

Abstract
This report is the second of a two-part evaluation of developmental differences in α–amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptor subunit expression in cell populations within white matter and cortex. In Part I, we report that in rat, developmental expression of Ca2+ permeable (GluR2-lacking) AMPARs correlated at the regional and cellular level with increased susceptibility to hypoxia/ischemia (H/I), suggesting an age-specific role of these receptors in the pathogenesis of brain injury. Part II examines the regional and cellular progression of AMPAR subunits in human white matter and cortex from midgestation through early childhood. Similar to the rodent, there is a direct correlation between selective vulnerability to H/I and expression of GluR2-lacking AMPARs in human brain. In midgestational cases aged 20-24 postconceptional weeks (PCW) and in premature infants (25-37 PCW), we found that radial glia, premyelinating oligodendrocytes and subplate neurons transiently expressed GluR2-lacking AMPARs. Notably, prematurity represents a developmental window of selective vulnerability for white matter injury, such as periventricular leukomalacia (PVL). During term (38-42 PCW) and post-term neonatal (43-46 PCW) periods, age windows characterized by increased susceptibility to cortical injury and seizures, GluR2 expression was low in the neocortex, specifically on cortical pyramidal and non-pyramidal neurons. This study indicates that Ca2+ permeable AMPAR blockade may represent an age-specific therapeutic strategy for potential use in humans. Furthermore, these data help validate specific rodent maturational stages as appropriate models for evaluation of H/I pathophysiology.
Keywords: glutamate receptor, perinatal, seizure, excitotoxicity, neuron, oligodendrocyte
INTRODUCTION
Hypoxia/ischemia (H/I) represents a major cause of perinatal brain damage, affecting both preterm and term infant populations (Volpe, 2001;Ferriero, 2004). Hypoxic/ischemic injury can occur at midgestation in utero with premature labor, maternal hypotension or placental insufficiency (Grafe, 1994). In preterm infants, H/I most often occurs in the setting of pulmonary immaturity, systemic hypoperfusion, inadequate cerebrovascular autoregulation, or as a complication of inflammation due to maternal-fetal infection (Rorke, 1992;Tsuji et al., 2000;Volpe, 2001;Wu, 2002;Kinney and Armstrong, 2002). At term, H/I is most commonly the result of perinatal asphyxia, respiratory insufficiency, sepsis, or as a complication of extracorporeal membrane oxygenation (ECMO) or cardiac corrective surgery with cardiopulmonary bypass (Bellinger et al., 1995;Ferriero, 2004).
The neuropathologic patterns, clinical presentations and sequelae of H/I injury are highly age-dependent, despite the similar initiating insult. Cerebral white matter injury, or periventricular leukomalacia (PVL), is prevalent following H/I injury in the very premature/midgestational cases (20-24 postconceptional weeks, PCW) and slightly older preterm infants (25-37 PCW) (Banker and Larroche, 1962;Barkovich and Sargent, 1995;Okumura et al., 1997;Rorke, 1998;Volpe, 2001). The neuropathological features of PVL include focal necrosis involving loss of all cellular elements, as well as diffuse injury to developing oligodendrocytes (OLs), gliosis and subsequent hypomyelination (Banker and Larroche, 1962;Dambska et al., 1989;Rorke, 1998;Kinney and Armstrong, 2002;Kinney et al., 2004). Although the cortex is relatively spared with respect to acute injury (Marin-Padilla, 1997), MRI imaging reveals secondary cortical thinning at birth and later infancy (Inder et al., 1999;Inder et al., 2003). PVL represents the major antecedent of the neuromotor deficit cerebral palsy and is also associated with cognitive deficits (Hack et al., 2000;Peterson et al., 2000;Volpe, 2001;Hack et al., 2002).
In contrast, term infants and neonates suffering from H/I encephalopathy exhibit predominantly grey matter lesions and seizures (Hauser et al., 1993;Okumura et al., 1997;Estan and Hope, 1997;Saliba et al., 1999;Volpe, 2001;Kinney and Armstrong, 2002;Cowan et al., 2003). The most affected brain regions around term are the perirolandic cortex, hippocampus, and subcortical grey matter structures including the thalamus and basal ganglia (Barkovich, 1992;Maller et al., 1998;Roland et al., 1998;Kinney and Armstrong, 2002). The neuropathologic substrate of these lesions is selective neuronal loss followed by gliosis (Marin-Padilla, 1999;Kinney and Armstrong, 2002). The resulting neurological deficits in this population include motor deficits, mental retardation and epilepsy (Okumura et al., 1997;Rivkin, 1997;Volpe, 2001;Mercuri et al., 2004).
The pathogenesis of perinatal H/I brain injury in humans is likely to involve multiple mechanisms. Regional and cellular vulnerability to H/I is governed by complex developmental factors, including regional metabolic and vascular factors (Volpe, 2001;Ferriero, 2004), as well as differential intrinsic cellular stressors (Follett et al., 2004;Folkerth et al., 2004). Experimental animal models reveal that critical factors in H/I injury are glutamate accumulation (Benveniste et al., 1984;Silverstein et al., 1991;Vannucci et al., 1999;Loeliger et al., 2003) followed by excessive glutamate receptor (GluR) activation (Hagberg et al., 1994;Jensen et al., 1995;Chen et al., 1998;Follett et al., 2000;Koh and Jensen, 2001;Follett et al., 2004). Clinically, elevated glutamate levels have been demonstrated in the cerebrospinal fluid of infants suffering perinatal H/I injury (Gucuyener et al., 1999), strongly suggesting that similar mechanisms may be implicated in H/I brain damage in the human infant.
The α–amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) glutamate receptor subtype is heteromeric in structure, comprised of a combination of the GluR1, GluR2, GluR3 and GluR4 subunits, and is permeable to Ca2+ when GluR2 is lacking (Hollmann and Heinemann, 1994). Ca2+ influx through GluR2-lacking AMPARs represents a major mechanism implicated in the normal processes of synaptic plasticity, learning and memory (Massicotte and Baudry, 2004). However, under pathologic conditions in which there is glutamate accumulation, these receptors become excessively activated, leading to elevated Ca2+ influx, which has been shown to be a major mechanism of H/I glial and neuronal injury and epilepsy (Friedman and Veliskova, 1999;Sanchez et al., 2001;Jensen et al., 2001;Deng et al., 2003;Follett et al., 2004).
In part I of this two-part study evaluating regional and cellular developmental regulation of AMPARs, we demonstrated that age-specific elevated expression of GluR2-lacking AMPARs in the immature rat brain correlates with developmental windows reported to exhibit high regional and cellular increases in susceptibility to H/I (Talos et al., 2005). In Part II we hypothesized that AMPAR subunit expression in the human perinatal brain would demonstrate an age- and cell-specific pattern similar to the rat. Furthermore, given age-related differences in regional vulnerability to H/I, we specifically hypothesized that GluR2 subunit is relatively lacking on cells within white matter in midgestational and preterm tissue and on cells within cortex in term and post-term neonatal brain. As in Part I of this study, we evaluated the developmental expression of each AMPAR subunit in human perinatal white matter and cortex by immunoblotting and by immunofluorescence double-labeling, to evaluate the differential expression of GluR2-lacking AMPARs in the component cell populations in these regions.
MATERIALS AND METHODS
Human subjects
Brain specimens were collected prospectively from the fetal, pediatric and adult autopsy populations of the Children's Hospital and Brigham and Women's Hospital, Boston (n=32). The samples were obtained from standard diagnostic postmortem examinations and all procedures and experiments were conducted under guidelines approved by the hospitals’ Institutional Review Boards. Neuropathologic examinations were done on all cases by one of us (R. D. F.), and included macroscopic evaluations of the brain in situ and after removal, as well as microscopic examination. For light microscopic diagnosis a minimum of 5 blocks, including cerebral cortex, white matter, deep grey matter structures, hippocampus, brainstem and cerebellum, were taken in each case. Tissues fixed in 4% paraformaldehyde were cut, embedded in paraffin, and processed in standard fashion. Slides were stained with hematoxylin-and-eosin. Additional control cases were obtained from University of Maryland Brain Tissue Bank for Developmental Disorders (n=14). All specimens included in this study were diagnosed as controls; since this is a baseline (normative) study, cases with evidence of white matter and /or cortical injury, as well as central nervous system malformations, were specifically excluded. The relevant clinical and neuropathological data of all 46 human brain specimens used in this study are summarized in the Results section (Table 1).
Table 1.
Clinical characteristics of sample population
| Case # | GA (weeks) | PNA (weeks) | PCA (weeks) | PMI (h) | Gender | Diagnosis |
|---|---|---|---|---|---|---|
| 1 | 18 | 0 | 18 | 4 | F | extreme prematurity |
| 2 | 20 | 0 | 20 | 11 | M | extreme prematurity |
| 3 | 20 | 0 | 20 | 34 | M | stillbirth |
| 4 | NA | NA | 20 | 6 | NA | extreme prematurity |
| 5 | 20 | 0 | 20 | 0 | F | therapeutic abortion |
| 6 | 21 | 0 | 21 | 52 | F | ACA, prematurity |
| 7 | 21 | 0 | 21 | 11 | F | extreme prematurity |
| 8 | 21 | 0 | 21 | 53 | M | stillbirth |
| 9 | 21 | 0 | 21 | 30 | F | stillbirth, ACA |
| 10 | 23 | 0 | 23 | 4 | F | therapeutic abortion |
| 11 | 23 | 0 | 23 | 28 | M | extreme prematurity |
| 12 | 23 | 0 | 23 | 42 | M | extreme prematurity |
| 13 | 24 | 0 | 24 | 32 | M | extreme prematurity |
| 14 | 24 | 0 | 24 | 18 | M | extreme prematurity, MCA |
| 15 | 25 | 0 | 25 | 25 | M | extreme prematurity |
| 16 | 25 | 1 | 26 | 24 | M | prematurity, perforated ileum, necrotizing BP, sepsis |
| 17 | 26 | 0 | 26 | 7 | F | prematurity |
| 18 | 27 | 0 | 27 | 14 | M | stillbirth |
| 19 | 29 | 0 | 29 | 23 | M | prematurity, pulmonary hypoplasia |
| 20 | 30 | 0 | 30 | 9 | M | prematurity, respiratory insufficiency, polycystic kidney |
| 21 | 31 | 0 | 31 | 4 | M | stillbirth, pulmonary hypoplasia |
| 22 | 31 | 0 | 31 | 6 | M | stillbirth |
| 23 | 32 | 0 | 32 | 51 | F | prematurity, subarachnoid hemorrhage |
| 24 | 29 | 3 | 32 | 22 | M | necrotizing enterocolitis |
| 25 | 34 | 0 | 34 | 31 | F | stillbirth |
| 26 | 34 | 0 | 34 | 9 | F | prematurity, lung hypoplasia, renal dysgenesis |
| 27 | 35 | 0 | 35 | 10 | M | stillbirth |
| 28 | 37 | 0 | 37 | 25 | M | stillbirth |
| 29 | 37 | 0 | 37 | 17 | M | ACA, massive pulmonary hemorrhage |
| 30 | 37 | 1 | 38 | 5 | M | CHD |
| 31 | 38 | 0 | 38 | 40 | F | MCA, pulmonary hypoplasia |
| 32 | 38 | 0 | 38 | 16 | M | stillbirth, alpha thalassemia major |
| 33 | 39 | 0 | 39 | 19 | M | CHD |
| 34 | 38 | 1 | 39 | 36 | F | pulmonary alveolar capillary dysplasia |
| 35 | 40 | 0 | 40 | NA | M | SIDS |
| 36 | 41 | 0 | 41 | 24 | M | CHD |
| 37 | 40 | 3 | 43 | 13 | F | CHD, viral myocarditis, pulmonary hypertension |
| 38 | 40 | 5 | 45 | 7 | M | suffocation |
| 39 | 40 | 6 | 46 | 22 | M | SIDS |
| 40 | 40 | 14 | 54 | 16 | M | CHD |
| 41 | 40 | 52 | 92 | 15 | M | drowning |
| 42 | 40 | 128 | 168 | 20 | F | trauma |
| 43 | 36 | 174 | 210 | 11 | F | drowning |
| 44 | 40 | 38 yrs | 38 yrs | 17 | M | myelogenous leukemia |
| 45 | 40 | 57 yrs | 57 yrs | 11 | M | leukemia |
| 46 | 40 | 63 yrs | 63 yrs | 19 | F | lung cancer |
Abbreviations: GA=gestational age; PNA=postnatal age; PCA=postconceptional age; PMI=postmortem interval; ACA=acute chorioamnionitis; MCA=multiple congenital anomalies; CHD=congenital heart disease; BP=bronchopneumonia; SIDS= sudden infant death syndrome; NA=information not available; M=male; F=female.
