

Abstract
Glycogen storage disease type II (GSD-II; Pompe disease; MIM 232300) stems from the inherited deficiency of acid-α-glucosidase (GAA; acid maltase; EC 3.2.1.20), which primarily involves cardiac and skeletal muscles. We hypothesized that hydrostatic isolated limb perfusion (ILP) administration of an adeno-associated virus (AAV) vector containing a muscle specific promoter could achieve relatively higher transgene expression in the hindlimb muscles of GAA-knockout (GAA-KO) mice, in comparison with intravenous (IV) administration. ILP adminstration of AAV2/8 vectors encoding alkaline phosphatase or human GAA transduced skeletal muscles of the hindlimb widely, despite the relatively low number of vector particles administered (1×1011), and IV administration of an equivalent vector dose failed to transduce skeletal muscle detectably. Similarly, ILP administration of fewer vector particles of the AAV2/9 vector encoding human GAA (3×1010) transduced skeletal muscles of the hindlimb widely and significantly reduced glycogen content to, in comparison with IV administration. The only advantage for IV administration was moderately high level transduction of cardiac muscle, which demonstrated compellingly that ILP administration sequestered vector particles within the perfused limb. Reduction of glycogen storage in the extensor digitorum longus demonstrated the potential advantage of ILP-mediated delivery of AAV vectors in Pompe disease, because type II myofibers are resistant to enzyme replacement therapy. Thus, ILP will enhance AAV transduction of multiple skeletal muscles while reducing the required dosages in terms of vector particle numbers.
Keywords: Glycogen storage disease type II, gene therapy, hydrostatic delivery, isolated limb perfusion, adeno-associated virus, acid alpha-glucosidase, acid maltase, Pompe disease
INTRODUCTION
The infantile form of Pompe disease (acid maltase deficiency; glycogen storage disease type II; MIM 232300) is a progressive, lethal metabolic myopathy. Pompe disease is characterized by the massive accumulation of lysosomal glycogen in striated muscle with an accompanying disruption of cellular functions. Infantile-onset Pompe disease causes death early in childhood from cardiorespiratory failure related to an underlying hypertrophic cardiomyopathy 1 Late-onset forms of Pompe disease feature progressive weakness without significant cardomyopathy, and patients with juvenile-onset Pompe disease typically become ventilator-dependent due to respiratory muscle involvement.
Pompe disease results from the inherited deficiency of lysosomal acid α-glucosidase (GA; acid maltase; EC 3.2.1.20). Currently enzyme replacement therapy (ERT) has received marketing approval in Pompe disease. ERT in Pompe disease is facilitated by mannose-6-phosphate (M6P) receptor-mediated uptake of the 110 kD precursor GAA.1–3. The limitations of ERT in Pompe disease include humoral immunity, the requirement for high dosages to achieve efficacy, and the high frequency of intravenous infusions. 4,5 In clinical trials of ERT in Pompe disease, up to 40 mg/kg of hGAA was required to improve clinical endpoints in clinical trials.6–8 High-titer anti-GAA antibody formation has been demonstrated in Pompe patients who lacked any residual GAA protein, termed cross-reacting immune material negative (CRIM-negative). Neutralizing antibodies occurred in CRIM-negative Pompe disease patients, and markedly reduced the efficacy of ERT in these subjects.6,7,9 Thus, other therapeutic approaches such as gene therapy are under development for Pompe disease.
