

Abstract
Objective
Mutations in the Progranulin gene (PGRN) recently have been discovered to be associated with frontotemporal dementia (FTD) linked to 17q21 without identified MAPT mutations. The range of mutations of PGRN that can result in the FTD phenotype and the clinical presentation of patients with PGRN mutations have yet to be determined.
Methods
In this study, we examined 84 FTD patients from families not known previously to have illness linked to chromosome 17 for identified PGRN and MAPT mutations and sequenced the coding exons and the flanking intronic regions of PGRN. We compared the prevalence, clinical characteristics, magnetic resonance imaging and 18-fluoro-deoxyglucose positron emission tomography results, and neuropsychological testing of patients with the PGRN R493X mutation with those patients without identified PGRN mutations.
Results
We discovered a new PGRN mutation (R493X) resulting in a stop codon in two patients. This was the only PGRN mutation identified in our sample. The patients with the PGRN R493X mutation had a rapid illness course and had predominant right-sided atrophy and hypometabolism on magnetic resonance imaging and 18-fluoro-deoxyglucose positron emission tomography. The affected father of one of the patients with the PGRN R493X mutation showed frontal and temporal atrophy without neurofibrillary tangles on neuropathological examination.
Interpretation
Known PGRN and MAPT mutations were rare and of similar prevalence in our sample (2 compared with 1/84). The patients with the PGRN R493X mutation had a clinical presentation comparable with other behavior-predominant FTD patients. The neuropathology of an affected family member of a patient with the PGRN R493X mutation appears not to be Alzheimer’s disease.
Frontotemporal dementia (FTD) is an umbrella term for a set of progressive disorders affecting predominantly the frontal and/or the anterior temporal lobes of the brain. FTD usually presents clinically with behavior or language changes, or both, and is associated with atrophy and decreased metabolism of the frontal and anterior temporal lobes on imaging.1,2 The mean age of onset for FTD is in the mid-50s.3,4 It is the second most common cause of dementia in people younger than 65 (after Alzheimer’s disease), comprising approximately one third of these cases.4,5 Between 35 and 50% of cases are reported to have a family history of FTD.4–10 In the majority of these cases, the illness is inherited in a manner consistent with an autosomal dominant mechanism.5,6,9 Fourteen percent of FTD patients have clinical motor neuron disease (MND),11 and up to about 40% of patients with behavioral variant FTD show MND-type inclusions.12
Several mutations have been associated with rare cases of FTD.13 Mutations in the MAPT gene at 17q21 coding for the tau protein are the most commonly associated with FTD.14–18 More than 30 MAPT mutations associated with the development of FTD have been discovered (a list of these mutations is available online at: http://www.molgen.ua.ac.be/ADMutations/default.cfm?MT=1&ML=1&Page=MutByQuery&Query=tblContexts.GeneSymbol%20In%20('MAPT')&Selection=Gene%20In%20(MAPT)&CFID=136313&CFTOKEN=48500543). These mutations are associated with intraneuronal and/or glial deposits that are immunopositive for tau on neuropathology.6 The reported prevalence of MAPT mutations in patients with FTD has ranged from 0 to 18%.4,6,8,19–22 In several families with inherited FTD, the illness has been linked to 17q21, but no MAPT mutations have been identified, and the cases are not associated with tau-positive inclusions.6,23,24 In a recent report, cases of FTD linked to 17q21 with a DR2-DR8 haplotype were seven times more common than FTD cases with identified MAPT mutations (7 vs 1% of the sample).6 On neuropathology, one of these cases had frontotemporal lobar degeneration (FTLD) with rare tau-negative, ubiquitin-positive inclusions.6
Recent reports show that, although it appears to be an unlikely coincidence, mutations in another gene at 17q21 (Progranulin or PGRN) are also associated with FTD.14–16 PGRN is a 593-amino acid (68.5kDa) multifunctional growth factor.25 Mutations in PGRN that likely result in premature termination and null alleles segregated with illness in several large families and were absent in 550 control patients.14 RNA extracted from patient cells consisted almost entirely of wild-type RNA with little of the mutant RNA detected and with a reduction in wild-type RNA, suggesting that the mutations result in degradation of the mutant RNA and a loss of functional PGRN (haploinsufficiency).14
In the original report,14 eight mutations predicted to cause premature termination of the coding sequence of PGRN were detected in patients with familial FTD without MAPT mutations. In a series of familial FTD patients, PGRN mutations were 3.5 times more frequent than MAPT mutations.26 In this study, we serially evaluated 84 FTD patients for known MAPT and PGRN mutations and sequenced the coding exons and the flanking intronic regions of PGRN. We compared the PGRN mutation carriers with the rest of the patients for demographic characteristics, clinical presentation, imaging, neuropsychological testing, and pathology.