Western Blot Analysis
Immunoblotting
Western blot analysis was performed on tissue homogenates of dissected white matter (n=16) and cortex (n=22) from cases ranging between 18 postconceptional (gestational plus postnatal) weeks (PCW) and 210 PCW (ca. 3.3 years), using adult cases as indices of maturity (n=2). At the time of autopsy, the unfixed brain was examined and parieto-occipital lobe tissue was snap-frozen immediately and stored at –80°C. Subsequently, grey and white matter was dissected and homogenized separately for protein extraction. The protocols used in this study were the same as those described for the rat in Part I (Talos et al., 2005). After membrane protein extraction, samples were run on 7.5% Tris HCl gels (Bio-Rad, Hercules, CA), transferred onto polyvinylidene difluoride (PVDF) membranes and subsequently immunostained with antibodies against AMPAR subunits GluR1-GluR4.
Data analysis
For each receptor subunit, values obtained from densitometry of bands were normalized to adult standards run on each blot. In addition, nonGluR2/GluR2 ratios were calculated as described in Part I (Talos et al., 2005). Relative protein levels for individual AMPAR subunits and their expression ratios were plotted as a function of postconceptional age (PCA). Regression analyses of PCA on relative GluR1-4 values and nonGluR2/GluR2 ratios were performed and linear or quadratic regression curves were fitted to the levels of individual AMPAR subunits and the nonGluR2/GluR2 ratios. Regression analyses were performed to determine if there was an effect of postmortem interval (PMI) upon the levels of GluR1-GluR4 subunits. An analysis of covariance was performed to determine if there was an effect of gender on GluR1-4 expression, controlling for PCA. In all analyses, a p<0.05 was considered significant. Because of the lack of cases between 54 and 92 PCW and the small number thereafter, these few older cases were not included in the statistical analysis.
Immunocytochemistry
Immunostaining of tissue sections
Immunofluorescence double-labeling was performed in 33 cases ranging between 20-210 PCW. Blocks of parietal lobe cortex and underlying white matter at the level of the atrium immediately adjacent to the blocks collected for western blot were taken at the time of autopsy and immersed in fresh 4% paraformaldehyde in PBS (pH=7.4). Tissue was maintained in the same fixative at 4°C for at least 48-72 h and then cryoprotected in 30% sucrose in PBS for 24h. Blocks (1 to 1.2 cm diameter) were cut at 50μm on a freezing microtome (HM 440 E, Microm International, Germany) and collected serially as free-floating sections. Additional immunocytochemistry experiments were performed on snap-frozen material. Tissue blocks (1 to 1.2 cm diameter) were removed from -80°C freezer dropped in a pre-cooled paraformaldehyde-containing fixative and maintained at -20°C for 1 week. Blocks were cryoprotected in 30% sucrose in PBS for 24h and cut on a freezing microtome at 80-100μm. The fixative used in these experiments contained 12g paraformaldehyde and 40g sucrose in100ml 0.1M PBS, 40ml 100% ethanol, 40ml 100% ethylene glycol and 20ml 100% glycerol (Ulfig et al., 1998).
The technique for immunocytochemistry was according to our prior protocols (Follett et al., 2004;Folkerth et al., 2004) and the same as that used in Part I (Talos et al., 2005). Sections were labeled with monoclonal glial and neuronal markers (vimentin, GFAP, O4, O1, MBP and NeuN) in combination with polyclonal GluR1, GluR2 and GluR4 antibodies, as well as with polyclonal cell markers (GFAP, GalC and NSE) in combination with monoclonal GluR3 antibodies. Negative controls, in which one or both primary antibodies were omitted, adding only the secondary antibodies, were run for each condition to exclude false-positive secondary antibody binding.
Data analysis
Slides were viewed using epifluorescence microscopy on a Zeiss Axioscope (Germany). At least 3 brains per age group and staining condition and a minimum of 2 sections per brain were qualitatively analyzed. The distribution patterns of each AMPAR subunit, including the level of expression, as well as regional and cellular localization were analyzed on tissue sections processed within the same immunocytochemical run. The expression levels were compared only within a single antibody, as binding characteristics vary between different antibodies. Positive controls were present in all circumstances of negative staining, either in adjacent regions or in additional tissue sections.
Preparation of illustrations
Images were captured with a Spot digital camera, using the Spot Software 4.5 (Diagnostic Instruments, Sterling Heights, MI) and then were transferred to Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA). The brightness, contrast and color were adjusted to achieve an optimal quality of the prints, without altering in any way the individual staining patterns of the antibodies.
Antibodies
Sensitivity and specificity of the GluR1-4 antibodies used here are reviewed in Part I (Talos et al., 2005). Notably, these antibodies have been successfully used in previous human studies, both in brain tissue homogenates, as well as immunostaining of tissue sections (Bernard et al., 1996;Meng et al., 1997;Moga et al., 2003;Murphy et al., 2005). Antibody specificity in our tissue samples was demonstrated by the presence of single bands in both cortex and white matter immunoblots, corresponding to the known molecular weight of each AMPAR subunit and in tissue sections by a staining patterns comparable with previous published studies. In case of GluR1 and GluR4, preabsorption with corresponding inhibitory peptides blocked all immunostaining with the respective antibodies, both in western blots and ICC.
The cell markers used in the human study were the same as the ones applied in the rat and their concentrations and specificity are described in Part I. Importantly, similar to AMPAR subunits antisera, most of these antibodies have been used in previous human studies (Osborn et al., 1984;Bernard et al., 1996;Sarnat et al., 1998;Spreafico et al., 2000;Back et al., 2001;Haynes et al., 2003;Follett et al., 2004;Kultas-Ilinsky et al., 2004;Folkerth et al., 2004)
RESULTS
Clinical and pathological evaluation
Normal subjects were included in this study based on combined clinical criteria (cause of death, survival interval and postmortem interval) and neuropathological diagnosis, as described below (Table 1), and consistent with previously published studies from our group (Follett et al., 2004;Folkerth et al., 2004).
Age
The age of the 46 analyzed cases ranged from midgestation through adulthood. In fetuses, the age was determined by foot measurement. All ages were expressed as postconceptional age in weeks (PCW), which was calculated as gestational age (GA) plus postnatal age (PNA), with the exception of the adult cases that were expressed in years of age. Subjects fell into the following clinically relevant age groups: midgestation (18-24 PCW, n=14), preterm period (25-37 PCW, n=15), term period (38-42 PCW, n=7), post-term neonatal period (43-46 PCW, n=3), infancy/early childhood (54-210 PCW, n=4) and adulthood (38-63 years, n=3).
Gender
Both genders (16 females, 29 males, 1 case without available gender information) were included in the analysis.
Cause of death
In all subjects, the death was due to non-neurological disorders. Excluding stillbirths (n=10) and pregnancy terminations (n=2), the cause of death in live born infants included extreme prematurity (n=11), congenital heart defects (n=5), multiple congenital anomalies (n=3), pulmonary dysplasia/ hypoplasia (n=2), sepsis (n=2), sudden infant death syndrome (n=2), drowning (n=2), trauma (n=1), suffocation (n=1), subarachnoid hemorrhage (n=1) and pulmonary hemorrhage (n=1). Among the adult population, causes of death included hematological malignancies (n=2) and lung cancer (n=1).
Survival interval
Fetuses and neonates died within the first week after birth, except for one case that had a survival interval of 3 weeks.
Postmortem interval (PMI)
In most cases the PMI was less than 24h (n=32), while in several cases the PMI ranged between 25-53 h (n=13). In one case this information was not available. For cases with PMI>24h fresh tissue quality at the time of autopsy was assessed by the neuropathologist, so that only well-preserved, non-macerated tissue was included.
Standard neuropathologic evaluations
No brain abnormalities or lesions were detected by macroscopic examinations of cerebral cortex, white matter, deep grey matter structures, hippocampus, brainstem and cerebellum. All cases were diagnosed as histologically normal or presented minimal prominence of white matter astrocytes, a finding that in isolation is of uncertain significance (Kinney et al., 2004). These cases (n=10) were grouped with controls, but were only used for analysis of cortical AMPARs development, and not for white matter development. Since the purpose of this study was to elucidate AMPAR expression in normal human brain development, cases with grey matter necrosis or white matter injury, such as PVL, brain malformations, or any other diagnostic lesion of the central nervous system, were specifically excluded, to be the subject of separate studies.
Developmental regulation of AMPAR subunit expression in human parietal white matter
The overall aim of this study was to determine the temporal and spatial sequence of individual AMPAR subunit expression in human parietal white matter and cortex, in a manner similar to that performed in rodent cerebral cortex in Part I of this study (Talos et al., 2005).