The strategy of liver-targeted gene therapy in Pompe disease depends upon the conversion of the liver into a depot for GAA production. The liver secreted GAA into circulating blood, similar to injections of recombinant GAA during ERT, which will be taken up by affected muscles to correct glycogen storage. Hepatic expression of human GAA has achieved a moderate degree of efficacy in mouse models.10–12 This strategy was limited in some experiments by anti-GAA antibody formation, accompanied by markedly reduced efficacy.10,11 However, AAV vectors containing a liver-specific promoter induced immune tolerance against hGAA in GAA-KO mice and efficiently cleared glycogen in cardiac muscle and the majority of skeletal muscles.10–12 The over-expression of hGAA in liver might not be ideal, because the liver is not a therapeutic target and biochemical correction of striated muscles has been inconsistent in proof-of-concept experiments. Uptake of GAA has been reduced at least in part by down-regulation of the cation-independent mannose-6-phospahte receptor in type II myofibers, which leads to uncomplete clearance of accumulated glycogen in the striated muscles by ERT or hepatic expression of GAA.5
Muscle-targeted gene therapy might be a more efficient approach to gene therapy in Pompe disease. Muscle-restricted transgene expression with a muscle-specific expression cassette has evaded transgene-directed immune responses; furthermore, this strategy reduced glycogen accumulations in the heart to near-normal levels, and to a lesser extent in skeletal muscle.13,14 The ideal approach might be to inject an AAV vector intravenously to transduce muscle widely with a muscle-specific transgene. While AAV vectors have delivered genes to striated muscle following intravenous administration, achieving efficacy without disrupting vascular integrity depended upon the administration of very high numbers of vector particles 14,15, which might not be feasible clinically due to the limits imposed by limitations of production and potential T cell responses against the vector capsid proteins.16
One strategy that might reduce the number of vector particles required to transduce skeletal muscle is isolated limb perfusion (ILP), which has the potential to correct multiple limb muscles with lower number of vector particles.17,18 ILP involves the rapid injection of a plasmid or viral vector while a proximal tourniquet occludes venous drainage of the limb, which creates sufficient pressure to disrupt vascular integrity and to release the vector solution into surrounding muscle. Originally developed in rat models with plasmid DNA, this method has been adapted to the delivery of viral vectors in larger animal models, and enhanced transduction of limb muscles was demonstrated with AAV vectors in comparison with low-pressure intravenous (IV) infusion. 17–19 The widespread transduction of skeletal muscle following hydrostatic ILP suggested that this approach might be advantageous for the development of muscle-targeted gene therapy in Pompe disease.
We have evaluated the relative efficacy of low dose AAV vector-mediated GAA expression in mice with Pompe disease, administered either by IV injection or hydrostatic ILP. The number of vector particles administered (1×1011) previously failed to achieve biochemical correction of hindlimb muscles following IV injection with the AAV2/8 vector evaluated, an highly efficacious AAV2/9 vector was administered at even lower dose (3×1010), to test the limits of efficacy for ILP in this study.
RESULTS
Systemic correction of skeletal muscles in GAA-KO mice required IV administration of a very high number of AAV vector particles.14 In order to increase transduction of hindlimb muscles with fewer vector particles, an AAV vector containing the highly active muscle-specific MHCK7 regulatory cassette to drive human placental alkaline phosphatase (AP) expression was administered by ILP.20 AP staining demonstrated widespread transduced myofibers in the quadriceps, gastrocnimus and extensor digitorum longus (EDL) muscle following ILP administration. IV administration of the equivalent number of vector particles resulted in much less transduction of skeletal muscles and much more transduction of cardiac muscle, in comparison with ILP administration (Figure 1).
Fig. 1. Transduction of myofibers following hydrostatic ILP or IV administration of an AAV2/8 vector.
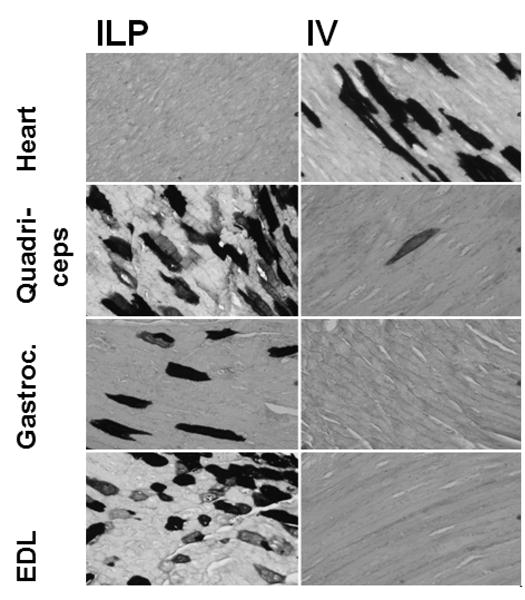
Histochemical staining of striated muscle from GAA-KO mice following IV or ILP administration of an AAV2/8 vector that contains the MHCK7 promoter driving human placental alkaline phosphatase.
