

Abstract
Background
The genetic diagnosis of mental retardation (MR) is difficult to establish and at present many cases remain undiagnosed and unexplained. Standard karyotyping has been used as one of the routine techniques for the last decades. The implementation of Array Comparative Genomic Hybridization (array-CGH) has enabled the analysis of copy number variants (CNVs) with high resolution. Major cohort studies attribute 11% of patients with unexplained mental retardation to clinically significant CNVs. Here we report the use of array-CGH for the first time in a Greek cohort. A total of 82 children of Greek origin with mean age 4.9 years were analysed in the present study. Patients with visible cytogenetic abnormalities ascertained by standard karyotyping as well as those with subtelomeric abnormalities determined by Multiplex Ligation-dependent Probe Amplification (MLPA) or subtelomeric FISH had been excluded.
Results
Fourteen CNVs were detected in the studied patients. In nine patients (11%) the chromosomal aberrations were inherited from one of the parents. One patients showed two duplications, a 550 kb duplication in 3p14.1 inherited from the father and a ~1.1 Mb duplication in (22)(q13.1q13.2) inherited from the mother. Although both parents were phenotypically normal, it cannot be excluded that the dual duplication is causative for the patient's clinical profile including dysmorphic features and severe developmental delay. Furthermore, three de novo clinically significant CNVs were detected (3.7%). There was a ~6 Mb triplication of 18q21.1 in a girl 5 years of age with moderate MR and mild dysmorphic features and a ~4.8 Mb duplication at (10)(q11.1q11.21) in a 2 years old boy with severe MR, multiple congenital anomalies, severe central hypotonia, and ataxia. Finally, in a 3 year-old girl with microcephaly and severe hypotonia a deletion in (2)(q31.2q31.3) of about ~3.9 Mb was discovered. All CNVs were confirmed by Fluorescence in situ hybridization (FISH). For the remaining 9 patients the detected CNVs (inherited duplications or deletions of 80 kb to 800 kb in size) were probably not associated with the clinical findings.
Conclusions
Genomic microarrays have within the recent years proven to be a highly useful tool in the investigation of unexplained MR. The cohorts reported so far agree on an around 11% diagnostic yield of clinically significant CNVs in patients with unexplained MR. Various publicly available databases have been created for the interpretation of identified CNVs and parents are analyzed in case a rare CNV is identified in the child. We have conducted a study of Greek patients with unexplained MR and confirmed the high diagnostic value of the previous studies. It is important that the technique becomes available also in less developed countries when the cost of consumables will be reduced.
Background
Mental retardation (MR) is a common disorder for which the genetic diagnosis in many instances is lacking. The detection rate of chromosomal abnormalities in patients with MR and dysmorphic features has increased due to the improvements of molecular cytogenetic methods. Standard cytogenetic methods cannot detect imbalances smaller than 5-10 Mb and the detection rate of visible chromosomal abnormalities in patients with moderate to severe MR is 3.7% [1]. The development of Fluorescence in situ hybridization (FISH) probes for the subtelomeric regions has led to the identification of cryptic unbalanced rearrangements in an additional 2.5-3% of patients with moderate/severe MR of unknown cause [2-4].
Molecular karyotyping (MK) through array-CGH or Single Nucleotide Polymorphisms array (SNP-array) is rapidly becoming the first tier clinical genetic test for patients with unexplained developmental delay/intellectual disability, autism spectrum disorders, and multiple congenital anomalies. Numerous studies have clearly demonstrated that MK offers a much higher diagnostic yield (15%-20%) for this group of patients in respect to conventional karyotyping with a G-banded karyotype (approximately 3%, excluding Down syndrome and other recognizable chromosomal syndromes), primarily because of its higher sensitivity for submicroscopic deletions and duplications [5].
The widespread application of this technique has lead to the identification of large-scale copy number polymorphisms (CNPs), shown to contribute substantially to genomic variation [6,7], and segmental duplications were found to define hotspots of chromosomal rearrangements [8].
Here we report the first Greek experience from a cohort of 82 children with learning disabilities and dysmorphism, in whom subtelomeric chromosomal abnormalities were excluded by FISH or MLPA techniques. All children presented with various degrees of unexplained MR or learning difficulties (MR/LD) and facial dysmorphism/congenital malformations, suggestive of chromosomal anomalies: a) not associated with congenital brain malformation (CBM) in either CT or MRI brain scan, b) associated with CBM in either CT or MRI brain scan without improvement and c) associated with CBM in either CT or MRI brain scan with improvement.