Patients and Methods
Patients
The 84 FTD patients in this study are characterized in Table 1. These patients were referred by outside providers to the Cognitive Neuroscience Section of the National Institute of Neurological Disorders and Stroke (NINDS) with a diagnosis of FTD and enrolled in an ongoing study to further characterize patients with FTD. They were accepted into the research program if the diagnosis by an outside provider of FTD was judged sufficient by the researchers and if there was no other contraindication to participation in the research (eg, too advanced to be able to tolerate imaging or neuropsychological testing). They were not selected to be enriched for family history (or to be from families with illness linked to chromosome 17), clinical presentation, or other characteristics. The patients came to the National Institutes of Health (NIH) for a single visit. They were evaluated clinically, and the diagnosis was confirmed according to published criteria.1 As part of the clinical evaluation, an extensive history and neurological examination was performed by a neurologist. All neuropsychological testing was performed by trained testers. A full family history was taken and a pedigree made for each patient. We required all subjects to have an assigned research Durable Power of Attorney before admission to the protocol, and the assigned individuals gave written informed consent. All aspects of the study and the consent procedure were approved by the NINDS institutional review board. Caregivers of patients found to have PGRN mutations were contacted to update the patient profile and family history.
Table 1.
Characteristics of Frontotemporal Dementia Patients with and without PGRN Mutations in Our Patient Population of 84 Patients with Frontotemporal Dementia Compared with Other Patient Groups
| Characteristics | NINDS FTD Patients |
Other Patient Groups |
||
|---|---|---|---|---|
| No PGRN Mutation Found | PGRN R493X Mutation | van der Zee and Colleagues6 | Baker and Colleagues14 | |
| Patients, N | 82 | 2 | 98 unrelated patients screened, 7 linked to 17q21 without identified MAPT mutation | Once PGRN mutation identified in 1 family, individuals from 41 additional families with familial FTD without known MAPT mutations screened for PGRN mutations; 7 additional mutations found in 8 families |
| Type of mutation/variation | No PGRN mutations identified; 1 MAPT mutation identified | 2 R493X predicted to cause early termination of the coding sequence of PGRN | See ref 26 | See ref 14; all mutations predicted to cause premature termination of the coding sequence of PGRN |
| Behavior- vs language- predominant subtype | 60 behavioral, 20 language, 2 unknown | All behavioral | Of 11 linked patients, 4 initially diagnosed with language predominant, 7 with behavior predominant | Not reported |
| Mean age of onset, yr | 56 | 56 | 63 | 57 (in 41 screened families) |
| Sex, M/F | 49/33 | 1/1 | 6/5 | Not reported |
| Family history of FTD | 14/82 | 2/3 | 7 | All families had a family history of FTD |
| Family history of MND | 1/82 | None | None noted | 10 probands had clinical MND |
| Family history of parkinsonism | 2/82 | 2/3 | None noted | 4 probands had parkinsonism |
| MND symptoms | 5 with MND | None | None noted | 10 probands had clinical MND |
| Parkinsonian symptoms | — | 1/3 | None noted | 4 probands had parkinsonism |
| Imaging | — | 2 patients with right-sided predominance of atrophy and reduced metabolism | Atrophy and/or hypo-perfusion lateralized to the left side in 7 of 11 patients | Not reported |
| Pathology | 6 patients have come to autopsy: 3 showed FTLD with ubiquitin- immunopositive inclusions, 2 had Pick’s disease, 1 had dementia lacking distinctive histopathology | Affected father of 1 patient with PGRN R493X mutation showed frontal and temporal atrophy without neurofibrillary tangles or neuritic plaques | One case FTLD tau- negative, rare ubiquitin-positive inclusions | All 41 families included individuals with FTLD with ubiquitin-positive inclusions; 11 families had cases with neuronal intranuclear inclusions |
NINDS = National Institute of Neurological Disorders and Stroke; FTD = frontotemporal dementia; MND = motor neuron disease; FTLD = frontotemporal lobar degeneration.