We next confirmed that isolated limb perfusion (ILP) of the AAV2/8 vector greatly increased AAV transduction and GAA expression in multiple hindlimb muscles of GAA-KO mice. AAV-MHCK7hGAApA, which contains the MHCK7 regulatory cassette to drive human GAA expression, was packaged as AAV2/8 and administered to 3-month-old GAA-KO mice by hydrostatic ILP or IV injection, and control mice were administered PBS by ILP injection. Eighteen weeks following vector administration, high level of GAA expression were detected in multiple limb muscles including the gastrocnemius, soleus, EDL and quadriceps, in ILP vector-injected mice (Figure 2A). The efficiency of ILP administration was higher in three out of 6 GAA-KO with lower glycogen content in the gastrocnemius, and the ILP results were sorted into two groups accordingly (Figure 2B; ILP, High vs Low; p=0.049). Biochemical correction was confirmed by demonstrating that glycogen reduction was marked following ILP in the hindlimb muscles, including gastrocnemius (64%), soleus (70%), EDL (38%), and quadriceps (43%), in comparison with PBS-injected GAA-KO mice.
Fig. 2. Biochemical evaluation of striated muscle transduction with an AAV2/8 vector delivered by hydrostatic ILP.

GAA activity (A) and glycogen content (B) in GAA-KO mice 18 weeks following AAV vector administration.. Mice were injected with AAV vector by ILP and sorted into high efficiency (ILP High n=3) or or low efficiency (ILP Low, n=3) transduction groups, or by IV injection (IV, n=3); whereas control mice were injected with PBS by ILP (PBS, n=4). Mean +/− standard deviation shown.
In contrast, the IV-injected mice were found to have elevated GAA activity only in the heart, not in skeletal muscles (Figure 2A). Glycogen content was not reduced in the hindlimb muscles following IV administration, in comparison with PBS-injected GAA-KO mice (Figure 2B). Furthermore, glycogen content was reduced to a greater extent following ILP in comparison with IV administration in quadriceps (p=0.02), soleus (p=0.01), and EDL (p=0.03) (Figure 2B). The mean glycogen content of gastrocnemius for the higher efficiency ILP group (ILP High) was significantly reduced in comparison with the IV group (p=0.008); however, combining the results for all ILP-treated mice did not reveal significantly reduced glycogen content in the gastrocnemius in comparison with the IV group (p=0.06). The differing response between ILP High and Low groups reflected variations in transduction efficiency with hydrostatic ILP over the course of the initial experiment, which did not preclude significantly improved biochemical correction in 75% of the skeletal muscles analyzed over that achieved by IV administration. Differences in anti-GAA antibody levels were detected by ELISA between groups of GAA-KO mice following ILP and IV injections. The antibody levels were higher in ILP injected mice when compared with IV injected mice, and did not interfere with efficacy in terms of biochemical correction of striated muscle (Figure 2C). Furthermore, efficacy in skeletal muscle did correlate with vector genome quantification. For gastrocnemius, vector genomes were increased following ILP administration, in comparison with IV administration (0.11±0.05 versus 0.001±0.001, respectively), as quantified by Realtime PCR.
The pattern of GAA expression was further confirmed by Western blot analysis of muscles from each group following AAV2/8 vector administration (Figure 3). Glycogen staining revealed glycogen clearance in most myofibers in the gastrocneminus of the ILP vector-injected mouse (Figure 4). Despite the presence of anti-GAA antibodies in all vector-injected mice (not shown), long-term GAA expression and glycogen clearance was achieved in AAV-transduced muscle cells.
Fig. 3. Western blot detection of GAA expression in striated muscle of GAA-KO mice with an AAV2/8 vector.

Each lane represents one mouse from each group. ILP-AAV mouse: lanes 1,4,7,10,13; IV-AAV mouse: lanes 2,5,8,11,14; ILP-PBS mouse: lanes 3, 6, 9, 12, 15; Lane 16: recombinant human GAA..
Fig. 4. Glycogen staining of skeletal muscle.

Periodic acid/Schiff staining staining of gastrocneminus 18 weeks following intravenous administration of the AAV2/8 vector (1×1011 vector particles) by ILP or IV administration to GAA-KO mice. Controls were sham-treated, age-matched GAA-KO (PBS) and C57BL/6 (Wildtype) mice. Arrows indicated clearance of glycogen in the myofibers.
Subsequently the number of vector particles was further reduced to evaluate the efficacy of ILP at a lower dose. Previously AAV2/9 was deemed a more efficient pseudotype for the correction of skeletal muscle than AAV2/8 in GAA-KO mice.14 The AAV2/9 vector was administered at a lower dose (3×1010 vp), either by ILP or IV administration.