Results
Eighty-two patients with unexplained MR and presence of features suggestive of a chromosomal anomaly were analyzed by array-CGH. All patients had an apparently normal karyotype when investigated by standard GTG-banding (> 550 band resolution per haploid karyotype). In addition, subtelomeric FISH and MLPA analyses were performed without revealing any rearrangements. Among the 82 patients analyzed, 13 (15.8%) were found to have cryptic chromosomal imbalances: 6 patients with duplications, 5 patients with deletions, one with triplication and one patient with two duplications. Array-CGH results and phenotype of these individuals are given in Table 1. For all 13 patients, array-CGH analysis has been extended to parental samples, so to establish if an aberration was inherited or de novo. In 3 out of the 13 patients the chromosomal rearrangements occurred de novo; these aberrations were classified as causative for the phenotype. The other 11 aberrations which were observed in the remaining of 10 patients, were considered as likely benign since they have not previously been reported and have been inherited from a healthy parent. In total, 3 de novo clinically significant CNVs were identified in 3 out of the 82 patients (3.6%) and 11 abnormalities with uncertain clinical significance were detected in 10 patients (12.2%). All three patients presented with CBM. Two patients were of subgroup c (Background) associated with CBM and improvement and one patient was of subgroup b associated with CBM and no improvement after intervention. The median de Vries score] of all 82 patients was 4.47 (range 0-9).
Table 1.
Clinical information and array-CGH results in Greek children with unexplained mental retardation
| Case/no | Gender | MR | Array-CGH results | Origin | Estimated size | Clinical Features |
|---|---|---|---|---|---|---|
| 1 | F | Moderate | tripl(18)(q21.1) 42,812 Mb-48,558 Mb |
de novo | 6 Mb | maxillary hypoplasia, small jaw, prominent occiput, hypertelorism, epicanthal folds, downward slanting palpebral fissures with sunken eyes and long eyelashes. Brain MRI scan suggested periventricular leukomalacia (PVL). |
| 2 | M | Severe | dup(10)(q11.1q11.21) 46,568 Mb-51,264 Mb |
de novo | 6 Mb | corpus callosum hypoplasia, mild dilatation of subarachnoid areas and frontotemporal atrophy, severe central hypotonia, ataxia, triangular face, enlarged cranium cerebrale, bifid scrotum, cryptorchidism, ulnar deviation of both elbows, deep palmar creases of hands and feet and syndactyly of 2nd and 3rd toes bilaterally. |
| 3 | F | Moderate | del(2)(q31.2q31.3) 178,393 Mb-182,296 Mb |
de novo | 3,9 Mb | severe hypotonia with microcephaly, Brain MRI scan showing dilated lateral ventricles and diminishing white matter at the level of the trigons bilaterally. Speech limited to simple vocalization with lack of meaning. Her overall developmental level was equivalent to 8 months, which corresponds to a developmental quotient (D.Q.) = 30. |
| 4 | M | Severe | del(6)(p21.2) 38,420 Mb-38,554 Mb |
paternal | 135 kb | mild dysmorphic facial features (epicanthial folds, hypertelorism and auricle abnormalities)normal growth parameters severe mental retardation DQ 45, behavioural disorders with autistic features. motor disorder, hypotonia of central origin, brain malformation,(pituitary cyst), no cognitive improvement after intervention |
| 5 | F | Moderate | dup(15)(q13.3) 29,809 Mb-30,298 Mb |
maternal | 0.5 Mb | mild dysmorphic facial and body features, such as hrinolalia with high-pitched voice, epicanthus, myopia, clinodactyly, and wide internipple distance |
| 6 | F | Moderate | dup(20)(p11.21) 25,375 Mb-25,420 Mb |
paternal | 80 kb | microcephaly, cleft palate, somatometric parameters below the 3rd centile, perimembranous ventricular septal defect, hyperopia, prominent forehead, synorphrys, long eyelashes, bulbous nasal tip, smooth philtrum, thin upper lip, hirsutism, and bilateral clinodactyly of the 5th finger |
