

Abstract
We report two patients with genetically-confirmed spinocerebellar ataxia type 7 (SCA-7), who presented with progressive central visual loss and dyschromatopsia. Ocular funduscopic changes were subtle, with only mild retinal artery attenuation and subtle macular changes. Despite this, the electroretinogram (ERG) was abnormal in both patients. Both patients also had slowing of saccades and partially limited ductions, although neither reported diplopia. Although the older patient had cerebellar ataxia, the younger only had an unsteady tandem gait. This constellation of signs should indicate SCA-7 as a diagnostic possibility, and prompt further investigation with ERG and genetic studies.
Search Terms: Spinocerebellar Ataxia Type-7, Retinopathy, Ophthalmoplegia, Electroretinography
Introduction
Spinocerebellar ataxia type 7 (SCA-7) is a rare neurodegenerative disease caused by a CAG triplet repeat expansion in the SCA-7 gene on chromosome 3,1–3 encoding for a protein called ataxin-7.4 It is inherited in an autosomal dominant fashion, although sporadic cases are rarely reported.5 SCA-7 characteristically produces a progressive cerebellar syndrome; its common ophthalmologic manifestations include visual loss from a pigmentary maculopathy, and ophthalmoplegia.6,7 We describe two unrelated patients with SCA-7 who had central visual loss and abnormal electroretinography (ERG), yet only subtle ocular fundus findings. Both patients also had slowed saccades and partial limitation of ductions. These early neuro-ophthalmologic features of SCA-7 emphasize the importance of assessing eye movements and obtaining ERG for timely diagnosis of this disorder.
Case Reports
Case 1
A 12-year-old African-American boy, with a history of attention deficit hyperactivity disorder, presented with progressive painless bilateral central visual loss over several years. His family had also noticed that his eyes became “crossed” when he looked laterally, but he denied diplopia or any other neurologic symptoms. Family history was significant for a similar syndrome in his mother and twin sister.
On examination, his best-corrected visual acuities were 20/200 in the right eye and 20/300 in the left eye. He could identify only the control Ishihara plate bilaterally. Confrontation visual fields revealed bilateral central scotomas. External and anterior segment examinations were normal. Ocular funduscopic examination revealed possible mild temporal disc pallor and attenuated retinal arteries (fig 1A). The foveal light reflexes were absent and there was subtle pigmentary mottling in the maculae, but no pigmentary changes were seen in the retinal peripheries (fig 1A). Pupil examination was normal. Ocular motor examination revealed limited abduction of each eye, which could be partially overcome with vestibular stimulation, and slow horizontal saccades (video 1). He did not have downbeat, gaze-evoked, or rebound nystagmus. He did not have saccadic dysmetria. Smooth pursuit and vergence eye movements were normal. The remainder of the neurologic examination revealed unsteady tandem gait, and was otherwise unremarkable.
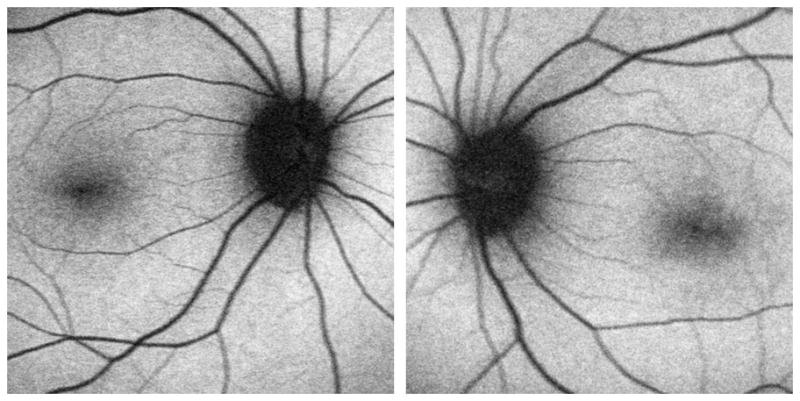
Figure 1.

A. Ocular fundus photographs from Case 1 demonstrate retinal artery attenuation, subtle pigmentary mottling in the maculae, and possible temporal optic disc pallor. B. Fundus photographs from Case 2 demonstrate only subtle retinal artery attenuation.
Humphrey visual fields confirmed bilateral central scotomas. Brain MRI revealed cerebellar atrophy (fig 2A). Multi-focal ERG showed severely attenuated central responses (fig 3). Genetic testing revealed an increased CAG repeat number of 65 (normal <18, borderline 19–36) in one SCA-7 allele, confirming the diagnosis of SCA-7.
Figure 2.