Immune responses that might reduce efficacy were prevented by treating the tolerant GAA-KO mouse strain. GAA-KO mice were previously rendered immune tolerant to human GAA through the insertion of a low-expressing liver-specific transgene that prevented antibody formation in response to long-term ERT 5, Immune tolerant GAA-KO mice would be anticipated to achieve a higher degree of efficacy from a given dose of AAV vector, due to the potential for secretion of GAA from transduced myofibers and the receptor-mediated uptake of GAA by nontransduced myofibers. Initially the absence of antibody formation was confirmed by persistent expression of GAA in plasma in vector treated mice as detected by Western blot (not shown). Consistent with earlier results, ILP administration significantly increased the GAA activity of hindlimb muscles other than quadriceps (p<0.05), in comparison with IV administration (Figure 5A). All mice treated with ILP responded demonstrated similarly reduced glycogen content in the hindlimb muscle during the second experiment, possibly related to increased experience with the method. Glycogen accumulations were significantly reduced in the gastrocnemius (p=0.0005), soleus (p=0.0004), and EDL (p=0.03) muscles by ILP administration, in comparison with IV (Figure 5B). However, IV administration significantly elevated GAA activity (p=3 ×10−8) and reduced glycogen content in the heart (p=0.0007) in comparison with ILP administration (Figure 5), confirming the advantage of systemic delivery with regard to correction of cardiac muscle.
Fig. 5. Biochemical correction following ILP administration of an AAV2/9 vector.

Transduction of striated muscle in tolerant GAA-KO mice 12 weeks following administration of the AAV2/9 vector. (A) GAA activity in the indicated striated muscles analyzed 12 weeks following administration of AAV vector by ILP (ILP-AAV, n=7), IV (IV-AAV, n=3) or mocktreated GAA-KO mice (ILP-PBS; n=5). (B) Glycogen content for tolerant GAA-KO mice in B. Mean +/− standard deviation shown.
DISCUSSION
Previously we have demonstrated that muscle targeted gene therapy is an effective approach for treatment of GAA-KO mice.13, 14 Muscle-restricted expression of hGAA with an AAV vector containing a muscle-specific promoter provoked only a humoral, but not a cellular immune response in GAA-KO mice by the evidence of a lack of lymphocytic infiltrates and absence of CD8+ lymphocytes in the injected muscle.13 However, systemic correction of muscle glycogen content requires intravenous administration of a very high number of AAV vector particles.14 The adaptation of ILP for muscle-targeted gene therapy in the mouse model has currently demonstrated biochemical correction of GAA deficiency and glycogen storage in hindlimb muscles at a much lower vector dose. Previously a 10-fold higher number of AAV2/8 vector particles achieved similar levels of biochemical correction in skeletal muscles, and the lower number of vector particles administered in this study has been consistently inefficacious when administered by the IV route.14 While the 3-fold lower number of AAV2/9 vector particles failed to express GAA as highly or reduce glycogen accumulations as effectively in a second experiment, the principle of higher efficiency transduction of skeletal muscle with fewer vector particles using ILP has now been convincingly established in the GAA-KO mouse model for Pompe disease. The main limitation of ILP is a lack of cardiac muscle transduction, which would be required to achieve efficacy in the severe form of Pompe disease, as it would be in many inherited metabolic myopathies and muscular dystrophies. The simple innovation of administering vector by both ILP and IV routes has been demonstrated with an AAV2/6 vector encoding a histochemical marker protein in a canine model.21
The immune tolerant mouse strain used for the AAV2/9 study was derived by introducing a low-expressing liver-specific transgene to achieve immune tolerance to human GAA.5 These immune tolerant GAA-KO mice are similar to a subgroup of patients with Pompe disease who synthesize a non-functional form of native enzyme that can be detected immunologically, called cross-reactive immunologic material (CRIM), and such patients are deemed CRIM-positive.9 CRIM-positive patients with Pompe disease do not form anti-GAA antibodies when exposed to human GAA via ERT. We did not intend to compare efficacy of AAV2/8 and AAV2/9 in this study; however, we attempted to optimize GAA transduction of skeletal muscle by using the immune tolerant GAA-KO mice in the AAV2/9 experiment to determine: 1) if secretion of hGAA would be achieved from AAV-transduced muscles, and 2) if the secreted GAA in blood would correct heart glycogen storage through enzyme uptake. Although the secretion of GAA in these mice was confirmed by Western blot analysis of plasma (data not shown), heart glycogen level was not reduced following hydrostatic ILP delivery of the AAV2/9 vector in immune tolerant GAA-KO mice. The latter data indicated that a high number of AAV2/9 vectors might be needed to convert the transduced skeletal muscle to a depot for GAA production in Pompe disease.