| 7 | M | Moderate | dup(16)(q22.1) 68,390 Mb-68,534 Mb |
maternal | 150 kb | dysmorphic facial features, motor disorder, epicanthus, hyperopia |
| 8 | M | Moderate | dup(16)(p11.2) 27,741 Mb -27,919 Mb |
paternal | 200 kb | mild microcephaly and dysmorphic facial features, maxillary hypoplasia, epicanthal folds, up-slanting palpebral fissures, long eyelashes and hypertelorism, auricle abnormalities and his mouth characterized by a long philtrum with gothic palate |
| 9 | F | Severe | del(1q)(31.3) 196,153 Mb-196,532 Mb |
maternal | 400 kb | somatometric parameters below the 3rd centile, mild dysmorphic facial features, auricle abnormalities, developmental and motor delay |
| 10 | M | Severe | del(11)(q21) 94,602, Mb-95,086, Mb |
paternal | 500 kb | multiple gestation (triplex-IVF), dysmorphic facial features, squint, flat filtrum, frontal bossing, epicanthus, auricle abnormalities, macrocephaly, fronto-temporal brain atrophy, motor disorder, hypotonia of central origin, severe mental delay, severe behavioral disorders |
| 11 | M | Severe | dup(17)(q25.1) 70,793 Mb-71,106 Mb |
paternal | 300 kb | neonatal hypotonia, macrocephaly, dolicocephaly, mild dilation of subarachnoid area, severe developmental and motor delay, cryptorchidism, mild dysmorphic, facial features, epicanthus, clinodactyly, short neck, large distance between nipples, short vraxionas |
| 12 | M | Moderate | del(1)(p12) 118,688 Mb-119,490 Mb |
paternal | 800 Kb | mild dysmorphic facial features, bulbous nose, short fingers, clinodactyly, upward slanting palpebral fissures, moderate learning difficulties, language delay with phonological problems and stuttering |
| 13 | M | Severe | dup(3)(p14.1) 65,912 Mb-66,462 Mb |
paternal | 550 kb | severe motor disorder (marked spastic tetraplegia), dysmorphic facial features (prominent forehead, low-set ears, epicanthal folds, flat philtrum, and long eyelashes), severe developmental delay as his developmental level was equivalent to 5 months. |
| dup(22)(q13.1q13.2) 38,255 Mb-39,383 Mb |
maternal | 1.1 Mb |
Discussion
Array-CGH has proven to be an important tool to detect submicroscopic chromosomal aberrations. We used DNA oligonucleotides to study 82 patients with normal karyotype in whom a chromosomal abnormality was suspected due to the combination of clinical features. All 82 patients presented with MR or LD, dysmorphic facial features and congenital malformations. In order to better define the clinical features correlated with chromosomal imbalance, we divided the cohort in three subgroups.
a) 24/82 (29.3%) patients with MR/LD, facial dysmorphism, congenital malformations not associated with CBM,
b) 40/82 (48.8%) patients with MR/LD, facial dysmorphism, congenital malformations associated with CBM and no improvement of cognitive skills after intervention,
c)18/82 (21.9%) patients with MR/LD, facial dysmorphism, congenital malformations associated with CBM and improvement of cognitive skills after intervention.
All patients showed a normal G-banded karyotype, and in all of the cases telomere rearrangements had been excluded by FISH or MLPA.
In the 82 patients, 14 chromosomal imbalances were detected (~17%). Three of the observed chromosomal aberrations were de novo and eleven aberrations were inherited from one of the phenotypically normal parents. The array-CGH results were confirmed by FISH technique in all three de novo cases (Figure 1).
Figure 1.
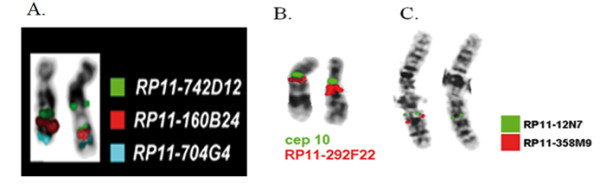
FISH analysis for the confirmation of a-CGH results in the three de novo imbalances. A. In patient 1 the analysis performed using BAC probes RP11-742D12 in 18q21.1 (triplicated), RP11-160B24 in 18q21.2 (normal) and RP11-704G4 in 18q23 (normal). B. In patient 2 it was used BAC probe RP11-292F22 in 10q11.22 (duplicated) and cep10 probe (normal) C. Lastly in patient 3 BAC probes RP11-12N7 in 2q31.1 (normal) and RP11-358M9 in 2q31.3 (deleted) were applied.