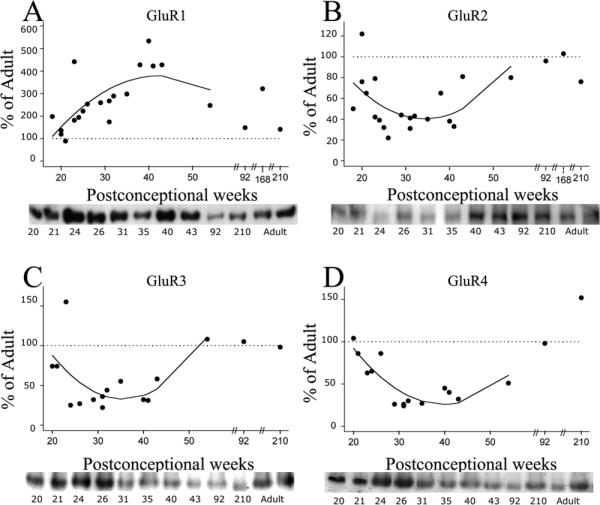
Relative protein levels of GluR1-4 subunit expression in the human white matter demonstrated differential developmental trends. Figure 1 shows the normalized GluR1-4 values (% of adult) for individual cases, from midgestation through early childhood, with either linear or quadratic regression lines superimposed, if significant. If there was no significant change with age, then a line was drawn through the mean value for expression across development. These fitted curves demonstrate that between18-46 PCW, GluR1 expression (Figure 1A) in the white matter did not significantly change (p >0.05), while there were statistically significant variations in GluR2 (p=0.022), GluR3 (p=0.003) and GluR4 (p=0.004) expression. GluR2 expression (Figure 1B) remained consistently below adult levels for the time period analyzed, despite a gradual increase with age. Predicted GluR2 values from the regression analysis of individual points demonstrated a modest increase from 38% of adult levels at midgestation (20 PCW) to 48% of adult during the preterm period (30 PCW), to 58% at term (40 PCW), and to 63% during post-term neonatal period (45 PCW). Unlike GluR2, GluR3 and GluR4 expression was higher than in adulthood, reaching maximal levels during the preterm period. GluR3 subunit expression (Figure 1C) superceded adult levels with estimated levels of 668% at 20 PCW, 1626% at 30PCW, and 1058% at 40 PCW, dropping to only 201% of adult at 45 PCW. Similarly, estimated GluR4 levels (Figure 1D) represented 453% of adult at 20 PCW, increased further to 1132% at 30 PCW, then modestly decreased to 758% at 40 PCW, and dropped to 175% at 45 PCW. There was no effect of PMI or gender on AMPAR subunit expression in human white matter (data not shown).
Figure 1.

AMPAR subunit expression in the developing white matter from midgestation (18 PCW) through early childhood (210 PCW), as compared to adult standard (100%), indicated as horizontal dotted line in all graphs. Relative protein levels for GluR1-4 demonstrate no change in GluR1 expression with maturation (A), but show a significant downregulation of GluR2 subunit (B), accompanied by an upregulation of GluR3 (C) and GluR4 (D) during the early fetal and preterm period. Linear or quadratic regression lines were fitted to the individual data points if significant; otherwise, a line was drawn through the mean. Insets are representative western blots for individual subunits.
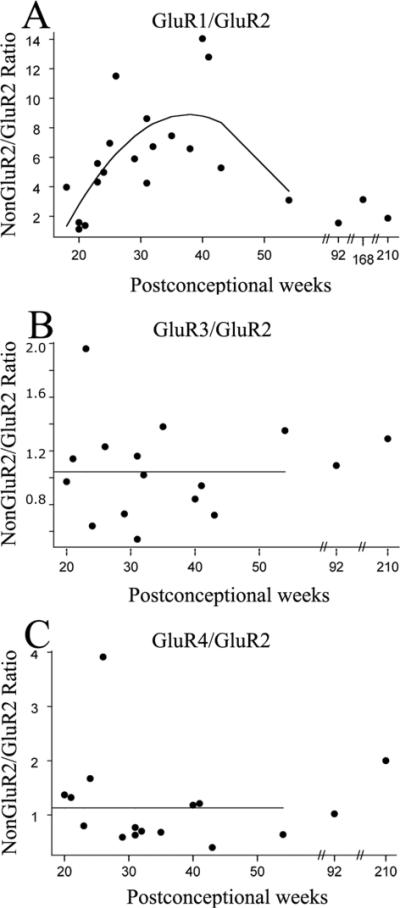
Since Ca2+ permeability of AMPARs depends on the expression level of GluR2 subunit relative to the levels of other GluR subunits (Pellegrini-Giampietro et al., 1992;Sanchez et al., 2001;Kumar et al., 2002), we next analyzed the ratios of nonGluR2/GluR2 subunit expression across development. Figure 2 shows the ratios of nonGluR2/GluR2 subunits for each individual case as a function of age, with superimposed regression lines. All nonGluR2/GluR2 ratios demonstrated significant developmental changes (p=0.038 for GluR1/GluR2 ratio; p=0.046 for GluR3/GluR2 ratio; p=0.01 for GluR4/GluR2 ratio), being much higher than adult (defined for comparison as 1:1) at younger ages and then decreasing progressively with maturation. Using predicted values from the regression analysis, the GluR1/GluR2 ratio (Figure 2A) decreased from 6.1:1 at midgestation (20 PCW), to 4.7:1 during preterm period (30 PCW), to 3.3:1 at term (40 PCW) and 2.6:1 during neonatal period (45 PCW). GluR3/GluR2 and GluR4/GluR2 ratios showed a slightly different pattern, with GluR3/GluR2 ratio (Figure 2B) underwent a transient increase from 19.8:1 at 20 PCW to 31.7:1 at 30 PCW, before decreasing again to 17.9:1 at 40 PCW and only 1.3:1 at 45 PCW. GluR4/GluR2 ratio (Figure 2C) similarly increased from 13.3:1 at 20 PCW, to 23.3:1 at 30 PCW, and decreased later to 14.1:1 at 40 PCW and to 2.2:1 at 45 PCW.
Figure 2.

NonGluR2/GluR2 ratios in the developing human white matter expressed as a function of postconceptional age. GluR1/GluR2 (A), GluR3/GluR2 (B) and GluR4/GluR2 (C) ratios demonstrate maximal values between 25-35 PCW, the window of vulnerability to PVL. Linear or quadratic regression lines were fitted to the individual data points, if significant.
Differential cellular distribution of AMPAR subunits in human parietal white matter
White matter western blots indicated that AMPARs are maximally expressed during midgestation and the preterm periods with a relative lack in GluR2 subunit expression (higher nonGluR2/GluR2 ratios), suggesting that these receptors are likely to be Ca2+ permeable. To further characterize the cellular specificity of GluR2-lacking AMPAR expression, immunocytochemical double labeling with GluR1-4 subunit antibodies and the glial markers vimentin, GFAP, O4, O1, GalC and MBP, was performed during the same interval analyzed by western blot.
Cellular changes in the developing white matter were first analyzed by differential staining patterns of stage-specific glial cell markers (Kinney and Back, 1998;Ulfig et al., 1998;Back et al., 2001;deAzevedo et al., 2003). At 20-24 PCW, radial glia represented the predominant cell type of the astrocytic lineage and was robustly labeled with vimentin and GFAP. Radial glial cell bodies were present within the periventricular zone and extended long parallel processes through the white matter up to the pial surface (Figure 3, A1-C1). Between 25-37 PCW, radial glial fibers gradually disappeared, coincident with a progressive increase in the number of mature vimentin-/GFAP+ astrocytes (Figure 3, D1-F1). During later development, astrocytes continued to increase in number and size, peaking at 38-42 PCW before decreasing again with maturation (data not shown). Between 20-37 PCW, premyelinating OLs (preOLs), including both O4+/GalC- OL precursors (Figure 3, A2-C2) and O4+/GalC+ immature OLs (Figure 3, D2-F2), represented the predominant OL population in the white matter. MBP immunoreactivity was first observed at 30-32 PCW (data not shown), as described in previous reports (Kinney and Back, 1998;Back et al., 2001) and increased progressively with subsequent development.
Figure 3.
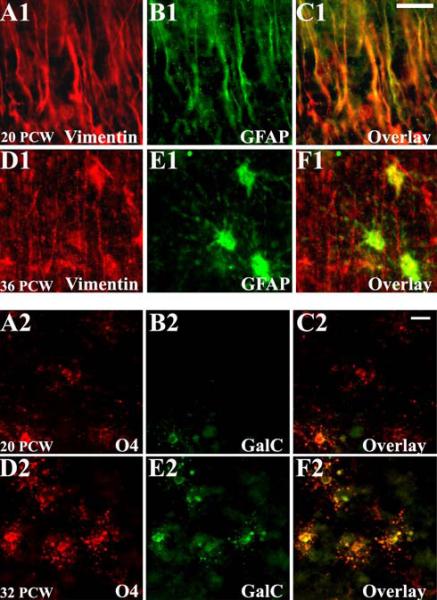
Human white matter cellular development demonstrated by differential expression of stage-specific glial markers. At midgestation (A1-C1), radial glia fibers are abundant in the white matter and display both vimentin and GFAP immunoreactivity, while shortly before term birth (D1-F1), mature astrocytes (vimentin-/GFAP+) predominate. Between 20-24 PCW (A2-C2), developing OLs are represented mostly by O4+/GalC-precursors, and at 25-37 PCW (D2-F2) there is a substantial increase in O4+/GalC- immature OLs. Scale bars 10μm.
Consistent with the western blot data, the most pronounced changes in AMPAR subunit expression in white matter were observed during midgestation and the preterm period. Developing astroglia expressed AMPARs as early as midgestation. At 20-24 PCW GluR4 subunit was highly expressed on radial glia fibers (Figure 4, A1-C1). Between 25-37 PCW, as radial glia matured, GluR4 immunoreactivity continued to increase in astrocytes (Figure 4, A2-C2) where it peaked around 38-42 PCW (Figure 4, A3-C3). GluR3 expression paralleled that of GluR4 in both radial glia and astrocytes (data not shown). GluR2 immunoreactivity was relatively lacking in radial glia between 20-24 PCW (Figure 4, D1-F1), but was visible on astrocytes between 25-37 PCW (Figure 4, D2-F2) and increased significantly at 38-42 PCW, in parallel with their morphological maturation (Figure 4, D3-F3). Relative to neuronal cell types, there was low or no GluR1 expression on astroglial cells at all ages studied (data not shown).
Figure 4.