Hydrostatic ILP has been evaluated in a number of experiments involving normal rodents, dogs, or primates 17–19,21–24 Generally ILP has been deemed well-tolerated, causing no apparent dysfunction and only transiently elevating serum creatine phosphokinase levels. 17,24 Transient edema and minimal levels of myofibers damage were detected following ILP in rats and in rhesus monkeys.(toumi 2006; vigen k 2007} One limitation of these experiments in normal animals was the inability to demonstrate any biochemical efficacy of transgene expression, because the models used lacked any disease phenotype. Recently a study involving the delivery of a plasmid encoding dystrophin in mdx mice demonstrated the widespread preservation of myofibers following multiple ILP administrations in association with up to 20% of normal levels of dystrophin.
The higher resistance of specific muscle groups to correction by ERT in GAA-KO mice has been attributed to both a lack of receptor-mediated uptake and abnormal trafficking of GAA by type II myofibers.5,25 The EDL muscle is comprised mainly of type II myofibers26, and the ability of the AAV vector administered herein to partially correct glycogen accumulations demonstrated a potential advantage for muscle-targeted gene therapy over ERT, in addition to decreased frequency and potentially decreased cost of gene therapy. Achieving a similar degree of correction with 30-fold lower number of AAV2/9 vector particles administered by ILP, in comparison with our earlier study of IV administration, represents an advance in the development of gene therapy for Pompe disease.14
Several obstacles to the translation of muscle-targeted gene therapy, especially with regard to ILP, remain, including the need to transduce skeletal muscles widely with minimal risk. Recent studies of ILP in primates have illustrated how this transition might be accomplished.24 The need for papaverine administration to achieve vasodilation and exsanguinations to eliminate blood from the vasculature prior to vector administration were deemed unnecessary, and neither method was used in the current study. In contrast to earlier studies, elevations of creatine phosphokinase reflecting muscle damage were much reduced by changing the anesthesia regimen. This latter consideration is reassuring with regard to the treatment of metabolic myopathies the predispose to myoglobinuria with accompanying risks for acute renal failure, although Pompe disease does not convey that risk.27 Finally, the ability to perform ILP on multiple limbs could potentially treat late-onset Pompe disease that lacks significant cardiac involvement1,24, even in absence of secretion of GAA from the transduced skeletal muscles or direct transduction of the cardiac muscle by muscle-targeted gene therapy.
In conclusion, we have established that muscle-restricted expression of GAA in transduced myofibers will achieve long-term biochemical correction in GAA-KO mice, and that ILP will enhance AAV transduction of multiple skeletal muscles while reducing the required dosages in terms of vector particle numbers. These data justify further preclinical experiments aimed toward the development of muscle-targeted gene therapy in Pompe disease.
MATERIALS AND METHODS
Preparation of AAV vectors
AAV-MHCK7hGAApA contains the MHCK7 promoter20, the human GAA cDNA, and a human growth hormone polyadenylation sequence. The vector plasmids, pAAV-MHCK7hGAApA14 and αMHCKChAP20 (provided by Dr. Stephen Hauschka, University of Washington, Seattle, WA) have been described. Briefly, 293 cells were transfected with an AAV vector, the AAV packaging plasmid 28 (courtesy of Dr. James M. Wilson, University of Pennsylvania, Philadelphia, PA), and pAdHelper (Stratagene, La Jolla, CA). Cell lysate was harvested 48 hours following infection and freeze-thawed 3 times, and isolated by sucrose cushion pelleting followed by 2 cesium chloride gradient centrifugation steps. AAV stocks were dialyzed against 3 changes of Hanks buffer, and aliquots were stored at −80°C. The number of vector DNA containing-particles was determined by DNase I digestion, DNA extraction, and Southern blot analysis. All viral vector stocks were handled according to Biohazard Safety Level 2 guidelines published by the NIH.
In vivo analysis of AAV vector
The vector stocks were administered by ILP as described17 or intravenously (via the retroorbital sinus) in 3 month-old GAA-KO mice.29 At the indicated time points post-injection, plasma or tissue samples were obtained and processed as described below. All animal procedures were done in accordance with Duke University Institutional Animal Care and Use Committee-approved guidelines.