The de novo triplication (Patient 1) in 18q is reported here for the first time. It involves chromosome band 18q21.1 and it spans a region of about 5.8 Mb. It is known that Edwards syndrome is mostly associated with trisomy 18, however several individuals with partial trisomy of the long arm of chromosome 18 have been reported. In these cases patients display manifestations ranging from a relatively mild to a severe phenotype. Genotype-phenotype correlations have suggested that duplication of regions (18)(q12.1q21.2) is critical for the trisomy 18 phenotype [9,10], while the relationship between duplication of the other 18q regions and mental retardation, growth delay, and dysmorphism is less clear. Our patient displayed only mild dysmorphic features and speech delay. Only one other case with duplication in this area but in a much larger region has been reported [11]. It concerns a 9 year-old boy with profound MR and growth delay. This individual was diagnosed with a duplication involving (18)(q12.3q21.31), which spans a region much wider than the one reported here. Neither the boy nor the girl in our study display the typical Edwards syndrome phenotype but both individuals share some clinical features like failure to thrive, slanting palpebral fissures and ventricular septal defect. The absence of seizures in our case and the difference in severity of MR and growth delay are probably related to the difference in the size of the duplicated region. It is possible that the mild phenotype in our case is due to the fact that the region does not include genes influencing physical development, or that the triplication does not alter significantly the expression pattern of the corresponding genes.
Proximal 10q duplication (Patient 2) is a well defined but rare genetic syndrome [12-19]. This represents the first case of partial proximal trisomy 10q characterized by array-CGH (Figure 2). The typical profile of partial proximal trisomy 10q syndrome includes postnatal growth retardation, microcephaly, and mild to moderate developmental delay. Frequent dysmorphic features are prominent forehead, small deep-set eyes, epicanthus, upturned nose, bow-shaped mouth, micrognathia, flat and thick ear helices, and long slender limbs. In concordance, our patient showed severe central hypotonia, ataxia, triangular face, enlarged cranium cerebrale, bifid scrotum, cryptorchidism, ulnar deviation of both elbows, deep palmar creases of hands and feet, and syndactyly of 2nd and 3rd toes bilaterally. It is noteworthy that the pericentromeric region 10p11.2 to 10q11.2 has been reported to contain unbalanced chromosomal abnormalities without phenotypic consequences [20].
Figure 2.
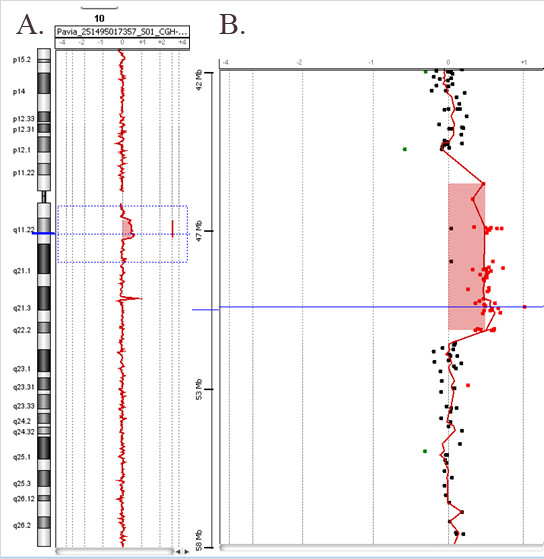
Array-CGH profile of chromosome 10 showing an interstitial duplication (patient 2). A. The whole chromosome 10 view. B. The enlarged view of the rearrangement as provided by Agilent Technologies, CGH Analytics 3.5.14. The proximal duplication breakpoint was between 45,478 Mb and 46,578 Mb. The distal duplication breakpoint was between 51,264 Mb and 51,676 Mb. The overall size of the duplication was about 4.8 Mb.
Deletions involving 2q31-q32 (Patient 3) (Figure 3) have been reported in more than 30 patients [21-26]. Most mutations, involving the 2q31 segment, comprised the HOXD gene cluster which plays an important role in limb development. In our case, the patient showed no limb malformation as the deletion did not involve the HOXD gene cluster.
Figure 3.