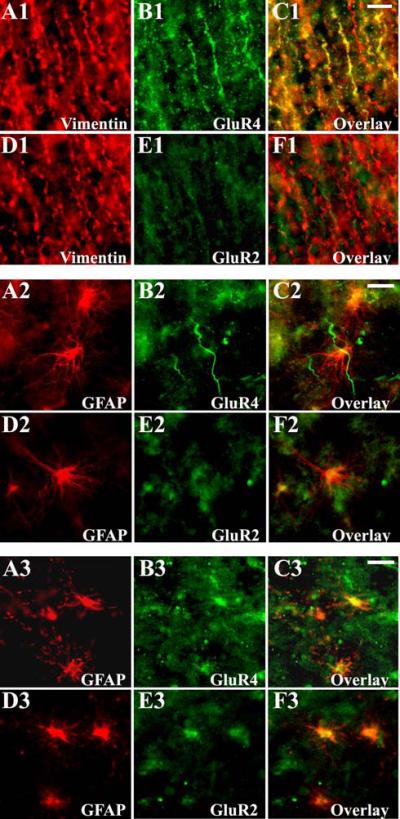
Cell specific AMPAR subunit expression in the developing white matter astroglia during prenatal life. At 20 PCW (A1-F1), vimentin + radial glial fibers express high levels of GluR4 (A1-C1), which is associated with a relative lack in GluR2 (D1-F1) staining. At 32 PCW (A2-F2), mature GFAP+ astrocytes express GluR4 (A2-C2), but in contrast to immature radial glia, astrocytes co-express GluR2 (D2-F2). At 39 PCW (A3-F3), the majority of GFAP expressing astrocytes are intensely immunolabeled with both GluR4 (A3-C3) and GluR2 (D3-F3). Scale bars 10μm.
AMPAR expression on preOLs was minimal at 20-24 PCW (Figure 5, A1-C1). In contrast, GluR4-containing AMPARs were highly expressed on preOLs between 25-37 PCW (Figure 5, A2-C2), consistent with earlier work from our laboratory (Follett et al., 2004). GluR4 expression subsequently declined in human white matter OLs at 38-42 PCW (Figure 5, A3-C3). Interestingly, there was a relative lack of GluR3 immunoreactivity in developing OLs at all ages analyzed (data not shown). GluR2 subunit was minimally expressed on preOLs and did not change with age (Figure 5, D1-F1, D2-F2, and D3-F3). GluR1 immunoreactivity was not detected on preOLs at any time (data not shown).
Figure 5.

Cell specific AMPAR subunit expression in O4+ OLs in white matter during the second half of gestation (20-42 PCW). At 20 PCW (A1-F1), immunohistochemical analysis of the human fetal white matter demonstrates little GluR4 (A1-C1) and GluR2 (D1-F1) expression in the O4+ OLs. At 32 PCW (A2-F2), intense GluR4 immunoreactivity co-localize with OL marker O4 (A2-C2), in contrast to GluR2, which remains downregulated in the O4+ OLs (D2-F2). At 39 PCW (A3-F3), O4+ preOLs in human fetal white matter show weak GluR4 (A3-C3) and GluR2 (D3- F3) immunoreactivity. Scale bars 10μm.
These data indicate that in developing human white matter GluR2-lacking (Ca2+permeable) AMPARs are expressed at midgestation selectively on radial glia, while during the preterm period these receptors are transiently expressed on preOLs, but not on differentiated white matter astrocytes.
Developmental regulation of AMPAR subunit expression in human parietal cortical grey matter
Similar to the white matter, we first analyzed the age-dependent variation of relative GluR1-4 protein levels. Figure 6 shows the individual normalized GluR1-4 values (% of adult) in the developing cortex, as a function of postconceptional age, with regression lines superimposed. In the cortex, between 18-54 PCW, all four AMPAR subunits demonstrated significant developmental changes (p=0.047 for GluR1, p=0.018 for GluR2, p=0.02 for GluR3 and p=0.001 for GluR4). GluR1 expression (Figure 6A) was higher than adult levels at all times, peaking around term birth. Predicted GluR1 values rose from 152% of adult at midgestation (20 PCW), to 310% of adult during the preterm period (30 PCW), reached maximal values of 376% at term (40 PCW) and decreased to 352% during infancy (50 PCW). In contrast to GluR1 expression, which largely superceded adult levels, expression of GluR2, GluR3 and GluR4 remained below or around adult levels at all time points analyzed. Estimated GluR2 expression (Figure 6B) represented only 67% of adult at 20 PCW, 43% at 30 PCW, 44% at 40 PCW, and reached 72% of adult levels at 50 PCW. Relative to adult, estimated GluR3 (Figure 6C) and GluR4 (Figure 6D) expression was 88% and 92% at 20 PCW, 40% and 42% at 30 PCW, 37% and 26% at 40 PCW, 81% and 44% of adult at 50 PCW. Similar to the white matter, there was no effect of PMI or gender upon the level of expression of any AMPAR subunits (data not shown).
Figure 6.
AMPAR subunit expression in the developing cortex from midgestation (18 PCW) through early infancy (210 PCW) as compared to adult standard (100%), indicated as horizontal dotted line. Relative protein levels for GluR1-4 demonstrate a significant upregulation of GluR1 subunit (A) and a downregulation of GluR2 (B), GluR3 (C) and GluR4 (D) subunits during the term/neonatal period. Insets are representative western blots for individual subunits. Regression lines were fitted to the individual data points, if significant.
Next, we analyzed the nonGluR2/GluR2 ratios in the cortex during the same developmental window (18-54 PCW). Individual nonGluR2/GluR2 ratios are represented in Figure 7 as a function of age with superimposed regression lines. Only GluR1/GluR2 ratio (Figure 7A) demonstrated significant developmental changes, being higher than the adult (defined for comparison as 1:1) during the entire time frame analyzed (p=0.007). In contrast, GluR3/GluR2 (Figure 7B) and GluR4/GluR2 (Figure 7C) ratios were low at all times and showed no significant variations with advancing postconceptional age (p>0.05). Predicted GluR1/GluR2 ratio rose from 2.8:1 at midgestation (20 PCW) to 7.8:1 during the preterm period (30PCW), peaking with a value of 8.8:1 at term (40 PCW) before decreasing to 5.9:1 during early infancy (50 PCW).
Figure 7.
NonGluR2/GluR2 ratios in the developing human cortex, expressed as a function of postconceptional age. Relative to adult, GluR1/GluR2 ratios (A) are significantly elevated between 35-45 PCW, the window of increased susceptibility to cortical infarctions and seizures. By contrast, GluR3/GluR2 (B) and GluR4/GluR2 (C) demonstrate no significant variations with age. Regression lines were fitted to the individual data points, if significant; otherwise, a line was drawn through the mean.
Differential cellular distribution of AMPAR subunits in human parietal grey matter
In contrast to white matter western blots that indicated maximal AMPAR expression in midgestational and premature infant brain, immunoblotting of human cortical tissue demonstrated highest AMPAR levels during the preterm, term and post-term neonatal period. As in the white matter, there was a simultaneous relative lack of GluR2 expression, suggesting the presence of Ca2+ permeable AMPARs on developing neurons. To identify differential cellular distribution of GluR2-lacking AMPARs in the human neocortex, immunocytochemical double labeling for GluR1-4, in combination with neuronal markers NeuN and NSE was performed during the same developmental window as that analyzed by western blot.
We first used cell-specific labeling to characterize the developmental changes in neuronal subpopulations in our tissue samples (Honig et al., 1996). Between 20-24 PCW, both NeuN and NSE staining revealed the presence of a wide subplate, located below the cortical plate and populated by dispersed and predominantly horizontally oriented neurons (data not shown). The subplate persisted between 25-37 PCW as a broad band of neurons underneath the developing cortex (Figure 8, B1), decreasing progressively from 38-46 PCW through infancy, as previously described (Kostovic and Rakic, 1990). At 92-210 PCW (1.0-3.3 years of age), the subplate virtually disappeared leaving only scattered interstitial white matter neurons (Figure 8, B2). In contrast to the subplate neurons, neurons of the cortical plate stained with NeuN or NSE, were densely packed at 20-24 PCW, exhibiting rounder cell bodies and fewer vertically oriented processes (data not shown). Between 25-37 PCW, large pyramidal neurons with long apical dendrites were aligned within the cortical plate (Figure 8, A1), and by 38-46 PCW all six cortical layers were distinguishable (data not shown). Between 92-210 PCW the thickness of all cortical layers increased substantially and both pyramidal and non-pyramidal neurons displayed long, elaborated processes (Figure 8, A2).
Figure 8.

Cortical and subcortical maturational changes, visualized by neuronal markers NeuN and NSE. At 31 PCW (A1-B1), NeuN staining demonstrates the presence of a broad subplate zone (B1) underneath the developing cortical plate (A1). By contrast, at 210 PCW /3.3 years (A2-B2), NSE staining reveals that only few interstitial neurons are still present in the subcortical white matter (B2), while there is an expansion of all six cortical layers (A2). Scale bar 20μm.
AMPAR expression on the subplate and cortical plate neurons underwent dramatic developmental changes during the preterm, term and post-term neonatal periods. Subplate neurons expressed AMPARs earlier than immature cortical neurons. However, during fetal and early postnatal development, GluR1 was the predominant subunit on both subplate and cortical neurons. GluR1 was expressed on subplate neurons as early as 20 PCW (data not shown), and continued to increase with age. Between 25-37 PCW the majority of subplate neurons were intensely GluR1 immunopositive (Figure 9, A1-C1). GluR1 began to decrease at 38-46 PCW (Figure 9, A2-C2) and between 92-210 PCW only a few of the remaining subplate neurons stained with GluR1 (Figure 9, A3-C3). In contrast, GluR2 expression was undetectable or minimal on subplate neurons at midgestation (20-24 PCW) and during the preterm period (25-37 PCW) (Figure 9, D1-F1). GluR2 expression became visible around term (38-46 PCW) (Figure 9, D2-F2) and increasing further on remaining subplate neurons through the first 3 years of life (92-210 PCW) (Figure 9, D3-F3). Similar to the pattern of expression of GluR2, GluR3 was low during gestation and early infancy on subplate neurons, but the staining became robust during early childhood (data not shown). Compared to developing glial cells, subplate neurons demonstrated low GluR4 expression at all ages (data not shown).
Figure 9.

Differential expression of AMPARs in the subplate neurons during perinatal and early postnatal development. During preterm period (31 PCW) subcortical neurons, stained with NeuN (A1-F1), demonstrate intense GluR1 expression on cell bodies and processes (A1-C1), in contrast to GluR2, which is relatively lacking (D1-F1). During post-term neonatal period (45 PCW), the majority of the NeuN+ subplate neurons (A2-F2) show high GluR1 expression (A2-C2), coincident with the appearance of GluR2 immunoreactivity (D2-F2). In early childhood (168 PCW) only few NeuN-labeled subplate neurons (A3-F3) are still GluR1 positive (A3-C3), in contrast to intense GluR2 staining in most subcortical neurons (D3-F3). Scale bars 10μm.