A. Brain MRI from Case 1 shows cerebellar atrophy, most marked in the vermis, and minimal brainstem atrophy. B. Brain MRI from Case 2 demonstrates marked cerebellar and brainstem atrophy. A cystic pineal lesion was incidentally detected and felt to be benign.
Figure 3.
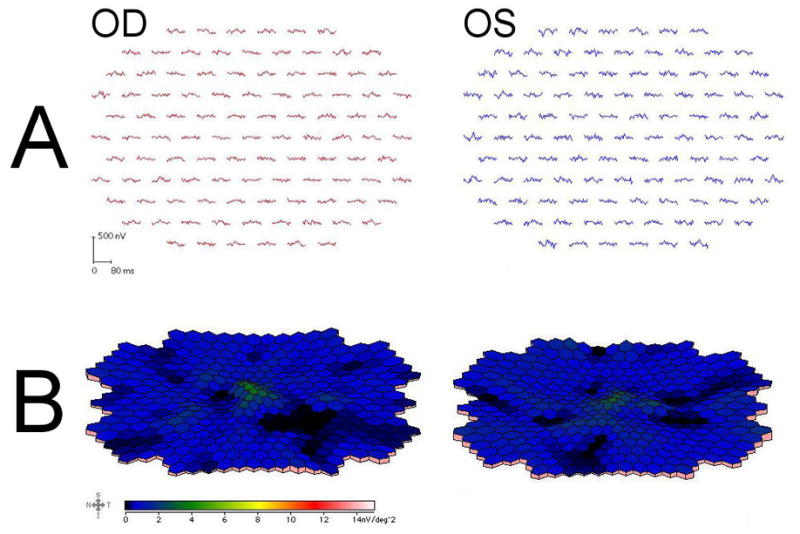
Multi-focal ERG from the right eye (left panel) and left eye (right panel) of Case 1. Trace arrays (A) and field plots (B) demonstrate severely attenuated central responses, with an absent foveal peak.
Case 2
A 21-year-old Caucasian woman presented with bilateral central visual loss and gait ataxia, progressive since age 14 years. By age 18 years, she had developed a wide-based gait and, over the following three years, increasing clumsiness of her upper limbs. She took no regular medications and denied alcohol consumption. Family history was significant for a similar syndrome occurring in her mother (diagnosed as “multiple sclerosis”) and her mother’s male cousin.
On examination, her best corrected visual acuities were 20/100 bilaterally. She could identify only the control Ishihara plate with each eye. Confrontation visual fields revealed bilateral central scotomas. External and anterior segment examinations were unremarkable. Ocular funduscopic examination revealed normal optic discs and slightly attenuated retinal arteries (fig 1B). The foveal light reflexes were absent, but there were no pigmentary changes in the maculae or retinal peripheries (fig 1B). Pupillary examination was normal. Ocular motor examination revealed slowing of horizontal and vertical saccades with limitation of abduction and elevation that could be partially overcome with vestibular stimulation (video 2). She did not have downbeat, gaze-evoked, or rebound nystagmus. There was no saccadic dysmetria, and her smooth pursuit and vergence eye movements were normal. The general neurologic examination revealed mild bilateral ptosis, facial weakness, head titubation, and appendicular, truncal, and gait ataxia. Her deep tendon reflexes were symmetrically brisk, but her plantar responses were flexor.
Ocular fundus autofluorescence was normal (fig 4). Humphrey and Goldmann visual fields confirmed bilateral central scotomas. Brain MRI was reported to show only a pineal lesion; on review, however, the presence of significant brainstem and cerebellar atrophy was confirmed (fig 2B). Full-field ERG showed abnormalities consistent with severe cone dysfunction and moderate rod dysfunction. Genetic testing revealed an increased CAG repeat number of 56 in one SCA-7 allele, confirming the diagnosis of SCA-7.
Figure 4.
Ocular fundus autofluorescence from Case 2 was unremarkable.