GAA activity and glycogen content were analyzed as described.30 Western blotting of hGAA was performed as described 31 using the hGAA monoclonal antibody (courtesy of Genzyme Corp., Framingham, MA). Alkaline phosphatase staining was performed as described.20 The ELISA was performed as described.10 All samples yielded absorbance values that were within the linear range of the assay at this dilution. Realtime PCR quantification of AAV-MHCK7hGAApA has been described.14
Statistical analyses
Comparison of two groups was assessed by a homoscedastic Student T-test. A P value of <0.05 indicated a significant difference between the observed values for each group.
Acknowledgments
This work was supported by NIH Grant R01 HL081122-01A1 from the National Heart, Lung, and Blood Institute. BS was supported by the Muscular Dystrophy Association. GAA-KO mice were provided courtesy of Dr. Nina Raben at the National Institutes of Health (Bethesda, MD). The AAV8 and AAV9 packaging plasmids were provided courtesy of Dr. James M. Wilson at the University of Pennsylvania (Philadelphia, PA).
Footnotes
CONFLICT OF INTEREST
The authors have no competing interests as defined by Nature Publishing Group, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
- 1.Hirschhorn R, Reuser AJJ. Glycogen Storage Disease Type II: Acid α-Glucosidase (Acid Maltase) Deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis for Inherited Disease. McGraw-Hill; New York: 2001. pp. 3389–3420. [Google Scholar]
- 2.Hoefsloot LH, Hoogeveen-Westerveld M, Reuser AJ, Oostra BA. Characterization of the human lysosomal alpha-glucosidase gene. Biochem Journal. 1990;1:493–437. doi: 10.1042/bj2720493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wisselaar HA, Kroos MA, Hermans MMP, Vanbeeumen J, Reuser AJJ. Structural and Functional-Changes of Lysosomal Acid Alpha-Glucosidase During Intracellular-Transport and Maturation. J Biol Chem. 1993;268:2223–2231. [PubMed] [Google Scholar]
- 4.Bijvoet AG, Van Hirtum H, Kroos MA, Van de Kamp EH, Schoneveld O, Visser P, et al. Human acid alpha-glucosidase from rabbit milk has therapeutic effect in mice with glycogen storage disease type II. Hum Mol Genet. 1999;8:2145–2153. doi: 10.1093/hmg/8.12.2145. [DOI] [PubMed] [Google Scholar]
- 5.Raben N, Danon M, Gilbert AL, Dwivedi S, Collins B, Thurberg BL, Mattaliano RJ, Nagaraju K, Plotz PH. Enzyme replacement therapy in the mouse model of Pompe disease. Mol Genet Metab. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 6.Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–138. doi: 10.109700125817-200103000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Van den Hout JMP, Kamphoven JHJ, Winkel LPF, Arts WFM, De Klerk JBC, Loonen MCB, et al. Long term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. 2004;113:E448–E457. doi: 10.1542/peds.113.5.e448. [DOI] [PubMed] [Google Scholar]
- 8.Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, et al. Recombinant human acid {alpha}-glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 9.Kishnani PS, Goldenberg PC, Dearmey SL, Heller J, Benjamin D, Young S, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99:26–36. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franco LM, Sun B, Yang X, Bird A, Zhang H, Schneider A, et al. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther. 2005;12:876–884. doi: 10.1016/j.ymthe.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 11.Ziegler RJ, Bercury SD, Fidler J, Zhao MA, Foley J, Taksir TV, et al. Ability of adeno-associated virus serotype 8-mediated hepatic expression of acid alpha-glucosidase to correct the biochemical and motor function deficits of presymptomatic and symptomatic Pompe mice. Hum Gene Ther. 2008;19:609–621. doi: 10.1089/hum.2008.010. [DOI] [PubMed] [Google Scholar]