In the cortical plate, GluR1 immunoreactivity was observed only as diffuse neuropil staining between 20-24 PCW (data not shown). A significant increase in GluR1 immunoreactivity was observed between 25-37 PCW, predominantly on neurons located in the deeper portion of the cortical plate (Figure 10, A1-C1). Between 38-46 PCW, the majority of pyramidal and non-pyramidal neurons in all cortical layers showed intense GluR1 immunoreactivity (Figure 10, A2-C2). Between 92-210 PCW, a marked decrease in GluR1 labeling was observed on pyramidal neurons (Figure 10, A3-C3), while most non-pyramidal neurons remained strongly labeled (data not shown). In contrast, GluR2 expression was not visible on cortical neurons between 20-37 PCW (Figure 10, D1-F1) and was present only at low levels at 38-46 PCW (Figure 10, D2-F2). During early childhood (92-210 PCW) GluR2 became highly expressed on layer III and V cortical pyramidal neurons (Figure 10, D3-F3), but remained low in the non-pyramidal neurons (data not shown). The GluR3 subunit paralleled the expression pattern of GluR2, increasing progressively with age, mostly during postnatal development, and predominantly on pyramidal neurons. GluR4 expression remained constantly low on cortical neurons at all ages, except for scattered large non-pyramidal neurons, which became robustly labeled between 92-210 PCW (data not shown).
Figure 10.

Cell-specific AMPAR subunits in cortical neurons of human parietal neocortex from prenatal period through early infancy. At 31 PCW (A1-F1), distinct cortical neurons strongly label with GluR1 (A1-C1), while GluR2 remains downregulated (D1-F1). At 45 PCW (A2-F2), the majority of neurons show robust GluR1 immunoreactivity (A2-C2), while only scattered neurons are GluR2 positive (D2-F2). At 168 PCW (A3-F3), pyramidal neurons demonstrate a marked decrease in GluR1 immunoreactivity (A3-C3), and a substantial increase in GluR2 expression (D3-F3). Scale bars 10μm.
Taken together, these data suggest that high levels of GluR2-lacking AMPARs are expressed on subplate neurons transiently during the preterm period and on cortical pyramidal and non-pyramidal neurons during term and the post-term neonatal period. In contrast to pyramidal neurons, which gradually acquire GluR2 subunits during early postnatal development, there is a relative GluR2 deficiency on non-pyramidal throughout early childhood.
DISCUSSION
The rationale for mapping AMPARs in both developing rat and human brain in this study was twofold: 1) to test the hypothesis that GluR2-lacking (Ca2+ permeable) AMPARs play an age-specific role in selective perinatal H/I vulnerability in the human and 2) to validate specific rodent stages as appropriate models to investigate the pathophysiology of H/I injury in the developing brain. The present study demonstrates that AMPAR subunits are developmentally regulated in glial and neuronal cell types in developing human white and grey matter. During the preterm period, a window of increased vulnerability for PVL, GluR2-lacking AMPARs are predominantly expressed on white matter radial glia, preOLs, and on closely apposed subplate neurons. Subsequently during later development at term, when infants are at risk for H/I induced encephalopathy and seizures, GluR2-lacking AMPARs are highly expressed on cortical neurons, coincident with decreased expression on white matter cells. Strikingly, the developmental regulation of the GluR2-lacking receptors on specific cell types follows similar temporal and regional order to that reported for the rat in Part I of this study. These findings are summarized in Figure 11, which specifically graphs the chronology of GluR2-lacking AMPARs, as opposed to total AMPARs. These parallel developmental patterns allow identification of analogous maturational stages between rat and human that is essential for the design and interpretation of pathophysiological rodent models of human perinatal H/I cerebral injury.
Figure 11.

Comparative analysis of human and rat developmental regulation of GluR2-lacking AMPARs, relative to the windows of increased susceptibility to H/I. In human, prematurity represents a developmental window of selective vulnerability for white matter injury, such as periventricular leukomalacia (PVL), while term and neonatal periods are characterized by increased susceptibility to cortical injury and seizures. In midgestational cases (20-24 PCW), radial glia expresses high levels of GluR2-lacking AMPARs, while in premature infants (25-37 PCW), these receptors are transiently expressed on premyelinating oligodendrocytes and subplate neurons. During term (38-42 PCW) and post-term neonatal (43-46 PCW) periods, GluR2-lacking AMPARs are specifically expressed on cortical pyramidal and non-pyramidal neurons, while during infancy and early childhood (92-210 PCW), these receptors can only be found in non-pyramidal neurons. Furthermore, there are striking similarities between rat and human regarding regional and temporal developmental regulation of glial and neuronal expression of GluR2-lacking AMPARs. In both Long Evans and Sprague Dawley rats, the periods of increased susceptibility to H/I correlates with a regional and cell-specific relative GluR2-deficiency. However, despite similar temporal and regional progression, developmental changes in GluR expression occurs 2-3 days earlier in Sprague Dawley rats, compared to Long Evans.
Differential regional and cell-type specific expression of GluR2-deficient AMPARs in the developing human brain
To date, there has been no comprehensive study of the regulation of AMPAR subunit protein in human cortex and white matter across fetal, neonatal and early childhood development. Previous studies in human brain tissue have characterized the developmental regulation of AMPARs during earlier (8-20 PCW) or later (0-6 years) developmental windows (Ritter et al., 2001;Murphy et al., 2005) or have focused on areas other than white matter and neocortex , such as the basal ganglia and the brainstem (Meng et al., 1997;Panigrahy et al., 2000). AMPARs have been previously identified by in situ hybridization and autoradiography in all cortical structures, including the cortical plate, marginal, intermediate and subventricular zones during the first half of gestation (8-20 PCW) (Ritter et al., 2001). Interestingly, during these early stages of fetal development, there was a relative lack in GluR2 transcripts compared to other subunits. Our present results are the first to evaluate AMPAR subunit protein expression during clinically relevant intervals with respect to differential susceptibility to H/I injury. Here we show that GluR2-lacking AMPARs are transiently expressed in the perinatal human brain, peaking in the white matter during early fetal development through the preterm period, and in the cortex during the term/neonatal period through infancy. We also found marked maturational differences in the cellular distribution of GluR2-lacking AMPARs. These receptors appear to be highly expressed specifically on radial glia at 20-24 PCW and are transiently upregulated on premyelinating OLs and subplate neurons from 25-37 PCW. GluR2-lacking AMPARs appear to be abundant in the neocortical pyramidal and non-pyramidal neurons at 38-46 PCW and also remain highly expressed on non-pyramidal neurons at 92-210 PCW (Figure 11). Furthermore, the developmental pattern appears to proceed in an “inside-out” manner, with the innermost cells, such as radial glia and preOLs, maturing and acquiring AMPARs prior to the more peripheral subcortical and cortical neurons.
Potential role for GluR2-lacking AMPARs in age-specific H/I injury in the human neonate
Perinatal H/I represents a major cause of brain injury in both preterm and term infants (Volpe, 2001). Improved neonatal intensive care has lead to survival of 85-90% of the more than 55,000 very low birth weight (1500 gm) premature infants born annually in the US (Volpe, 2001). The incidence of H/I brain injury in these survivors is relatively high, with 10% developing major spastic motor deficits and an additional 25-50% exhibiting cognitive and behavioral deficits (Hack et al., 2002) . PVL represents the prevalent form of H/I brain injury in fetuses and preterm infants (Banker and Larroche, 1962;Dambska et al., 1989;Rorke, 1998;Volpe, 2001;Kinney and Armstrong, 2002;Kinney et al., 2004), involving death of precursor cells such as OL precursors and immature OLs. While at this developmental period the cortex is relatively spared with respect to acute injury (Marin-Padilla, 1997;Haynes et al., 2003), clinical and neuroimaging follow-up studies of premature infants show cognitive and sensory deficits (Cioni et al., 1997;Piecuch et al., 1997;Hack et al., 2002) and a reduction of cerebral cortical grey matter volume (Inder et al., 1999;Inder et al., 2003), especially in association with white matter injury.
In term infants, H/I causes cortical neuronal necrosis with neonatal seizures and subsequent neurodevelopmental disabilities in 1-2 per 1000 live term births (Hauser et al., 1993;Estan and Hope, 1997;Saliba et al., 1999;Volpe, 2001;Kinney and Armstrong, 2002). Neonatal seizures can be extremely refractory to therapy, and conventional antiepileptic drugs used in older children and adults are often ineffective at suppressing neonatal seizure activity (Painter et al., 1999;Volpe, 2001), suggesting that the pathogenesis of these seizures may be age-specific.
The present study demonstrates that the differential regional and cellular distribution of GluR2-lacking, and likely Ca2+ permeable, AMPARs in perinatal human brain correlates with the age-specific vulnerability to H/I injury. In the earliest developmental interval studied (20-24 PCW), we show that radial glia transiently express GluR2-lacking AMPARs, suggesting a possible basis for vulnerability of these cells to fetal H/I injury and excitotoxicity. The radial glial cells are transiently present in the developing fetal brain where they guide neuronal migration but can also proliferate to give rise to both glia and neurons (Rakic, 1972;Rakic, 1974;Noctor et al., 2001). After the neuronal migration is complete, radial glia gradually transform into astrocytes and start migrating into the cortex between 18-35 PCW (Rakic, 2003;deAzevedo et al., 2003). Hence selective excitotoxic injury of radial glia mediated by Ca2+ permeable AMPARs could impair normal cortical development by disrupting the late phases of neuronal migration and by reducing the astrocytic precursor pool destined for the upper cortex (Gressens et al., 1992). By contrast, astrocytes have receptors with low Ca2+ permeability and are more resistant to H/I injury (Yamaya et al., 2002).
Our data demonstrate that in premature infants (25-37 PCW), an age characterized by enhanced susceptibility to PVL, white matter OLs express high levels of AMPARs with increased Ca2+ permeability. In contrast, by term (38-42 PCW) there is little to no AMPAR expression on white matter OLs. As demonstrated in the rat (Follett et al., 2000;Follett et al., 2004), Ca2+ influx through GluR2-lacking AMPARs may represent a prominent mechanism of H/I injury to developing OLs in human.
We show that during the preterm period subplate neurons possess AMPAR subunit configurations consistent with Ca2+ permeability, supporting a possible mechanism for their selective vulnerability in the human. Subplate neurons are only transiently present in the developing human, where the subplate zone peaks at 22-35 PCW and diminishes during early infancy and childhood (Kostovic and Rakic, 1990;Samuelsen et al., 2003). Subplate neurons play an essential role in the development of connections between thalamus and visual cortex and of connections within the cortex (Ghosh and Shatz, 1993;Allendoerfer and Shatz, 1994;Kanold et al., 2003). Subplate neurons have been shown to be selectively vulnerable to H/I in the developing rat brain (McQuillen et al., 2003). AMPAR-mediated injury to subplate neurons in humans may thus contribute to the high incidence of cortical visual impairments (Cioni et al., 1997) and the widespread abnormalities of cerebral development (Piecuch et al., 1997;Inder et al., 1999;Miller et al., 2002;Inder et al., 2003) that are observed in preterm infants suffering from H/I encephalopathy.
