregma, 2.0 mm lateral, 3.5 or 2.8 mm ventral with 3 μl injected at each depth and for the SN, 5.3 mm bregma, 2.1 mm lateral, 7.6 mm ventral with 4 μl injected [30]. The needle remained in place at the injection site for 1 additional min before the cannula was removed slowly (over 2 min). The skin was sutured, and the animal was placed on a heating pad until it began to recover from the surgery, before being returned to its individual cage. All animal care and procedures were in accordance with institutional IACUC and NIH guidelines.
Primary cultures
Mixed neuron/glia cultures were prepared from cerebral cortex of newborn Sprague–Dawley rats (Harlan) as described [31]. Cells (1 × 105) were plated on 35-mm dishes. The cultures were treated with vectors 8 days after plating and evaluated for GFP expression 1–10 days later. Based on previous experience, the AAV preparations typically have a particle-to-infectivity ratio of about 50. We tested infections with up to 1 × 1011 vector genomes and estimated multiplicity of infection of 2 × 104 at the highest concentrations tested.
Western blots
Primary cultures were harvested in RIPA buffer (1% Nonidet P-40/0.5% sodium deoxycholate/0.1% SDS/PBS) with protease inhibitors (Halt protease inhibitor cocktail kit from Pierce, Rockford, IL, USA) and the soluble fraction was prepared by polytron homogenization and centrifugation. The dorsal hippocampus was dissected and the soluble fraction was prepared in the same buffer by Dounce homogenization and centrifugation. Samples were normalized for protein content by Bradford assay and subjected to 12% SDS–polyacrylamide gel electrophoresis. The primary antibody for immunoblots was GFP monoclonal from Chemicon (Temecula, CA, USA; 1:1000). Secondary antibody and ECL reagents were from Amersham (Buckinghamshire, UK).
Immunostaining
Primary cultures were washed with PBS, fixated with 4% paraformaldehyde/4% sucrose/PBS for 15 min, washed, and permeabilized with 0.3% Triton X-100 for 5 min. Primary antibodies were NeuN (monoclonal from Chemicon; 1:250) or GFAP (polyclonal from Chemicon; 1:250). Cy3-conjugated secondary antibodies were from Jackson ImmunoResearch (West Grove, PA, USA; 1:300).
Anesthetized animals were perfused with PBS followed by cold 4% paraformaldehyde in PBS. The brain was removed and immersed in fixative overnight at 4°C. Brains were equilibrated in a cryoprotectant solution of 30% sucrose/PBS at 4°C. Coronal sections (50 μm thick) were cut on a sliding microtome with a freezing stage. Antigen detection was conducted on free-floating sections.
Primary antibody incubations were overnight at 4°C on a shaking platform. For immunoperoxidase staining, endogenous peroxidase activity was quenched with 0.1% H2O2/PBS for 10 min. The sections were washed in PBS and incubated for 5 min in 0.3% Triton X-100/PBS and washed before primary antibody was applied. Primary antibodies for immunostaining included NeuN monoclonal, parvalbumin monoclonal, GFAP polyclonal, TH polyclonal, CD 11b monoclonal (all from Chemicon and used at dilutions of 1:500), and GFP polyclonal from Molecular Probes/Invitrogen (Carlsbad, CA, USA; 1:100,000 in Fig. 2 or 1:5000 in Supplemental Fig. 1). Biotinylated secondary antibodies for peroxidase staining were from DAKO Cytomation (Carpinteria, CA, USA; 1:2000) and incubated for 1 h at room temperature. The sections were washed with PBS and labeled with horseradish peroxidase-conjugated extravidin (Sigma, St. Louis, MO, USA; 1:2000) for 30 min at room temperature. The chromogen was diaminobenzidine (0.67 mg; Sigma) in 0.3% H2O2, 80 mM sodium acetate buffer containing 8 mM imidazole and 2% NiSO4. After being mounted on slides, the sections were dehydrated in a series of alcohol and xylene and coverslipped with Eukitt (Electron Microscopy Sciences, Hatfield, PA, USA). For immunofluorescence, sections were incubated in primary antibody overnight, washed, and incubated with Cy3-conjugated secondary antibodies (Jackson ImmunoResearch; 1:300) for 2 h, followed by DAPI counterstaining (1 μg/ml), washes, and coverslipping with glycerol/gelatin (Sigma).