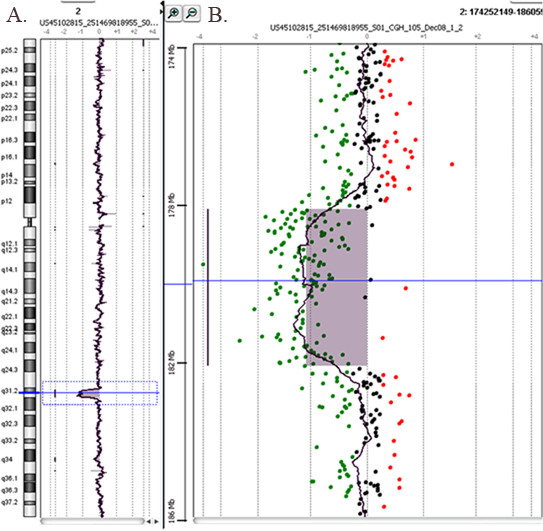
Array-CGH profile of chromosome 2 showing an interstitial deletion (average log2 ratio: -0.83) (patient 3). A. The whole chromosome 2 view. B. The enlarged view of the rearrangement as provided by Agilent Technologies, CGH Analytics 3.5.14. The proximal deletion breakpoint was between 178,370 Mb and 178,393 Mb, and the distal deletion breakpoint was between 182,296 Mb and 182,369 Mb. The overall size of the deletion was about 3.9 Mb.
Dual chromosomal duplication (Patient 13) is a rarely reported genetic condition. To our knowledge this is the first case of simultaneous duplication involving 3p14.1 and 22q13.1-q13.2. Pramparo et al. (2008) [27] identified for the first time a 7 Mb duplication of (22)(q13.1q13.2) in a 10-year-old girl with dysmorphic features (prominent forehead, small low-set ears, hypertelorism, epicanthal folds, small palpebral fissures, short philtrum, and syndactyly), brain-MRI clinical findings (signal abnormalities in the periventricular white matter, hypoplastic corpus callosum, under-rotated hippocampus on the left and atrophic hippocampus on the right side), moderate MR, and severely disorganized mood and behavior with periodic manic episodes. The authors showed that the duplication was the result of a maternal intrachromosomal insertion. The 7 Mb duplicated region is gene-rich, carrying > 120 known genes and ~50 novel transcripts, presumably including genes whose copy number increase is most likely associated with the phenotypic features. Our patient's chromosome 22 duplication is smaller in size (~1.1 Mb) but still contains 9 genes of known function (8 fully-sized and one partial gene sequence at the proximal breakpoint). The clinical features of our patient have common characteristics with those of the patient published by Pramparo et al. (2008), mainly with regard to malformations. We anticipate that the ~1.1 Mb duplicated chromosome 22 region is associated with the patient's phenotype, while some contribution from the 550 kb 3p14.1 duplication should also be considered. The latter assumption is supported by the fact that the chromosome 22 duplication alone was detected in the phenotypically normal mother. It cannot be excluded that the dual duplication in this patient is causative for the patient's phenotype, as also suggested by the double hit model proposed recently, saying that two events (two CNVs) act in concert to produce a more severe phenotype [28].
Nine other patients (Patients 4-12) with developmental delay and various clinical features were found to have deletions or duplications inherited from one of the parents (Table 1). The size of these chromosomal aberrations ranged between 80 kb and 500 kb. However, none of those appears to be associated with the patients' phenotype since they were inherited from a healthy parent. Therefore, these CNVs can be classified as likely benign [29,30] and the underlying causes for the abnormal phenotypes remain unknown.
Methods
Patients
A total of 82 Greek children were referred to the Department of Pediatrics, ''Aglaia Kyriakou'' Children's Hospital, Athens for developmental assessment from 2007 to 2009. All patients were scored using a clinical scoring system [8]. Ages varied between one and thirteen years, with a mean age of 4.9 years. A total of 46 boys and 36 girls were analyzed. All patients had normal karyotype (G-banding analysis at resolution of 550 bands per haploid karyotype, ISCN 2005) and subtelomeric abnormalities determined by MLPA or FISH techniques had been excluded. Written informed consent was obtained from the parents of all patients.
DNA isolation
Blood samples with EDTA were collected from patients and parents. DNA extraction was carried out using the Qiagen QIAamp® DNA blood mini kit (QIAGEN, Valencia, CA, USA).