Family histories were evaluated using a consistent method.10 Pedigrees were classified as a positive family history of FTD, amyotrophic lateral sclerosis, or Parkinson’s disease if they met one of the following criteria: (1) at least three members in two generations affected with the illness (FTD, amyotrophic lateral sclerosis, or Parkinson’s disease), with one person being a first-degree relative of the other two; (2) three relatives with the illness and the first criterion not met; (3) a single first-degree relative with dementia, amyotrophic lateral sclerosis, or Parkinson’s disease.
One additional FTD patient provided a blood sample and was found to have the PGRN R493X mutation, but had not been evaluated at NIH because she was unable to travel (Patient 3). We telephoned the caregiver and obtained clinical and family information, but we did not perform a physical examination, imaging, or neuropsychological testing on this patient. Her clinical presentation and family history is discussed in this article, but she is not included in the known PGRN mutation prevalence determination.
All of the patients with a discovered PGRN mutation were alive at the time of this study. However, the affected father of one of the patients discovered to have a PGRN mutation (Patient 1) had died in 1993. We obtained the report and neuropathology slides from the autopsy that was performed at the time of his death.
Sequencing
DNA was prepared from whole blood using standard protocols. The coding exons and the flanking intronic sequences were amplified and sequenced as described in the initial report of PGRN mutations14 using genomic DNA. MAPT exons 1, 9, 10, 11, 12, and 13 and their surrounding intronic borders were sequenced in patients, as described previously.18,27 The polymerase chain reaction (PCR) products were purified with Multiscreen plates (Millipore, Billerica, CA). The purified PCR amplicons were sequenced from both directions on automated sequencers (ABI 3700; Applied Biosystems, Foster City, CA) using ABI Prism BigDye Terminator Cycle sequencing kit, as recommended by the manufacturer.
Linkage Analysis
The microsatellite markers D17S1814, D17S1299, D17S951, D17S934, D17S950, TAU PROM (from centromere to telomere) were genotyped using the fluorescent-labeled primers and standard PCR protocols. The fluorescent-labeled PCR products were mixed with Form-amide and ABI LIZ 500 size standard. The samples subsequently were loaded on ABI 3100 Genetic Analyzer. The results were analyzed using Genotyper program (Applied Biosystems).
Neuropsychological Testing
The patients received clinical neuropsychological testing that included the measures: general cognition (the Wechsler Adult Intelligence Scale [3rd edition]28 and the Mattis Dementia Rating Scale 229), language (the Boston Naming Test [2nd edition]30 and the Token test31), memory (the Wechsler Memory Scale [3rd edition]32), executive function (the Delis Kaplan Executive Function System33), and behavioral symptoms (the Neurobehavioral Rating Scale34 and the Beck Depression Inventory-235).