Volume measures
Cavalieri probes estimated hippocampal volumes using the MicroBrightfield, Inc. (Williston, VT, USA), StereoInvestigator program with microscope/motorized stage. Grids were randomly oriented and spaced to yield ca. 150 markers and Gundersen error <0.05. For each subject, we analyzed six sections evenly spaced within 1.8 to 4.6 mm posterior to bregma. This region started at the septal pole of the hippocampus and ended before the CA fields of the dorsal hippocampus fused with the ventral portion. For each subject, we estimated the entire volume of this portion of the hippocampus on the side of the injection as well as the transduced volume, which we defined as regions containing GFP-immunostained cell bodies and processes. Regions that contained only GFP-positive processes without discernable GFP-positive perikarya were not included in the transduced volume.
Biophotonic imaging
Rats were anesthetized as above and perfused with 100 ml PBS. The brains were then extracted and stored in PBS. The order in which brains were harvested equalized the average time between extraction and imaging session for all the groups. Within 30 min, the brains were placed in the Xenogen (Alameda, CA, USA) IVIS 100/XFO-12 apparatus and imaged with the GFP filter set. Exposure times were constant at 0.5 s. The region of interest (above the injected hippocampus) was encircled and quantified for photons/s by the IVIS system.
Stereological estimates
The number of SN pars compacta neurons expressing TH immunoreactivity was estimated by unbiased stereology using the MicroBrightfield, Inc., system as previously described [11]. Eight sections evenly spaced throughout the SN pars compacta structure were analyzed for each probe. Optical dissectors were 50 × 50 × 16 μm cubes spaced in a systematic random manner 150 × 150 μm apart and offset 2 μm from the section surface. The fractionator sampling was optimized to yield about 150 counted cells per animal for Gundersen error coefficients <0.10.
Rotational behavior
Animals were challenged with d-amphetamine (free base, 2 mg/kg in saline, im; Sigma) 4 weeks after gene transfer. The amphetamine was injected 20 min before the animals were placed in an automated rotometer system from San Diego Instruments (San Diego, CA, USA) for 10 min.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Gene Therapy Program, University of Pennsylvania, for packaging plasmids for AAV8 production, and Jared Latiolais, Talicia Domonique Johnson, Tracee’ Terry, Lory Tubbs, Dr. Shayne Barlow, and Dr. Kathryn Hamilton for assistance. NIH/NINDS R01 NS048450, The Society for Progressive Supranuclear Palsy, The Parkinson’s Disease Resource of Northwest Louisiana, and The Louisiana Gene Therapy Research Consortium supported the work.
Footnotes
APPENDIX A. SUPPLEMENTARY DATA Supplementary data associated with this article can be found in the online version at doi:10.1016/j.ymthe. 2005.10.008.
REFERENCES
- 1.Nakai H, Fuess S, Storm TA, Muramatsu S, Nara Y, Kay MA. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J. Virol. 2005;79:214–224. doi: 10.1128/JVI.79.1.214-224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Z, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 3.Mandel RJ, Burger C. Clinical trials in neurological disorders using AAV vectors: promises and challenges. Curr. Opin. Mol. Ther. 2004;6:482–490. [PubMed] [Google Scholar]
- 4.Klein RL, et al. Neuron-specific transduction in the rat septohippocampal or nigrostriatal pathway by recombinant adeno-associated virus vectors. Exp. Neurol. 1998;150:183–194. doi: 10.1006/exnr.1997.6736. [DOI] [PubMed] [Google Scholar]
- 5.Klein RL, et al. Dose and promoter effects of adeno-associated viral vector for GFP expression in the rat brain. Exp. Neurol. 2002;176:66–74. doi: 10.1006/exnr.2002.7942. [DOI] [PubMed] [Google Scholar]
- 6.Ikawa M, Yamada S, Nakanishi T, Okabe M. ‘Green mice’ and their potential usage in biological research. FEBS Lett. 1998;430:83–87. doi: 10.1016/s0014-5793(98)00593-6. [DOI] [PubMed] [Google Scholar]
- 7.Liu HS, Jan MS, Chou CK, Chen PH, Ke NJ. Is green fluorescent protein toxic to living cells? Biochem. Biophys. Res. Commun. 1999;260:712–717. doi: 10.1006/bbrc.1999.0954. [DOI] [PubMed] [Google Scholar]
- 8.Detrait ER, et al. Reporter gene transfer induces apoptosis in primary cortical neurons. Mol. Ther. 2002;5:723–730. doi: 10.1006/mthe.2002.0609. [DOI] [PubMed] [Google Scholar]