Array-CGH
Array-CGH analysis was performed using 4 × 44 K, 2 × 105 K and 4 × 180 K commercial arrays (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer's instructions. This platform contains 60-mer oligonucleotide probes spanning the entire human genome with an overall median probe spacing of 22 Kb (19 Kb in Refseq genes). A sex-matched reference DNA (NA10851, NA15510, Coriell Cell Repositories) has been used for each subject tested. Previously reported benign CNVs listed in the Database of Genomic Variants http://projects.tcag.ca/variation/ were excluded from further analysis. After hybridization, the arrays were scanned in a dual-laser scanner (DNA Microarray Scanner with Sure Scan High-Resolution Technology, Model G2565CA, Agilent Technologies) and the images were extracted and analyzed through Agilent Feature Extraction software (v10.5.1.1) and DNA Analytics software (v4.0.73), respectively. Changes in test DNA copy number at a specific locus are observed as the deviation of the log2ratio value from the value of 0 of at least three consecutive probes. The quality of each experiment was assessed by using a parameter provided by Agilent software (QC metric) and on the basis of DNA quality.
Bioinformatics
Copy number changes identified in the samples were compared to the Database of Genomic Variants http://projects.tcag.ca/variation/ and also visualized by using the UCSC Genome Browser website http://genome.ucsc.edu/. The positions of oligomers refer to the Human Genome March 2006 assembly (hg18).
FISH analysis
All three significant de novo aberrations were confirmed by metaphase FISH using BAC clones (RP11-742D12, RP11-160B24 and RP11-704G4 for patient 1, RP11-292F22, RP11-463P17 and RP11-164N7 for patient 2 and RP11-12N7 and RP11-358M9 for patient 3) in the same region as the deletions or duplications identified by microarray analysis [31]. Metaphase chromosomes were obtained from blood lymphocytes according to a standard protocol [32].
MLPA analysis
MLPA analysis was carried out using P036 and P070 probes purchased commercially from MRC-Holland (Amsterdam, Netherlands). The procedure was carried out according to the manufacturer's protocol. Briefly, 100 ng DNA was denatured at 98°C and hybridized overnight at 60°C with the SALSA probe mix P036 and P070. The next day, samples were treated with Ligase 65 for 15 min at 54°C. The reaction was stopped by incubating the samples at 98°C for 5 min. Finally, the amplification step was carried out using the SALSA PCR FAM-labeled primers. The amplification products were analyzed on an ABI 3130 Genetic Analyzer (Applied Biosystems, Carlsbad, CA, USA) using 36 cm capillaries and POP-7 polymer. The obtained data were analyzed using Genemapper 4 Software. The final analysis of the MLPA data was carried out using the Coffalyser Software.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
EM wrote the manuscript; AG, LV, PP and LT referred the patients for study; LT coordinated the clinical analysis of the patients; EM performed the cytogenetic analysis; AV, KK, SR, and EL signed out the array-CGH analysis results; SO and PT were responsible for the MLPA analysis; GK performed the ophthalmologic examination; TL was responsible for the FISH analyses; MBP, ME and EM coordinated the study. All authors have read and approved the manuscript.
Contributor Information
Emmanouil Manolakos, Email: emanolakos@bioiatriki.gr.
Annalisa Vetro, Email: annalisavetro@gmail.com.
Konstantinos Kefalas, Email: kkefalas@biol.uoa.gr.
Stamatia-Maria Rapti, Email: molecular@bioiatriki.gr.
Eirini Louizou, Email: molecular@bioiatriki.gr.
Antonios Garas, Email: garasant@yahoo.gr.
George Kitsos, Email: gkitsos@cc.uoi.gr.
Lefteris Vasileiadis, Email: mpetersen@ich.gr.
Panagiota Tsoplou, Email: molecular@bioiatriki.gr.
Makarios Eleftheriades, Email: makarios@hotmail.co.uk.
Panagiotis Peitsidis, Email: panagiotis_pp@yahoo.com.
Sandro Orru, Email: s.orru@libero.it.
Thomas Liehr, Email: Thomas.Liehr@mti.uni-jena.de.
Michael B Petersen, Email: mpetersen@ich.gr.
Loretta Thomaidis, Email: dr_thomaidis@yahoo.gr.