Neuropathology
An autopsy had been performed on the affected father of one of the patients discovered in this study to have a PGRN mutation. The brain was harvested after embalming of the entire body and fixed in formalin for 6 weeks at the time of the patient’s death in 1993. The brain was grossly and microscopically examined at that time, but not stained for tau or ubiquitin. At that time, neuropathology showed frontal and temporal atrophy with neither neurofibrillary tangles nor Alzheimer’s plaques. Slides from this initial autopsy were attempted to be restained for ubiquitin in 2006 by the Neuropathology Core of the Indiana Alzheimer Disease Center. Blocks of tissue were obtained from the cortex (six samples) and brainstem (one sample). Histological sections were originally stained with hematoxylin and eosin, Bielschowsky, and Congo red. The Neuropathology Core of the Indiana Alzheimer Disease Center received seven hematoxylin and eosin–stained sections. After removing the cover slips from four of these sections, immunohistochemistry for ubiquitin was conducted. Diffuse immunopositivity was detected in some cell processes and in the cytoplasm of a few neurons. Clearly distinguishable intracytoplasmic or intranuclear ubiquitin-immunopositive inclusions were not seen; however, it should be noted that the quality of the material was not optimal for tissue fixation and preservation.
Results
Prevalence of Known PGRN Mutations
Of our group of 84 FTD patients, 2 were found to have a mutation of the PGRN gene (R493X). Patient 3 has the same PGRN mutation. The mutation changes the coding from an amino acid to a stop codon at position 493 (Fig, A). This mutation has not been previously reported. One additional patient was found to have a P301S MAPT mutation. None of the patients was found to have the mutations described in the original report of PGRN mutations.14
Fig.
PGRN mutation.
Linkage Analysis and Common Haplotype
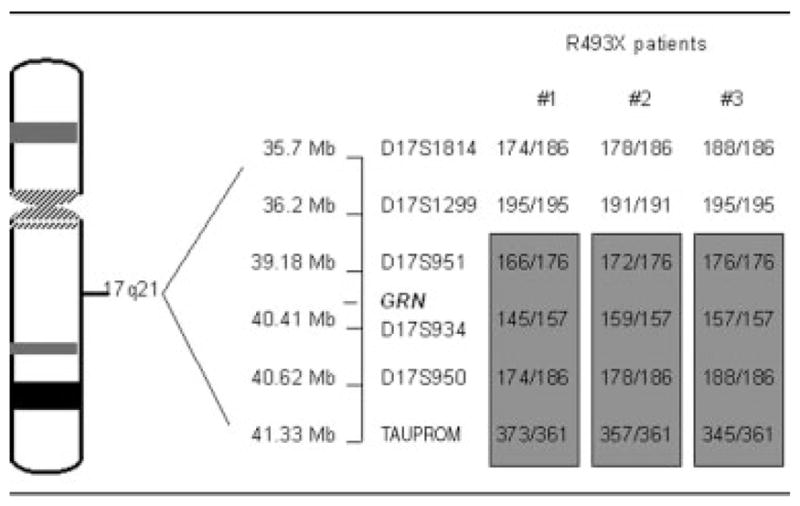
We genotyped the three apparently unrelated patients with the length polymorphic markers (see Patients and Methods) to examine the ancestral effect in these mutation carriers. The patients carry a common haplotype spanning a region of chromosome 17, D17S951 at the centromeric end and TAU PROM marker at the telomeric end, encompassing the PGRN gene. Patients 1 and 2 showed complete linkage for all the markers. Patient 3 (FTD 81) had consistent data for all markers except D17S1299, suggesting a recombination event. The PGRN gene is located between the markers D17S951 and D17S934, which are on the shared haplotype between the patients. These data suggest that the mutation originates from a common, probably ancestral, haplotype.
Neuropsychological Data
For the purposes of comparing the neuropsychological data among our patients, two control FTD patients without PGRN mutations were matched to each patient with a PGRN mutation by age of onset, duration of illness at time of testing, and education (see supplementary material online).