- 12.Sun B, Zhang H, Benjamin DK, Jr, Brown T, Bird A, Young SP, et al. Enhanced Efficacy of an AAV Vector Encoding Chimeric, Highly Secreted Acid alpha-Glucosidase in Glycogen Storage Disease Type II. Mol Ther. 2006;14:822–380. doi: 10.1016/j.ymthe.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun B, Zhang H, Franco LM, Brown T, Bird A, Schneider A, et al. Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol Ther. 2005;11:889–898. doi: 10.1016/j.ymthe.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 14.Sun B, Young SP, Li P, Di C, Brown T, Salva MZ, et al. Correction of multiple striated muscles in murine Pompe disease through adeno-associated virus-mediated gene therapy. Mol Ther. 2008;16:1366–1371. doi: 10.1038/mt.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG, et al. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 17.Hagstrom JE, Hegge J, Zhang G, Noble M, Budker V, Lewis DL, et al. A facile nonviral method for delivering genes and siRNAs to skeletal muscle of mammalian limbs. Molecular Therapy. 2004;10:386–398. doi: 10.1016/j.ymthe.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Su LT, Gopal K, Wang Z, Yin X, Nelson A, Kozyak BW, et al. Uniform scale-independent gene transfer to striated muscle after transvenular extravasation of vector. Circulation. 2005;112:1780–1788. doi: 10.1161/CIRCULATIONAHA.105.534008. [DOI] [PubMed] [Google Scholar]
- 19.Qiao C, Li J, Zheng H, Bogan J, Li J, Yuan Z, et al. Hydrodynamic limb vein injection of AAV8 canine myostatin propeptide gene in normal dogs enhances muscle growth. Hum Gene Ther. 2009;20:1–10. doi: 10.1089/hum.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salva MZ, Himeda CL, Tai PW, Nishiuchi E, Gregorevic P, Allen JM, et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol Ther. 2007;15:320–329. doi: 10.1038/sj.mt.6300027. [DOI] [PubMed] [Google Scholar]
- 21.Gregorevic P, Schultz BR, Allen JM, Halldorson JB, Blankinship MJ, Meznarich NA, et al. Evaluation of vascular delivery methodologies to enhance rAAV6-mediated gene transfer to canine striated musculature. Mol Ther. 2009;17:1427–1433. doi: 10.1038/mt.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toumi H, Hegge J, Subbotin V, Noble M, Herweijer H, Best TM, et al. Rapid intravascular injection into limb skeletal muscle: a damage assessment study. Mol Ther. 2006;13:229–236. doi: 10.1016/j.ymthe.2005.07.699. [DOI] [PubMed] [Google Scholar]
- 23.Vigen KK, Hegge JO, Zhang G, Mukherjee R, Braun S, Grist TM, et al. Magnetic resonance imaging-monitored plasmid DNA delivery in primate limb muscle. Hum Gene Ther. 2007;18:257–268. doi: 10.1089/hum.2006.115. [DOI] [PubMed] [Google Scholar]
- 24.Hegge JO, Wooddell CI, Zhang G, Hagstrom JE, Braun S, Huss T, et al. Evaluation of Hydrodynamic Limb Vein Injections in Non-human Primates. Hum Gene Ther. 2010 doi: 10.1089/hum.2009.172. epub ahead of press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukuda T, Roberts A, Ahearn M, Zaal K, Ralston E, Plotz PH, et al. Autophagy and lysosomes in Pompe disease. Autophagy. 2006;2:318–320. doi: 10.4161/auto.2984. [DOI] [PubMed] [Google Scholar]
- 26.Haida N, Fowler WM, Jr, Abresch RT, Larson DB, Sharman RB, Taylor RG, et al. Effect of hind-limb suspension on young and adult skeletal muscle. I. Normal mice. Exp Neurol. 1989;103:68–76. doi: 10.1016/0014-4886(89)90187-8. [DOI] [PubMed] [Google Scholar]
- 27.Wortmann RL, DiMauro S. Differentiating idiopathic inflammatory myopathies from metabolic myopathies. Rheum Dis Clin N Am. 2002;28:759–778. doi: 10.1016/s0889-857x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 28.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raben N, Nagaraju K, Lee E, Kessler P, Byrne B, Lee L, et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem. 1998;273:19086–19092. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- 30.Amalfitano A, McVie-Wylie AJ, Hu H, Dawson TL, Raben N, Plotz P, et al. Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid-alpha-glucosidase. Proc Natl Acad Sci U S A. 1999;96:8861–8866. doi: 10.1073/pnas.96.16.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun B, Zhang H, Franco LM, Young SP, Schneider A, Bird A, et al. Efficacy of an adeno-associated virus 8-pseudotyped vector in glycogen storage disease type II. Mol Ther. 2005;11:57–65. doi: 10.1016/j.ymthe.2004.10.004. [DOI] [PubMed] [Google Scholar]