References
- Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, Majnemer A, Noetzel M, Sheth RD. Quality Standards Subcommittee of the American Academy of Neurology; Practice Committee of the Child Neurology Society. Practice parameter: evaluation of the child with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology. 2003;60:367–380. doi: 10.1212/01.wnl.0000031431.81555.16. [DOI] [PubMed] [Google Scholar]
- Knight SJ, Regan R, Nicod A, Horsley SW, Kearney L, Homfray T, Winter RM, Bolton P, Flint J. Subtle chromosomal rearrangements in children with unexplained mental retardation. Lancet. 1999;354(9191):1676–81. doi: 10.1016/S0140-6736(99)03070-6. [DOI] [PubMed] [Google Scholar]
- Ravnan JB, Tepperberg JH, Papenhausen P, Lamb AN, Hedrick J, Eash D, Ledbetter DH, Martin CL. Subtelomere FISH analysis of 11 688 cases: an evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J Med Genet. 2006;43:478–489. doi: 10.1136/jmg.2005.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries BB, Winter R, Schinzel A, van Ravenswaaij-Arts C. Telomeres: a diagnosis at the end of the chromosomes. J Med Genet. 2003;40:385–398. doi: 10.1136/jmg.40.6.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, Martin CL, Ledbetter DH. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Månér S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, González JR, Gratacòs M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, Oseroff VV, Albertson DG, Pinkel D, Eichler EE. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff DJ, Schwartz MF, Cohen MM, Schwartz S. Precise mapping of a de novo duplication 18(q21-->q22) utilizing cytogenetic, biochemical, and molecular techniques. Am J Med Genet. 1993;46:520–523. doi: 10.1002/ajmg.1320460512. [DOI] [PubMed] [Google Scholar]
- Boghosian-Sell L, Mewar R, Harrison W, Shapiro RM, Zackai EH, Carey J, Davis-Keppen L, Hudgins L, Overhauser J. Molecular mapping of the Edwards syndrome phenotype to two noncontiguous regions on chromosome 18. Am J Hum Genet. 1994;55:476–483. [PMC free article] [PubMed] [Google Scholar]
- Mewar R, Kline AD, Harrison W, Rojas K, Greenberg F, Overhauser J. Clinical and molecular evaluation of four patients with partial duplications of the long arm of chromosome 18. Am J Hum Genet. 1993;53:1269–1278. [PMC free article] [PubMed] [Google Scholar]
- Vogel W, Back E, Imm W. Serial duplication of 10(q11->q22) in a patient with minor congenital malformations. Cli Genet. 1978;13:159–163. doi: 10.1111/j.1399-0004.1978.tb04244.x. [DOI] [PubMed] [Google Scholar]
- Fryns JP, Kleczkowska A, Igodt-Ameye L, van den Berghe H. Proximal duplication of the long arm of chromosome 10 (10q11.2---10q22): a distinct clinical entity. Clin Genet. 1987;32:61–65. doi: 10.1111/j.1399-0004.1987.tb03325.x. [DOI] [PubMed] [Google Scholar]
- De Michelana MI, Campos PJ. A new case of proximal 10q partial trisomy. J Med Genet. 1991;28:205–206. doi: 10.1136/jmg.28.3.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aalfs CM, Hoovers JM, Nieste-Otter MA, Mannens MM, Hennekam RC, Leschot NJ. Further delineation of the partial proximal trisomy 10q syndrome. J Med Genet. 1995;32:968–971. doi: 10.1136/jmg.32.12.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Buggenhout G, Decock P, Fryns JP. A distinct phenotype associated with partial trisomy 10q due to proximal direct duplication 10q11->q22.3? Genet Couns. 1996;7:53–59. [PubMed] [Google Scholar]
- Lam FWF, Chan WK, Lam STS, Chu WP, Knong NS. proximal 10q trisomy: a new case with anal atresia. J Med Genet. 2000;37:E 24. doi: 10.1136/jmg.37.9.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucaro A, Faedda A, Cao A, Boccone L. Partial proximal trisomy 10q syndrome: a new case. Genet Couns. 2002;13:411–416. [PubMed] [Google Scholar]