Cases
PATIENT 1
At 51 years old, Patient 1 began exhibiting “childish” behavior including playfully pushing people. He had increased food intake. His alcohol intake increased, and he was arrested twice for driving while intoxicated. His emotional reaction to events was blunted and inappropriate. His father had frontal lobe dementia with parkinsonism and died at the age of 76 (see Fig, B). The patient’s father had an autopsy at the time of his death in 1993, which showed frontal and temporal atrophy with neither neurofibrillary tangles nor Alzheimer’s plaques. The patient was seen at NINDS 1 year after the onset of his symptoms. At that time, his neurological examination was normal with the exception of a slow gait. His magnetic resonance imaging showed 9 to 10 small cavernous angiomas and atrophy of the frontal, temporal, and parietal lobes, which were worse on the right side (see Fig, C). His 18-fluoro-deoxyglucose positron emission tomography results showed marked reductions in glucose metabolism in the frontal, parietal, and temporal lobes, with the right side more affected than the left (see Fig, D). In the 2 years since he was seen at NINDS, he has had decreased verbal output and now rarely speaks.
PATIENT 2
Patient 2 first started having symptoms at 61 years old, including rudeness and making mistakes when performing complex activities. She developed apathy, emotional blunting, and had increased food consumption with weight gain. No one else in her family was diagnosed with FTD, but her father had “memory problems” (see Fig, E). She was seen at NINDS 2.5 years into the course of her illness. Her neurological examination showed a slow gait, excessive cooperation with passive movement (mitgehen), and a bilateral grasp reflex. Her magnetic resonance imaging showed prominent atrophy in both frontal and temporal lobes, with the right side more affected than the left. She had leukomalacia of the white matter of both cerebral hemispheres observed on fluid-attenuated inversion recovery (see Fig, F). On 18-fluoro-deoxyglucose positron emission tomography, she had marked reductions of glucose metabolism in the frontal, temporal, and parietal cortices of the right hemisphere and the left frontal cortex (see Fig, G). She is currently alive at the age of 65, and her symptoms have progressively worsened.
PATIENT 3
The patient started having symptoms at 45 years old. Her initial symptoms included bizarre behavior such as watching children play at a nearby school for several hours a day, apathy, executive dysfunction, and increased food intake with carbohydrate craving. Her symptoms worsened with decreased speech output, bowel and bladder incontinence, and the development of a parkinsonian gait and tremor. She has two siblings with similar symptoms (see Fig, H). She is currently alive at the age of 47, requires assistance with her activities of daily living, and is mostly nonverbal.
Discussion
Prevalence
The prevalence of both known MAPT and PGRN mutations are sufficiently low in our samples, and thus statistical comparison is not meaningful. However, the number of PGRN mutations is similar to the prevalence of MAPT mutations (2 compared with 1/84). Because the association between PGRN and FTD was discovered recently, there are likely more PGRN mutations that can result in FTD that have not yet been identified. Note that the positive family history for FTD (17%) and MAPT mutations in our patient population is lower than that reported in the literature in most,4–9 but not all,6 groups. It may reflect a referral bias that patients with family histories of FTD are more likely to be retained in local academic centers and not referred to the NIH; or, FTD patients with positive family histories and MAPT mutations might have been overrepresented at the academic medical centers where they have been studied compared with the community.
Age of Onset
The mean age of onset in the patients in our sample with PGRN mutations is similar to patients without PGRN mutations (52 vs 56 years old, including Patient 3). This agrees with previous reports.6,14,26 The identified PGRN mutations appear to cause haploinsufficiency (see earlier). Thus, the age of symptom development and/or the rate of disease progression could depend on the levels of PGRN protein produced by the functional allele.