- 9.Krestel HE, Mihaljevic AL, Hoffman DA, Schneider A. Neuronal co-expression of EGFP and β-galactosidase in mice causes neuropathology and premature death. Neurobiol. Dis. 2004;17:310–318. doi: 10.1016/j.nbd.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 10.Klein RL, King MA, Hamby ME, Meyer EM. Dopaminergic cell loss induced by human A30P α-synuclein gene transfer to the rat SN. Hum. Gene Ther. 2002;13:605–612. doi: 10.1089/10430340252837206. [DOI] [PubMed] [Google Scholar]
- 11.Klein RL, Dayton RD, Lin WL, Dickson DW. Tau gene transfer, but not α-synuclein, induces both progressive dopamine neuron degeneration and rotational behavior in the rat. Neurobiol. Dis. 2005;20:64–73. doi: 10.1016/j.nbd.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider JA, Bienias JL, Gilley DW, Kvarnberg DE, Mufson EJ, Bennett DA. Improved detection of SN pathology in Alzheimer’s disease. J. Histochem. Cytochem. 2002;50:99–106. doi: 10.1177/002215540205000111. [DOI] [PubMed] [Google Scholar]
- 13.Duda JE, et al. Concurrence of α-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol. 2002;104:7–11. doi: 10.1007/s00401-002-0563-3. [DOI] [PubMed] [Google Scholar]
- 14.Mirra SS, et al. Tau pathology in a family with dementia and a P301L mutation in tau. J. Neuropathol. Exp. Neurol. 1999;58:335–345. doi: 10.1097/00005072-199904000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Poorkaj P, et al. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann. Neurol. 2002;52:511–516. doi: 10.1002/ana.10340. [DOI] [PubMed] [Google Scholar]
- 16.Wakabayashi K, et al. Corticobasal degeneration: etiopathological significance of the cytoskeletal alterations. Acta Neuropathol. 1994;87:545–553. doi: 10.1007/BF00293314. [DOI] [PubMed] [Google Scholar]
- 17.Di Maria E, et al. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann. Neurol. 2000;47:374–377. doi: 10.1002/1531-8249(200003)47:3<374::aid-ana15>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Gong Y, et al. Recombinant adeno-associated virus serotype 2 effectively transduces primary rat brain astrocytes and microglia. Brain Res. Brain Res. Protoc. 2004;14:18–24. doi: 10.1016/j.brainresprot.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Burger C, et al. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol. Ther. 2004;10:302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 20.Kaspar BK, Erickson D, Schaffer D, Hinh L, Gage FH, Peterson DA. Targeted retrograde gene delivery for neuronal protection. Mol. Ther. 2002;5:50–56. doi: 10.1006/mthe.2001.0520. [DOI] [PubMed] [Google Scholar]
- 21.Passini MA, et al. AAV vector-mediated correction of brain pathology in a mouse model of Niemann–Pick A disease. Mol Ther. 2005;11:754–762. doi: 10.1016/j.ymthe.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 22.Paterna JC, Feldon J, Bueler H. Transduction profiles of recombinant adeno-associated virus vectors derived from serotypes 2 and 5 in the nigrostriatal system of rats. J. Virol. 2004;78:6808–6817. doi: 10.1128/JVI.78.13.6808-6817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutton M, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 24.Hefti F, Melamed E, Wurtman RJ. Partial lesions of the dopaminergic nigrostriatal system in rat brain: biochemical characterization. Brain Res. 1980;195:123–137. doi: 10.1016/0006-8993(80)90871-9. [DOI] [PubMed] [Google Scholar]
- 25.Hudson JL, et al. Correlation of apomorphine- and amphetamine-induced turning with nigrostriatal dopamine content in unilateral 6-hydroxydopamine lesioned rats. Brain Res. 1993;626:167–174. doi: 10.1016/0006-8993(93)90576-9. [DOI] [PubMed] [Google Scholar]
- 26.Klein RL, et al. Rapid neurofibrillary tangle formation after localized gene transfer of mutated tau. Am. J. Pathol. 2004;164:347–353. doi: 10.1016/S0002-9440(10)63124-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zolotukhin S, et al. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 28.Grimm D, Kern A, Rittner K, Kleinschmidt JA. Novel tools for production and purification of recombinant adeno-associated virus vectors. Hum. Gene Ther. 1998;9:2745–2760. doi: 10.1089/hum.1998.9.18-2745. [DOI] [PubMed] [Google Scholar]
- 29.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1986. [Google Scholar]
- 31.Herring D, Huang R, Singh M, Dillon GH, Leidenheimer NJ. PKC modulation of GABA receptor endocytosis and function is inhibited by mutation of a dileucine AP2 adaptin motif within the receptor beta2 subunit. Neuropharmacology. 2005;48:181–194. doi: 10.1016/j.neuropharm.2004.09.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.