- Lysy PA, Sibille C, Gillerot Y, Smets F, Sokal EM. Partial proximal 10q trisomy: a new case associated with biliary atresia. Hereditas. 2007;144:191–194. doi: 10.1111/j.2007.0018-0661.02032.x. [DOI] [PubMed] [Google Scholar]
- Liehr T, Stumm M, Wegner RD, Bhatt S, Hickmann P, Patsalis PC, Meins M, Morlot S, Klaschka V, Ewers E, Hinreiner S, Mrasek K, Kosyakova N, Cai WW, Cheung SW, Weise A. 10p11.2 to 10q11.2 is a yet unreported region leading to unbalanced chromosomal abnormalities without phenotypic consequences. Cytogenet Genome Res. 2009;124:102–105. doi: 10.1159/000200094. [DOI] [PubMed] [Google Scholar]
- Ramer JC, Ladda RL, Frankel CA, Beckford A. A review of phenotype-karyotype correlations in individuals with interstitial deletions of the long arm of chromosome 2. Am J Med Genet. 1989;32:359–363. doi: 10.1002/ajmg.1320320318. [DOI] [PubMed] [Google Scholar]
- Ramer JC, Mowrey PN, Robins DB, Ligato S, Towfighi J, Ladda RL. Five children with del (2)(q31q33) and one individual with dup (2)(q31q33) from a single family: review of brain, cardiac, and limb malformations. Am J Med Genet. 1990;37:392–400. doi: 10.1002/ajmg.1320370320. [DOI] [PubMed] [Google Scholar]
- Slavotinek A, Schwarz C, Getty JF, Stecko O, Goodman F, Kingston H. Two cases with interstitial deletions of chromosome 2 and sex reversal in one. Am J Med Genet. 1999;86:75–81. doi: 10.1002/(SICI)1096-8628(19990903)86:1<75::AID-AJMG15>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Van Buggenhout G, Van Ravenswaaij-Arts C, Mc Maas N, Thoelen R, Vogels A, Smeets D, Salden I, Matthijs G, Fryns JP, Vermeesch JR. The del(2)(q32.2q33) deletion syndrome defined by clinical and molecular characterization of four patients. Eur J Med Genet. 2005;48:276–289. doi: 10.1016/j.ejmg.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Mencarelli MA, Caselli R, Pescucci C, Hayek G, Zappella M, Renieri A, Mari F. Clinical and molecular characterization of a patient with a 2q31.2-32.3 deletion identified by array-CGH. Am J Med Genet A. 2007;143:858–865. doi: 10.1002/ajmg.a.31602. [DOI] [PubMed] [Google Scholar]
- Prontera P, Bernardini L, Stangoni G, Capalbo A, Rogaia D, Ardisia C, Novelli A, Dallapiccola B, Donti E. 2q31.2q32.3 deletion syndrome: report of an adult patient. Am J Med Genet A. 2009. pp. 706–712. [DOI] [PubMed]
- Pramparo T, de Gregori M, Gimelli S, Ciccone R, Frondizi D, Liehr T, Pellacani S, Masi G, Brovedani P, Zuffardi O, Guerrini R. A 7 Mb duplication at 22q13 in a girl with bipolar disorder and hippocampal malformation. Am J Med Genet A. 2008;146A:1754–1760. doi: 10.1002/ajmg.a.32326. [DOI] [PubMed] [Google Scholar]
- Veltman JA, Brunner HG. Understanding variable expressivity in microdeletion syndromes. Nat Genet. 2010;42:192–193. doi: 10.1038/ng0310-192. [DOI] [PubMed] [Google Scholar]
- Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007;39:S48–54. doi: 10.1038/ng2092. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Revenga L, Mila M, Rosenberg C, Lamb A, Lee C. Structural variation in the human genome: the impact of copy number variants on clinical diagnosis. Genet Med. 2007;9:600–606. doi: 10.1097/GIM.0b013e318149e1e3. [DOI] [PubMed] [Google Scholar]
- Liehr T, Weise A, Heller A, Starke H, Mrasek K, Kuechler A, Weier HU, Clausen U. Multicolor chromosome banding (MCB) with YAC/BAC-based probes and region-specific microdissection DNA libraries. Cytogenet Genome Res. 2002;97:43–50. doi: 10.1159/000064043. [DOI] [PubMed] [Google Scholar]
- Manolakos E, Orru S, Neroutsou R, Kefalas K, Louizou E, Papoulidis I, Thomaidis L, Peitsidis P, Sotiriou S, Kitsos G, Tsoplou P, Petersen MB, Metaxotou A. Detailed molecular and clinical investigation of a child with a partial deletion of chromosome 11 (Jacobsen syndrome) Mol Cytogenet. 2009;2:26. doi: 10.1186/1755-8166-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]