Family History
Of our three patients with PGRN mutations, two have a clear family history of FTD. The third (Patient 2) has four unaffected siblings, but her father had “memory problems” in his 70s and may have had the R493X mutation. This mutation may have incomplete penetrance in this family. In the family of Patient 1, the index patient does not have parkinsonism, with the exception of slow gait, whereas his father had parkinsonism, suggesting variable expression.
Clinical Presentation
All of our patients with PGRN mutations presented with predominantly behavioral symptoms. This is in contrast with the high rate of language-predominant FTD noted in patients linked to 17q21 without MAPT mutations previously reported (see Table 1).6 The patients from our sample with PGRN mutations display a symptom profile typical of behavior-predominant FTD (inappropriate social behaviors and affect, increased consumption of sweet foods, impaired executive functions). Parkinsonian symptoms occur in two of the three families. Larger numbers of patients with PGRN mutations will be required to determine the incidence of motor symptoms in FTD patients with PGRN mutations.
Imaging
Both of the patients with PGRN mutations that we imaged showed frontal and temporal and, to a lesser extent, parietal atrophy and hypometabolism with a right-sided predominance. This corresponds with the behavioral predominance of symptoms observed.36 The report of patients linked to 17q21 without MAPT mutations showed a left-sided predominance of atrophy on imaging consistent with their report of a high rate of language symptoms in their patients.6
Neuropsychology
Given the small number of patients with PGRN mutations, we did not examine the neuropsychological data statistically. The patients with PGRN mutations scored similarly to our control group of FTD patients on measures of overall cognitive symptoms (Mattis Dementia Rating Scale 2 and the Wechsler Adult Intelligence Scale [3rd edition]). Although we attempted to match the patients with PGRN mutations and the control patients on symptom duration, there is a discrepancy between the patients with PGRN mutations (1.8 years) and the control patients (3.2 years). Thus, although the two groups are at similar levels of overall cognitive dysfunction, the patients with PGRN mutations reached this point in half the time of the FTD control patients. A relatively rapid symptom course for patients with the R493X PGRN mutation is supported by Patient 3, although we do not have formal neuropsychological testing on her; in 2 years since symptom onset, she has progressed to severe dementia.
The patients with PGRN mutations scored a little higher on language tests (the Boston Naming and Token tests) than the control patients. This reflects the behavioral predominance of symptoms in the patients with PGRN mutations. On memory testing, the patients with PGRN mutations scored consistently higher than the control patients with the exception of working memory, which is especially dependent on frontal lobe function, suggesting a relative preservation of posterior memory structures in the patients with the R493X PGRN mutation. The two patient groups scored similarly on other measures of executive function (the Delis Kaplan battery).33
Pathology
The affected father of Patient 1 died and had an autopsy performed that showed atrophy of the frontal and temporal lobes without neurofibrillary tangles or neuritic plaques on microscopy. In the original report associating PGRN with FTD, the authors found that PGRN mutations result in FTLD with ubiquitin-positive inclusions (FTLD-MND).14 Our neuropathology findings are consistent with FTLD-MND, but we cannot confirm this neuropathological diagnosis because of the poor quality of the stored slides.
Conclusion
We describe a novel PGRN mutation associated with FTD (R493X) discovered in patients from families not previously known to have illness linked to chromosome 17. In our sample, none of the patients had previously identified PGRN mutations. Known PGRN and MAPT mutations are rare causes of FTD in our patients. The patients with the R493X PGRN mutation phenotypically present similarly to other behavioral-predominant FTD patients. Compared with patients without known PGRN mutations, patients with the R493X PGRN mutation appear to have a relatively rapid illness course and a right-sided predominance of atrophy and hypometabolism on imaging. The affected family member of a patient with the PGRN R493X mutation had atrophy of the frontal and temporal lobes without neurofibrillary tangles neuritic plaques on neuropathology. Future directions for research on this topic include the search for other PGRN mutations associated with FTD; elucidation of the mechanisms with which PGRN mutations result in FTD; and collection of epidemiological, clinical, and pathological data on the relation among PGRN mutations, behavioral and motor symptoms (especially symptoms of MND), and neuropathology.