Discussion
In contrast to the other autosomal dominant spinocerebellar ataxias, SCA-7 is characterized by progressive irreversible bilateral visual loss due to a degenerative retinopathy that initially affects cone photoreceptors, but progresses towards a cone-rod dystrophy phenotype.8,9 Early in the disease, there is central visual loss with decreased visual acuity and central scotomas, and relative sparing of the peripheral visual field and night vision.6,10 Dyschromatopsia in the blue-yellow axis might be detected years prior to symptom onset in affected individuals.6 The retinopathy classically results in granular pigmentary changes in the maculae,6,10 occasionally producing a “bull’s eye” appearance,11 often with associated secondary optic atrophy.6,10 However, this ocular fundus appearance is not usually seen until late in the course of the disease. Indeed, it has been noted that the ocular fundus may initially appear normal, even after significant central visual loss has occurred,6 potentially causing diagnostic delay. In both our patients, despite being symptomatic with central visual loss for several years, there was only subtle retinal artery attenuation, more noticeable in patient 1, and absent foveal light reflexes (fig 1). In our first patient, who had more severe visual loss, there was very subtle pigmentary mottling in the macular regions and possibly mild temporal optic disc pallor. Review of fundus photographs from previously reported SCA-7 patients, in which the ocular fundi were considered normal, suggests that subtle abnormalities, such as retinal arterial attenuation, may have been present but overlooked.12 A prospective study of individuals with the SCA-7 mutation is required to further delineate the sequence of ocular fundus changes in relation to symptom onset and visual function parameters.
Retinal imaging may help in the investigation of suspected SCA-7. Pigmentary changes in the macular regions can be more prominent on fluorescein angiography and retinal thinning that is more extensive than suggested by the funduscopic appearance can be detected with optical coherence tomography (OCT).9 A potential role for fundus autofluorescence photography,13 a non-invasive imaging technique for topographical mapping of lipofuscin in the retinal pigment epithelium, has not been evaluated in SCA-7. Lipofuscin is a fluorescent pigment that accumulates in the retinal pigment epithelium cells as a consequence of photoreceptor degradation.13 Although lipofuscin accumulation can be seen with a variety of inherited and acquired retinal diseases, fundus autofluorescence was normal in our second patient, suggesting that it might not be a useful tool for detecting retinal degeneration in the early stages of SCA-7.
The presence of photoreceptor disease can be confirmed using ERG. While full-field ERG is widely available and is abnormal once funduscopic changes have become apparent,10 multi-focal ERG may be more sensitive for detecting macular dysfunction in the early stages of the disease when there is only central visual loss.11,12 The multi-focal ERG findings suggest early cone photoreceptor dysfunction in the macular regions.12 Indeed, the multi-focal ERG can be grossly abnormal even when funduscopic changes are absent or subtle,12 as in our first case. Thus, ERG – preferentially multi-focal ERG – is the investigation of choice to document photoreceptor dysfunction in degenerative retinopathy from SCA-7.
Ocular motor abnormalities are also commonly seen in association with SCA-7. Slowing of saccades is an early sign, suggesting involvement of the brainstem reticular formation,10,14 later followed by progressive ophthalmoplegia. The ophthalmoplegia is often partially overcome with vestibular stimulation in the earlier stages of the disease, but later becomes consistent with an external ophthalmoplegia.10 In both of our cases, there were no symptoms, such as diplopia, to suggest ocular motor dysfunction. However, there was slowing of saccades in both, most marked for horizontal saccades, and limitation of ductions that could be partially overcome with vestibular stimulation. Ocular motor abnormalities that are commonly seen in association with spinocerebellar degeneration, such as downbeat, gaze-evoked, and rebound nystagmus, dysmetric saccades, and impaired smooth pursuit,15 were not present in either of our patients, consistent with previous reports.6,7,10 Unfortunately, there are few series10,14 in which quantitative eye movement recordings were obtained, and therefore a more systematic study of ocular motor abnormalities in this disease is required.
Other neurologic signs, especially those consistent with cerebellar dysfunction, can be found in almost all cases as the disease progresses,7 but may be subtle or absent in the early stages of the disease, as in our first case. Cerebellar and brainstem atrophy is often seen on MRI,10,14 but can also be subtle and easily overlooked, as in our second patient (see fig 2B).
In summary, SCA-7 is a rare inherited neurodegenerative disease that produces progressive irreversible bilateral visual loss, from a degenerative retinopathy, and ophthalmoplegia. In the early stages of the disease, the visual loss is central and there may be only subtle ocular funduscopic changes, such as retinal artery attenuation and absence of foveal light reflexes. Characteristic early ocular motor findings, such as slowing of saccades, may be asymptomatic and must be specifically sought to be detected. We suggest that SCA-7 should be considered a diagnostic possibility whenever this combination of neuro-ophthalmologic signs is present; recognition of this possibility may help establish the diagnosis in cases where other neurologic signs are absent, family history is unavailable, or the disease is sporadic.
Acknowledgments
This study was supported, in part, by a departmental grant (Department of Ophthalmology) from Research to Prevent Blindness, Inc, New York, New York, and by core grants P30-EY06360 (Department of Ophthalmology) from the National Institute of Health, Bethesda, Maryland. Dr. Newman is a recipient of a Research to Prevent Blindness Lew R. Wasserman Merit Award. Drs. Leigh and Thurtell are supported by NIH grant EY06717, the Department of Veterans Affairs, and the Evenor Armington Fund.