Unlike subplate neurons, cortical neurons demonstrate abundant expression of Ca2+ permeable AMPARs during the term (38-42 PCW) and post-term neonatal period (43-46 PCW), correlating with the predominant grey matter pathology known to occur at this age. Experimental data suggest that AMPARs may play a critical role in H/I induced neuronal injury, synaptic plasticity and epileptogenesis in perinatal human brain. In the developing rat, both hypoxia-induced seizures and neuronal cortical injury are highly dependent of AMPAR activation (Hagberg et al., 1994;Jensen et al., 1995;Koh and Jensen, 2001). Moreover, activation of Ca2+permeable AMPARs on pyramidal neurons by hypoxia induces further increases in neuronal excitability, synaptic plasticity and later seizure-induced neuronal injury (Sanchez et al., 2001;Koh et al., 2004;Sanchez et al., 2005).
Taken together, these findings support the hypothesis that GluR2-lacking AMPARs are present in vulnerable cell populations during discrete developmental windows. Importantly, these receptors may provide a therapeutic target for treatment of H/I injury in human infants, as has been suggested in rodent models of PVL and term H/I encephalopathy.
While these studies were performed to help elucidate factors important for age- and region-specific susceptibility to H/I and excitotoxicity, our findings also provide important normative data for human brain development. AMPARs on specific radial glia, such as the Bergmann glia, have been postulated to be important for cell-cell signaling and trophism during neuronal migration (Ishiuchi et al., 2001). However, mature astrocytes actually possess AMPARs with low Ca2+ permeability, and intracellular Ca2+ homeostasis and signaling depends mostly on release from intracellular stores (Yoshida et al., 2003;Zur and Deitmer, 2005). Ca2+permeable AMPARs regulate OL proliferation and differentiation in vitro (Gallo et al., 1996;Yuan et al., 1998), while in neurons, these receptors have been postulated to provide signaling that is essential to the development of normal cortical networks (Gu et al., 1996;Debray et al., 1997;Sanchez et al., 2001;Kumar et al., 2002;Sanchez et al., 2005). A role for Ca2+ permeable AMPARs on subplate neurons has not yet been studied, although it is established that excitatory neurotransmission mediated by these cells modulates the development of cortical networks (Ghosh and Shatz, 1993;Kanold et al., 2003). Thus, our findings have relevance beyond the pathologic implications in the setting of H/I.
Developmental profile of AMPARs in human versus experimental rodent models
The present study describes striking similarities in the regional progression of AMPAR subunit development between rodent and human cerebral white and grey matter. In addition, the regional expression of AMPARs in both species correlates with age-specific regional vulnerability to H/I injury. Our data (see Part I) show that in the rat GluR2-lacking AMPARs are present on radial glia, white matter OLs and subplate neurons during the first postnatal week, which is a window of development characterized by enhanced susceptibility to selective H/I white matter injury (Follett et al., 2000;Back et al., 2002;Liu et al., 2002;McQuillen et al., 2003). Likewise, we show that GluR2-lacking AMPARs are present in the cortical neurons during the second week of life in rat , when H/I causes neuronal necrosis and seizures (Jensen et al., 1991;Owens et al., 1997;Towfighi et al., 1997;Jensen et al., 1998;Chen et al., 1998).
The age-specificity of regional susceptibility has been confirmed in other non-rodent species. Prenatal H/I in preterm fetal sheep results in subcortical white matter injury, while the incidence of grey matter pathology, including cortical infarction and seizures, increases near term (Williams et al., 1990;Mallard et al., 1994;Reddy et al., 1998). Consistently, AMPARs are expressed at high levels in the white matter OLs and subplate neurons during the second half of gestation, while they reach their maximal expression levels on cortical neurons just before birth (Furuta and Martin, 1999).
This parallel and comparative study of human white matter and cortex (Part II) reveals similar patterns in the temporal sequence of expression of GluR subunits on specific cell populations to those of the rodent, although the time course is proportionally prolonged in the human relative to the rat (Figure 11). In both rat and human, the time period which corresponds to increased vulnerability of white matter to H/I injury is characterized by high levels of GluR2-lacking AMPARs on OL precursors compared to other ages. In both rat and human, H/I elicit cortical injury and seizures at a time that corresponds to prominent GluR2-deficiency in cortical neurons. While these OL and cortical neuronal relationships are consistent with well-studied clinical entities, the potential roles in disease for GluR2-lacking AMPARs on radial glia and subplate neurons are yet to be elucidated. H/I injury mediated by GluR2-lacking AMPARs on radial glia in midgestation could result in disorders of terminal neuronal migration and cortical development. Similarly, these receptors could contribute to selective subplate neuron injury in the preterm period, resulting in neuronal network excitability and cognitive disorders. Such hypotheses based on observations from human data, together with the knowledge of the appropriate rodent stages for AMPAR development, will allow further modeling to examine mechanisms and potential therapies for these age-specific forms of perinatal brain injury.
Acknowledgements
This work was supported by grants from the William Randolph Hearst Foundation (DMT), Epilepsy Foundation (DMT), HD01359 (PLF), Charles H. Hood Foundation (PLF), NIH:NINDS NS31718 (FEJ, REF), NS38475 (JJV, FEJ, RDF), United Cerebral Palsy Foundation (FEJ), and Mental Retardation Research Center Grant (NIH:NICHHD P30 HD18655). Additional tissue samples were generously provided by the University of Maryland Brain Tissue Bank for Developmental Disorders, Baltimore, MD, supported by N01-HD-4-3368 and N01-HD-4-3383. We thank Dr. Hannah Kinney for advice in conducting this study and Dr. Robin Hynes for help with tissue sample collection.
Reference List
- Allendoerfer KL, Shatz CJ. The subplate, a transient neocortical structure: its role in the development of connections between thalamus and cortex. Annu Rev Neurosci. 1994;17:185–218. doi: 10.1146/annurev.ne.17.030194.001153. [DOI] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. Journal of Neuroscience. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker BQ, Larroche JC. Periventricular leukomalacia of infancy. A form of neonatal anoxic encephalopathy. Arch Neurol. 1962;7:386–410. doi: 10.1001/archneur.1962.04210050022004. [DOI] [PubMed] [Google Scholar]
- Barkovich AJ. MR and CT evaluation of profound neonatal and infantile asphyxia. AJNR Am J Neuroradiol. 1992;13:959–972. [PMC free article] [PubMed] [Google Scholar]
- Barkovich AJ, Sargent SK. Profound asphyxia in the premature infant: imaging findings. AJNR Am J Neuroradiol. 1995;16:1837–1846. [PMC free article] [PubMed] [Google Scholar]
- Bellinger DC, Jonas RA, Rappaport LA, Wypij D, Wernovsky G, Kuban KC, Barnes PD, Holmes GL, Hickey PR, Strand RD. Developmental and neurologic status of children after heart surgery with hypothermic circulatory arrest or low-flow cardiopulmonary bypass. N Engl J Med. 1995;332:549–555. doi: 10.1056/NEJM199503023320901. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem. 1984;4:1369–1374. doi: 10.1111/j.1471-4159.1984.tb05396.x. [DOI] [PubMed] [Google Scholar]
- Bernard V, Gardiol A, Faucheux B, Bloch B, Agid Y, Hirsch EC. Expression of glutamate receptors in the human and rat basal ganglia: effect of the dopaminergic denervation on AMPA receptor gene expression in the striatopallidal complex in Parkinson's disease and rat with 6-OHDA lesion. J Comp Neurol. 1996;368:553–568. doi: 10.1002/(SICI)1096-9861(19960513)368:4<553::AID-CNE7>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Chen HS, Wang YF, Rayudu PV, Edgecomb P, Neill JC, Segal MM, Lipton SA, Jensen FE. Neuroprotective concentrations of the N-methyl-D-aspartate open-channel blocker memantine are effective without cytoplasmic vacuolation following post-ischemic administration and do not block maze learning or long-term potentiation. Neuroscience. 1998;86:1121–1132. doi: 10.1016/s0306-4522(98)00163-8. [DOI] [PubMed] [Google Scholar]
- Cioni G, Fazzi B, Coluccini M, Bartalena L, Boldrini A, Hof-van Duin J. Cerebral visual impairment in preterm infants with periventricular leukomalacia. Pediatr Neurol. 1997;17:331–338. doi: 10.1016/s0887-8994(97)00152-5. [DOI] [PubMed] [Google Scholar]
- Cowan F, Rutherford M, Groenendaal F, Eken P, Mercuri E, Bydder GM, Meiners LC, Dubowitz LM, de Vries LS. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet. 2003;361:736–742. doi: 10.1016/S0140-6736(03)12658-X. [DOI] [PubMed] [Google Scholar]
- Dambska M, Laure-kamionowska M, Schmidt-Sidor B. Early and late neuropathological changes in perinatal white matter damage. Journal of Child Neurology. 1989;4:291–298. doi: 10.1177/088307388900400408. [DOI] [PubMed] [Google Scholar]
- deAzevedo LC, Fallet C, Moura-Neto V, Daumas-Duport C, Hedin-Pereira C, Lent R. Cortical radial glial cells in human fetuses: depth-correlated transformation into astrocytes. J Neurobiol. 2003;55:288–298. doi: 10.1002/neu.10205. [DOI] [PubMed] [Google Scholar]
- Debray C, Diabira D, Gaiarsa JL, Ben Ari Y, Gozlan H. Contributions of AMPA and GABA(A) receptors to the induction of NMDAR-dependent LTP in CA1. Neurosci Lett. 1997;238:119–122. doi: 10.1016/s0304-3940(97)00865-3. [DOI] [PubMed] [Google Scholar]