Acknowledgments
This research was supported by the Intramural Research Programs of the NIH (National Institute of Neurological Disorders and Stroke, E.D.H., J.G., E.M.V., P.P., M.C.T., and National Institute on Aging, PHS P30 AG10133, B.G., S.S.), and the Department of Neurological and Behavioral Sciences, University of Siena, Siena, Italy (S.S.).
We thank A. Cavanagh and K. Detucci for their patient care, testing, and data preparation, and the NINDS nurses for their patient care. We thank T. E. Huang for his assistance obtaining neuropathological material.
References
- 1.Lund/Manchester. Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. J Neurol Neurosurg Psychiatry. 1994;57:416–418. doi: 10.1136/jnnp.57.4.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bozeat S, Gregory CA, Ralph MA, Hodges JR. Which neuro-psychiatric and behavioural features distinguish frontal and temporal variants of frontotemporal dementia from Alzheimer’s disease? J Neurol Neurosurg Psychiatry. 2000;69:178–186. doi: 10.1136/jnnp.69.2.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- 4.Rosso SM, Donker Kaat L, Baks T, et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126(pt 9):2016–2022. doi: 10.1093/brain/awg204. [DOI] [PubMed] [Google Scholar]
- 5.Neary D, Snowden J, Mann D. Frontotemporal dementia. Lancet Neurol. 2005;4:771–780. doi: 10.1016/S1474-4422(05)70223-4. [DOI] [PubMed] [Google Scholar]
- 6.van der Zee J, Rademakers R, Engelborghs S, et al. A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21-linked tau-negative FTLD. Brain. 2006;129(pt 4):841–852. doi: 10.1093/brain/awl029. [DOI] [PubMed] [Google Scholar]
- 7.Stevens M, van Duijn CM, Kamphorst W, et al. Familial aggregation in frontotemporal dementia. Neurology. 1998;50:1541–1545. doi: 10.1212/wnl.50.6.1541. [DOI] [PubMed] [Google Scholar]
- 8.Poorkaj P, Grossman M, Steinbart E, et al. Frequency of tau gene mutations in familial and sporadic cases of non-Alzheimer dementia. Arch Neurol. 2001;58:383–387. doi: 10.1001/archneur.58.3.383. [DOI] [PubMed] [Google Scholar]
- 9.Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol. 1999;56:817–822. doi: 10.1001/archneur.56.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldman JS, Farmer JM, Wood EM, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology. 2005;65:1817–1819. doi: 10.1212/01.wnl.0000187068.92184.63. [DOI] [PubMed] [Google Scholar]
- 11.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–1079. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 12.Kertesz A, McMonagle P, Blair M, et al. The evolution and pathology of frontotemporal dementia. Brain. 2005;128(pt 9):1996–2005. doi: 10.1093/brain/awh598. [DOI] [PubMed] [Google Scholar]
- 13.Hardy J, Momeni P, Traynor BJ. Frontal temporal dementia: dissecting the aetiology and pathogenesis. Brain. 2006;129(pt 4):830–831. doi: 10.1093/brain/awl035. [DOI] [PubMed] [Google Scholar]
- 14.Baker M, Mackenzie IR, Pickering-Brown S, et al. Mutations in Progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006 doi: 10.1038/nature05016. (in press) [DOI] [PubMed] [Google Scholar]
- 15.Rowland LP. Frontotemporal dementia, chromosome 17, and progranulin. Ann Neurol. 2006;60:275–277. doi: 10.1002/ana.20962. [DOI] [PubMed] [Google Scholar]
- 16.Mukherjee O, Pastor PN, Cairns NJ, et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006;60:314–322. doi: 10.1002/ana.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat. 2004;24:277–295. doi: 10.1002/humu.20086. [DOI] [PubMed] [Google Scholar]