Footnotes
Disclosure: The authors report no conflicts of interest
References
- 1.Gouw LC, Kaplan CD, Haines JH, et al. Retinal degeneration characterizes a spinocerebellar ataxia mapping to chromosome 3p. Nat Genet. 1995;10:89–93. doi: 10.1038/ng0595-89. [DOI] [PubMed] [Google Scholar]
- 2.Benomar A, Krols L, Stevanin G, et al. The gene for autosomal dominant cerebellar ataxia with pigmentary macular dystrophy maps to chromosome 3p12-p21.1. Nat Genet. 1995;10:84–8. doi: 10.1038/ng0595-84. [DOI] [PubMed] [Google Scholar]
- 3.Holmberg M, Johansson J, Forsgren L, et al. Localization of autosomal dominant cerebellar ataxia associated with retinal degeneration and anticipation to chromosome 3p12-p21.1. Hum Mol Genet. 1995;4:1441–5. doi: 10.1093/hmg/4.8.1441. [DOI] [PubMed] [Google Scholar]
- 4.Kaytor MD, Duvick LA, Skinner PJ, et al. Nuclear localization of the spinocerebellar ataxia type 7 protein, ataxin-7. Hum Mol Genet. 1999;8:1657–64. doi: 10.1093/hmg/8.9.1657. [DOI] [PubMed] [Google Scholar]
- 5.Stevanin G, Giunti P, Belal GD, et al. De novo expansion of intermediate alleles in spinocerebellar ataxia 7. Hum Mol Genet. 1998;7:1809–13. doi: 10.1093/hmg/7.11.1809. [DOI] [PubMed] [Google Scholar]
- 6.Gouw LG, Digre KB, Harris CP, et al. Autosomal dominant cerebellar ataxia with retinal degeneration: clinical, neuropathologic, and genetic analysis of a large kindred. Neurology. 1994;44:1441–7. doi: 10.1212/wnl.44.8.1441. [DOI] [PubMed] [Google Scholar]
- 7.David G, Durr A, Stevanin G, et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7) Hum Mol Genet. 1998;7:165–70. doi: 10.1093/hmg/7.2.165. [DOI] [PubMed] [Google Scholar]
- 8.Aleman TS, Cideciyan AV, Volpe NJ, et al. Spinocerebellar ataxia type 7 (SCA7) shows a cone-rod dystrophy phenotype. Exp Eye Res. 2002;74:737–45. doi: 10.1006/exer.2002.1169. [DOI] [PubMed] [Google Scholar]
- 9.Michalik A, Martin JJ, Van Broeckhoven C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet. 2004;12:2–15. doi: 10.1038/sj.ejhg.5201108. [DOI] [PubMed] [Google Scholar]
- 10.Enevoldson TP, Sanders MD, Harding AE. Autosomal dominant cerebellar ataxia with pigmentary macular dystrophy: a clinical and genetic study of eight families. Brain. 1994;117:445–60. doi: 10.1093/brain/117.3.445. [DOI] [PubMed] [Google Scholar]
- 11.Anh JK, Seo JM, Chung H, et al. Anatomical and functional characteristics in atrophic maculopathy associated with spinocerebellar ataxia type 7. Am J Ophthalmol. 2005;139:923–5. doi: 10.1016/j.ajo.2004.10.055. [DOI] [PubMed] [Google Scholar]
- 12.Hugosson T, Granse L, Ponjavic V, et al. Macular dysfunction and morphology in spinocerebellar ataxia type 7 (SCA 7) Ophthalmic Genet. 2009;30:1–6. doi: 10.1080/13816810802454081. [DOI] [PubMed] [Google Scholar]
- 13.Schmitz-Valckenberg S, Holz FG, Bird AC, et al. Fundus autofluorescence imaging: review and perspectives. Retina. 2008;28:385–409. doi: 10.1097/IAE.0b013e318164a907. [DOI] [PubMed] [Google Scholar]
- 14.Oh AK, Jacobson KM, Jen JC, et al. Slowing of voluntary and involuntary saccades: an early sign in spinocerebellar ataxia type 7. Ann Neurol. 2001;49:801–4. doi: 10.1002/ana.1059. [DOI] [PubMed] [Google Scholar]
- 15.Leigh RJ, Zee DS. The neurology of eye movements. 4. New York: Oxford University Press; 2006. [Google Scholar]