- Deng W, Rosenberg PA, Volpe JJ, Jensen FE. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc Natl Acad Sci U S A. 2003;100:6801–6806. doi: 10.1073/pnas.1136624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estan J, Hope P. Unilateral neonatal cerebral infarction in full term infants. Arch Dis Child Fetal Neonatal Ed. 1997;76:F88–F93. doi: 10.1136/fn.76.2.f88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferriero DM. Neonatal brain injury. N Engl J Med. 2004;351:1985–1995. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- Folkerth RD, Haynes RL, Borenstein NS, Belliveau RA, Trachtenberg F, Rosenberg PA, Volpe JJ, Kinney HC. Developmental lag in superoxide dismutases relative to other antioxidant enzymes in premyelinated human telencephalic white matter. J Neuropathol Exp Neurol. 2004;63:990–999. doi: 10.1093/jnen/63.9.990. [DOI] [PubMed] [Google Scholar]
- Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, Volpe JJ, Jensen FE. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: A protective role for topiramate. J Neurosci. 2004;24:4412–4420. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–9241. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LK, Veliskova J. GluR2 antisense knockdown produces seizure behavior and hippocampal neurodegeneration during a critical window. Ann N Y Acad Sci. 1999;868:541–545. doi: 10.1111/j.1749-6632.1999.tb11324.x. [DOI] [PubMed] [Google Scholar]
- Furuta A, Martin LJ. Laminar segregation of the cortical plate during corticogenesis is accompanied by changes in glutamate receptor expression. J Neurobiol. 1999;39:67–80. [PubMed] [Google Scholar]
- Gallo V, Zhou JM, McBain CJ, Wright P, Knutson PL, Armstrong RC. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. The Journal of Neuroscience. 1996;16(8):2659–2670. doi: 10.1523/JNEUROSCI.16-08-02659.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Shatz CJ. A role for subplate neurons in the patterning of connections from thalamus to neocortex. Development. 1993;117:1031–1047. doi: 10.1242/dev.117.3.1031. [DOI] [PubMed] [Google Scholar]
- Grafe MR. The correlation of prenatal brain damage with placental pathology. J Neuropathol Exp Neurol. 1994;53:407–415. doi: 10.1097/00005072-199407000-00013. [DOI] [PubMed] [Google Scholar]
- Gressens P, Richelme C, Kadhim HJ, Gadisseux JF, Evrard P. The germinative zone produces the most cortical astrocytes after neuronal migration in the developing mammalian brain. Biol Neonate. 1992;61:4–24. doi: 10.1159/000243526. [DOI] [PubMed] [Google Scholar]
- Gu JG, Albuquerque CJ, Lee CJ, MacDermott AB. Synaptic strengthening through activation of Ca2+-permeable AMPA receptors. Nature. 1996;381:793–796. doi: 10.1038/381793a0. [DOI] [PubMed] [Google Scholar]
- Gucuyener K, Atalay Y, Aral YZ, Hasanoglu A, Turkyilmaz C, Biberoglu G. Excitatory amino acids and taurine levels in cerebrospinal fluid of hypoxic ischemic encephalopathy in newborn. Clin Neurol Neurosurg. 1999;101:171–174. doi: 10.1016/s0303-8467(99)00035-9. [DOI] [PubMed] [Google Scholar]
- Hack M, Flannery DJ, Schluchter M, Cartar L, Borawski E, Klein N. Outcomes in young adulthood for very-low-birth-weight infants. N Engl J Med. 2002;346:149–157. doi: 10.1056/NEJMoa010856. [DOI] [PubMed] [Google Scholar]
- Hack M, Wilson-Costello D, Friedman H, Taylor GH, Schluchter M, Fanaroff AA. Neurodevelopment and predictors of outcomes of children with birth weights of less than 1000 g: 1992-1995. Arch Pediatr Adolesc Med. 2000;154:725–731. doi: 10.1001/archpedi.154.7.725. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Gilland E, Deimer N, Andine P. Hypoxic-ischemic damage in the neonatal rat brain: histopathology after post-treament with NMDA and non-NMDA receptor antagonists. Biology of the Neonate. 1994;66:213. doi: 10.1159/000244109. [DOI] [PubMed] [Google Scholar]
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota:1935-1984. Epilepsia. 1993;34:453–468. doi: 10.1111/j.1528-1157.1993.tb02586.x. [DOI] [PubMed] [Google Scholar]
- Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA, Volpe JJ, Kinney HC. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. 2003;62:441–450. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annual Review of Neuroscience. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Honig LS, Herrmann K, Shatz CJ. Developmental changes revealed by immunohistochemical markers in human cerebral cortex. Cereb Cortex. 1996;6:794–806. doi: 10.1093/cercor/6.6.794. [DOI] [PubMed] [Google Scholar]
- Inder TE, Huppi PS, Warfield S, Kikinis R, Zientara GP, Barns PD, Jolesz F, Volpe JJ. Periventricular white matter injury inthe premature infant is followed by reduced cerebral cortical gray matter volume at term. Annals of Neurology. 1999;46:755–760. doi: 10.1002/1531-8249(199911)46:5<755::aid-ana11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Inder TE, Wells SJ, Mogridge NB, Spencer C, Volpe JJ. Defining the nature of the cerebral abnormalities in the premature infant: a qualitative magnetic resonance imaging study. J Pediatr. 2003;143:171–179. doi: 10.1067/S0022-3476(03)00357-3. [DOI] [PubMed] [Google Scholar]
- Ishiuchi S, Tsuzuki K, Yamada N, Okado H, Miwa A, Kuromi H, Yokoo H, Nakazato Y, Sasaki T, Ozawa S. Extension of glial processes by activation of Ca2+-permeable AMPA receptor channels. NeuroReport. 2001;12:745–748. doi: 10.1097/00001756-200103260-00026. [DOI] [PubMed] [Google Scholar]
- Jensen FE, Alvarado S, Firkusny IR, Geary C. NBQX blocks the acute and late epileptogenic effects of perinatal hypoxia. Epilepsia. 1995;36(10):966–972. doi: 10.1111/j.1528-1157.1995.tb00954.x. [DOI] [PubMed] [Google Scholar]
- Jensen FE, Applegate CD, Holtzman D, Belin T, Burchfiel J. Epileptogenic effect of hypoxia in the immature rodent brain. Ann of Neurol. 1991;29:629–637. doi: 10.1002/ana.410290610. [DOI] [PubMed] [Google Scholar]
- Jensen FE, Wang C, Stafstrom CE, Liu Z, Geary C, Stevens MC. Acute and chronic increases in excitability in rat hippocampal slices after perinatal hypoxia in vivo. J Neurophysiol. 1998;79:73–81. doi: 10.1152/jn.1998.79.1.73. [DOI] [PubMed] [Google Scholar]
- Jensen JB, Lund TM, Timmermann DB, Schousboe A, Pickering DS. Role of GluR2 expression in AMPA-induced toxicity in cultured murine cerebral cortical neurons. J Neurosci Res. 2001;65:267–277. doi: 10.1002/jnr.1150. [DOI] [PubMed] [Google Scholar]
- Kanold PO, Kara P, Reid RC, Shatz CJ. Role of subplate neurons in functional maturation of visual cortical columns. Science. 2003;301:521–525. doi: 10.1126/science.1084152. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Armstrong DL. Perinatal Neuropathology. In: Graham DI, Lantos PE, editors. Greenfield's Neuropathology. Arnold; London: 2002. pp. 557–559. [Google Scholar]
- Kinney HC, Back SA. Human oligodendrocyte development: relationship to periventricular leukomalacia. Seminars in Pediatric Neurology. 1998;5:180–189. doi: 10.1016/s1071-9091(98)80033-8. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Haynes RL, Folkerth RD. Golden JA, Harding B, editors. White matter dissorders in the perinatal period. Pediatric Neuropathology. 2004. In Press.
- Koh S, Jensen FE. Topiramate blocks perinatal hypoxia-induced seizures in rat pups. Ann Neurol. 2001;50:366–372. doi: 10.1002/ana.1122. [DOI] [PubMed] [Google Scholar]
- Koh S, Tibayan FD, Simpson J, Jensen FE. NBQX or topiramate treatment following perinatal hypoxia-induced seizures prevents later increases in seizure-induced neuronal injury. Epilepsia. 2004;45:569–575. doi: 10.1111/j.0013-9580.2004.69103.x. [DOI] [PubMed] [Google Scholar]
- Kostovic I, Rakic P. Developmental history of the transient subplate zone in the visual and somatosensory cortex of the macaque monkey and human brain. J Comp Neurol. 1990;297:441–470. doi: 10.1002/cne.902970309. [DOI] [PubMed] [Google Scholar]
- Kultas-Ilinsky K, Fallet C, Verney C. Development of the human motor-related thalamic nuclei during the first half of gestation, with special emphasis on GABAergic circuits. J Comp Neurol. 2004;476:267–289. doi: 10.1002/cne.20216. [DOI] [PubMed] [Google Scholar]
- Kumar SS, Bacci A, Kharazia V, Huguenard JR. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci. 2002;22:3005–3015. doi: 10.1523/JNEUROSCI.22-08-03005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Silverstein FS, Skoff R, Barks JD. Hypoxic-ischemic oligodendroglial injury in neonatal rat brain. Pediatr Res. 2002;51:25–33. doi: 10.1203/00006450-200201000-00007. [DOI] [PubMed] [Google Scholar]
- Loeliger M, Watson CS, Reynolds JD, Penning DH, Harding R, Bocking AD, Rees SM. Extracellular glutamate levels and neuropathology in cerebral white matter following repeated umbilical cord occlusion in the near term fetal sheep. Neuroscience. 2003;116:705–714. doi: 10.1016/s0306-4522(02)00756-x. [DOI] [PubMed] [Google Scholar]
- Mallard EC, Williams CE, Johnston BM, Gluckman PD. Increased vulnerability to neuronal damage after umbilical cord occlusion in fetal sheep with advancing gestation. Am J Obstet Gynecol. 1994;170:206–214. doi: 10.1016/s0002-9378(94)70409-0. [DOI] [PubMed] [Google Scholar]
- Maller AI, Hankins LL, Yeakley JW, Butler IJ. Rolandic type cerebral palsy in children as a pattern of hypoxic-ischemic injury in the full-term neonate. J Child Neurol. 1998;13:313–321. doi: 10.1177/088307389801300702. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. II: white matter lesions of the neocortex. J Neuropathol Exp Neurol. 1997;56:219–235. doi: 10.1097/00005072-199703000-00001. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. III: gray matter lesions of the neocortex. J Neuropathol Exp Neurol. 1999;58:407–429. doi: 10.1097/00005072-199905000-00001. [DOI] [PubMed] [Google Scholar]
- Massicotte G, Baudry M. Brain plasticity and remodeling of AMPA receptor properties by calcium-dependent enzymes. Genet Eng (N Y ) 2004;26:239–254. doi: 10.1007/978-0-306-48573-2_12. [DOI] [PubMed] [Google Scholar]