- 18.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 19.Houlden H, Baker M, Adamson J, et al. Frequency of tau mutations in three series of non-Alzheimer’s degenerative dementia. Ann Neurol. 1999;46:243–248. doi: 10.1002/1531-8249(199908)46:2<243::aid-ana14>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 20.Rizzu P, Van Swieten JC, Joosse M, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet. 1999;64:414–421. doi: 10.1086/302256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fabre SF, Forsell C, Viitanen M, et al. Clinic-based cases with frontotemporal dementia show increased cerebrospinal fluid tau and high apolipoprotein E epsilon4 frequency, but no tau gene mutations. Exp Neurol. 2001;168:413–418. doi: 10.1006/exnr.2000.7613. [DOI] [PubMed] [Google Scholar]
- 22.Kowalska A, Asada T, Arima K, et al. Genetic analysis in patients with familial and sporadic frontotemporal dementia: two tau mutations in only familial cases and no association with apolipoprotein epsilon4. Dement Geriatr Cogn Disord. 2001;12:387–392. doi: 10.1159/000051285. [DOI] [PubMed] [Google Scholar]
- 23.Rademakers R, Cruts M, Dermaut B, et al. Tau negative frontal lobe dementia at 17q21: significant finemapping of the candidate region to a 4.8 cM interval. Mol Psychiatry. 2002;7:1064–1074. doi: 10.1038/sj.mp.4001198. [DOI] [PubMed] [Google Scholar]
- 24.Rosso SM, Kamphorst W, de Graaf B, et al. Familial frontotemporal dementia with ubiquitin-positive inclusions is linked to chromosome 17q21–22. Brain. 2001;124(pt 10):1948–1957. doi: 10.1093/brain/124.10.1948. [DOI] [PubMed] [Google Scholar]
- 25.He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med. 2003;81:600–612. doi: 10.1007/s00109-003-0474-3. [DOI] [PubMed] [Google Scholar]
- 26.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006 doi: 10.1038/nature05017. (in press) [DOI] [PubMed] [Google Scholar]
- 27.Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 28.Wechsler D. Administration and scoring manual. San Antonio, TX: The Psychological Corporation; 1997. WAIS-III. [Google Scholar]
- 29.Mattis S. Mental Status examination for organic mental syndrome in the elderly patient. In: Bellack L, Karusu TB, editors. Geriatric psychiatry. New York: Grune & Stratton; 1976. pp. 77–121. [Google Scholar]
- 30.Kaplan H, Goodglass H, Weintraub S. Boston Naming Test. Philadelphia: Lea & Febiger; 1983. [Google Scholar]
- 31.De Renzi E, Vignolo LA. The token test: a sensitive test to detect receptive disturbances in aphasics. Brain. 1962;85:665–678. doi: 10.1093/brain/85.4.665. [DOI] [PubMed] [Google Scholar]
- 32.Wechsler D. Manual for the Wechsler Memory Scale–Revised. San Antonio, TX: Psychological Corporation; 1997. [Google Scholar]
- 33.Delis DC, Kaplan E, Kramer J. Delis Kaplan Executive Function System. San Antonio, TX: The Psychological Corporation; 2001. [Google Scholar]
- 34.Levin HS, High WM, Goethe KE, et al. The neurobehavioural rating scale: assessment of the behavioural sequelae of head injury by the clinician. J Neurol Neurosurg Psychiatry. 1987;50:183–193. doi: 10.1136/jnnp.50.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beck AT, Steer RA, Brown GK. Beck Depression Inventory-II. New York: Psychological Corporation; 1996. [Google Scholar]
- 36.Rosen HJ, Allison SC, Schauer GF, et al. Neuroanatomical correlates of behavioural disorders in dementia. Brain. 2005;128(pt 11):2612–2625. doi: 10.1093/brain/awh628. [DOI] [PMC free article] [PubMed] [Google Scholar]