- McQuillen PS, Sheldon RA, Shatz CJ, Ferriero DM. Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J Neurosci. 2003;23:3308–3315. doi: 10.1523/JNEUROSCI.23-08-03308.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng SZ, Obonai T, Isumi H, Takashima S. A developmental expression of AMPA-selective glutamate receptor subunits in human basal ganglia. Brain Dev. 1997;19:388–392. doi: 10.1016/s0387-7604(97)00038-7. [DOI] [PubMed] [Google Scholar]
- Mercuri E, Barnett A, Rutherford M, Guzzetta A, Haataja L, Cioni G, Cowan F, Dubowitz L. Neonatal cerebral infarction and neuromotor outcome at school age. Pediatrics. 2004;113:95–100. doi: 10.1542/peds.113.1.95. [DOI] [PubMed] [Google Scholar]
- Miller SP, Vigneron DB, Henry RG, Bohland MA, Ceppi-Cozzio C, Hoffman C, Newton N, Partridge JC, Ferriero DM, Barkovich AJ. Serial quantitative diffusion tensor MRI of the premature brain: development in newborns with and without injury. J Magn Reson Imaging. 2002;16:621–632. doi: 10.1002/jmri.10205. [DOI] [PubMed] [Google Scholar]
- Moga DE, Janssen WG, Vissavajjhala P, Czelusniak SM, Moran TM, Hof PR, Morrison JH. Glutamate receptor subunit 3 (GluR3) immunoreactivity delineates a subpopulation of parvalbumin-containing interneurons in the rat hippocampus. J Comp Neurol. 2003;462:15–28. doi: 10.1002/cne.10710. [DOI] [PubMed] [Google Scholar]
- Murphy KM, Beston BR, Boley PM, Jones DG. Development of human visual cortex: a balance between excitatory and inhibitory plasticity mechanisms. Dev Psychobiol. 2005;46:209–221. doi: 10.1002/dev.20053. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- Okumura A, Hayakawa F, Kato T, Kuno K, Watanabe K. MRI findings in patients with spastic cerebral palsy. I: Correlation with gestational age at birth. Dev Med Child Neurol. 1997;39:363–368. doi: 10.1111/j.1469-8749.1997.tb07447.x. [DOI] [PubMed] [Google Scholar]
- Osborn M, Debus E, Weber K. Monoclonal antibodies specific for vimentin. Eur J Cell Biol. 1984;34:137–143. [PubMed] [Google Scholar]
- Owens J, Robbins CA, Wenzel J, Schwartzkroin PA. Acute and chronic effects of hypoxia on the developing hippocampus. Ann Neurol. 1997;41:187–199. doi: 10.1002/ana.410410210. [DOI] [PubMed] [Google Scholar]
- Painter MJ, Scher MS, Stein AD, Armatti S, Wang Z, Gardiner JC, Paneth N, Minnigh B, Alvin J. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. New England Journal of Medicine. 1999;341:485–489. doi: 10.1056/NEJM199908123410704. [DOI] [PubMed] [Google Scholar]
- Panigrahy A, Rosenberg PA, Assmann S, Foley EC, Kinney HC. Differential expression of glutamate receptor subtypes in human brainstem sites involved in perinatal hypoxia-ischemia. J Comp Neurol. 2000;427:196–208. doi: 10.1002/1096-9861(20001113)427:2<196::aid-cne3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Bennett MVL, Zukin RS. Are Ca2+-permeable kainate/AMPA receptors more abundant in immature brain? Neuroscience Letters. 1992;144:65–69. doi: 10.1016/0304-3940(92)90717-l. [DOI] [PubMed] [Google Scholar]
- Peterson BS, Vohr B, Staib LH, Cannistraci CJ, Dolberg A, Schneider KC, Katz KH, Westerveld M, Sparrow S, Anderson AW, Duncan CC, Makuch RW, Gore JC, Ment LR. Regional brain volume abnormalities and long-term cognitive outcome in preterm infants. JAMA. 2000;284:1939–1947. doi: 10.1001/jama.284.15.1939. [DOI] [PubMed] [Google Scholar]
- Piecuch RE, Leonard CH, Cooper BA, Sehring SA. Outcome of extremely low birth weight infants (500 to 999 grams) over a 12-year period. Pediatrics. 1997;100:633–639. doi: 10.1542/peds.100.4.633. [DOI] [PubMed] [Google Scholar]
- Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145:61–83. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- Rakic P. Neurons in rhesus monkey visual cortex: systematic relation between time of origin and eventual disposition. Science. 1974;183:425–427. doi: 10.1126/science.183.4123.425. [DOI] [PubMed] [Google Scholar]
- Rakic P. Developmental and evolutionary adaptations of cortical radial glia. Cereb Cortex. 2003;13:541–549. doi: 10.1093/cercor/13.6.541. [DOI] [PubMed] [Google Scholar]
- Reddy K, Mallard C, Marks K, Bennet L, Gunning M, Gunn A, Gluckman PD, Williams CE. Maturational change in the cortical response to hypoperfusion injury in the fetal sheep. Pediatric Research. 1998;43:674–682. doi: 10.1203/00006450-199805000-00017. [DOI] [PubMed] [Google Scholar]
- Ritter LM, Unis AS, Meador-Woodruff JH. Ontogeny of ionotropic glutamate receptor expression in human fetal brain. Brain Res Dev Brain Res. 2001;127:123–133. doi: 10.1016/s0165-3806(01)00126-2. [DOI] [PubMed] [Google Scholar]
- Rivkin MJ. Hypoxic-ischemic brain injury in the term newborn. Neuropathology, clinical aspects, and neuroimaging. Clin Perinatol. 1997;24:607–625. [PubMed] [Google Scholar]
- Roland EH, Poskitt K, Rodriguez E, Lupton BA, Hill A. Perinatal hypoxic-ischemic thalamic injury: clinical features and neuroimaging. Ann Neurol. 1998;44:161–166. doi: 10.1002/ana.410440205. [DOI] [PubMed] [Google Scholar]
- Rorke LB. Anatomical features of the developing brain implicated in pathonogenesis of hupoxic-ischemic injury. Brain Pathology. 1992;2:211–221. doi: 10.1111/j.1750-3639.1992.tb00694.x. [DOI] [PubMed] [Google Scholar]
- Rorke LB. Pathology of Perinatal Brain Injury. Raven Press; New York: 1998. [Google Scholar]
- Saliba RM, Annegers JF, Waller DK, Tyson JE, Mizrahi EM. Incidence of neonatal seizures in Harris County, Texas, 1992-1994. Am J Epidemiol. 1999;150:763–769. doi: 10.1093/oxfordjournals.aje.a010079. [DOI] [PubMed] [Google Scholar]
- Samuelsen GB, Larsen KB, Bogdanovic N, Laursen H, Graem N, Larsen JF, Pakkenberg B. The changing number of cells in the human fetal forebrain and its subdivisions: a stereological analysis. Cereb Cortex. 2003;13:115–122. doi: 10.1093/cercor/13.2.115. [DOI] [PubMed] [Google Scholar]
- Sanchez RM, Dai W, Levada RE, Lippman JJ, Jensen FE. AMPA/kainate receptor-mediated downregulation of GABAergic synaptic transmission by calcineurin after seizures in the developing rat brain. J Neurosci. 2005;25:3442–3451. doi: 10.1523/JNEUROSCI.0204-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez RM, Koh S, Rio C, Wang C, Lamperti ED, Sharma D, Corfas G, Jensen FE. Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia-induced seizures. J Neurosci. 2001;21:8154–8163. doi: 10.1523/JNEUROSCI.21-20-08154.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnat HB, Nochlin D, Born DE. Neuronal nuclear antigen (NeuN): a marker of neuronal maturation in early human fetal nervous system. Brain Dev. 1998;20:88–94. doi: 10.1016/s0387-7604(97)00111-3. [DOI] [PubMed] [Google Scholar]
- Silverstein FS, Naik B, Simpson J. Hypoxia-ischemia stimulates hippocampal glutamate efflux in perinatal rat brain: an in vivo microdialysis study. Pediatr Res. 1991;30:587–590. doi: 10.1203/00006450-199112000-00021. [DOI] [PubMed] [Google Scholar]
- Spreafico R, Tassi L, Colombo N, Bramerio M, Galli C, Garbelli R, Ferrario A, Lo RG, Munari C. Inhibitory circuits in human dysplastic tissue. Epilepsia. 2000;41(Suppl 6):S168–S173. doi: 10.1111/j.1528-1157.2000.tb01576.x. [DOI] [PubMed] [Google Scholar]
- Talos DM, Fishman RE, Park HK, Folkerth RD, Follett PL, Volpe JJ, Jensen FE. Developmental regulation of AMPA receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury: Part I. Rodent cerebral white matter and cortex. J Comp Neurol. 2005 doi: 10.1002/cne.20972. Submittet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towfighi J, Mauger D, Vannucci RC, Vannucci SJ. Influence of age on the cerebral lesions in an immature rat model of cerebral hypoxia-ischemia: a light microscopic study. Brain Research Developmental Brain Research. 1997;100:149–160. doi: 10.1016/s0165-3806(97)00036-9. [DOI] [PubMed] [Google Scholar]
- Tsuji M, Saul JP, du PA, Eichenwald E, Sobh J, Crocker R, Volpe JJ. Cerebral intravascular oxygenation correlates with mean arterial pressure in critically ill premature infants. Pediatrics. 2000;106:625–632. doi: 10.1542/peds.106.4.625. [DOI] [PubMed] [Google Scholar]
- Ulfig N, Nickel J, Bohl J. Monoclonal antibodies SMI 311 and SMI 312 as tools to investigate the maturation of nerve cells and axonal patterns in human fetal brain. Cell Tissue Res. 1998;291:433–443. doi: 10.1007/s004410051013. [DOI] [PubMed] [Google Scholar]
- Vannucci RC, Brucklacher RM, Vannucci SJ. CSF glutamate during hypoxia-ischemia in the immature rat brain. Brain Research. 1999;118:147–151. doi: 10.1016/s0165-3806(99)00142-x. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Neurology of the Newborn. Saunders; Philadelphia: 2001. [Google Scholar]
- Williams CE, Gunn AJ, Synek B, Gluckman PD. Delayed seizures occurring with hypoxic-ischemic encephalopathy in the fetal sheep. Pediatr Res. 1990;27:561–565. doi: 10.1203/00006450-199006000-00004. [DOI] [PubMed] [Google Scholar]
- Wu YW. Systematic review of chorioamnionitis and cerebral palsy. Ment Retard Dev Disabil Res Rev. 2002;8:25–29. doi: 10.1002/mrdd.10003. [DOI] [PubMed] [Google Scholar]
- Yamaya Y, Yoshioka A, Saiki S, Yuki N, Hirose G, Pleasure D. Type-2 astrocyte-like cells are more resistant than oligodendrocyte-like cells against non-N-methyl-D-aspartate glutamate receptor-mediated excitotoxicity. J Neurosci Res. 2002;70:588–598. doi: 10.1002/jnr.10425. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Matsumoto N, Tsuchiya R, Morita M, Miyakawa H, Kudo Y. Expression of group I metabotropic glutamate receptors in rat hippocampal cells in culture and their characterization by intracellular calcium ion dynamics. J Pharmacol Sci. 2003;92:245–251. doi: 10.1254/jphs.92.245. [DOI] [PubMed] [Google Scholar]
- Yuan X, Eisen AM, McBain CJ, Gallo V. A role for glutamate and its receptors in the regulation of oligodendrocyte development in cerebellar tissue slices. Development. 1998;125:2901–2914. doi: 10.1242/dev.125.15.2901. [DOI] [PubMed] [Google Scholar]
- Zur NR, Deitmer JW. The Role of Metabotropic Glutamate Receptors for the Generation of Calcium Oscillations in Rat Hippocampal Astrocytes In Situ. Cereb Cortex. 2005 doi: 10.1093/cercor/bhj013. [DOI] [PubMed] [Google Scholar]