

Abstract
RNA folding occurs via a series of transitions between metastable intermediate states. It is unknown whether folding intermediates are discrete structures folding along defined pathways or heterogeneous ensembles folding along broad landscapes. We use cryo-electron microscopy and single particle image reconstruction to determine the structure of the major folding intermediate of the specificity domain of a ribonuclease P ribozyme. Our results support the existence of a discrete conformation of this folding intermediate.
A major challenge in RNA folding is to define the role of folding intermediates. Folding to the native state occurs in a series of transitions. Populated intermediate states have been characterized by a variety of biophysical and biochemical methods.[1-8] Folding intermediates contain a majority of the native secondary structure and some tertiary structure. These intermediates may act as structural checkpoints, serving to minimize the propensity of RNA to misfold.
Whether intermediate states represent rapidly interconverting heterogeneous ensembles or discrete structures remains an open issue. Compared to native states, intermediate states are less compact, have more solvent exposed residues and contain fewer tertiary interactions. The majority of techniques used in RNA folding studies are ensemble methods which generate an average description of structures, pathways and landscapes. Single molecule studies can track properties of individual RNAs, for example, by following either the dynamics of FRET pairs or the force-versus-extension traces for two points on the RNA. However, these techniques do not provide structural information away from the probed sites, and have limited time-resolution, leaving the issue of structural heterogeneity unresolved. Electron cryo-microscopy (cryo-EM), by virtue of aligning individual molecules and averaging structurally identical molecules in the reconstruction process, is well poised to address this issue.
We determined the structure of a folding intermediate of the specificity domain of Bacillus subtilis RNase P ribozyme (S-domain) using cryo-EM. The native structure of this 154 residue RNA was solved by X-ray crystallography,[9] and its folding behavior in solution has been studied extensively.[8, 10, 11] In equilibrium folding, S-domain folds with well-separated Unfolded-to-Intermediate (U-to-I) and Intermediate-to-Native (I-to-N) transitions according to circular dichroism (CD) spectroscopy and small-angle X-ray scattering (SAXS) [10, 11]. Under the experimental condition for cryo-EM (~19 μM RNA, 1 mM MgCl2, 20 mM TrisHCl, pH 8.0, 22°C; red data point in Fig. 1A), the intermediate state comprised over 95% of the total population. The [Mg2+] used here to populate the intermediate state was significantly higher than previous CD (0.3 μM RNA, 0.1 mM Mg2+) and SAXS (6.3 μM RNA, 0.4 mM Mg2+) studies because of the higher RNA concentration required in cryo-EM experiment (19 μM RNA, 1 mM Mg2+). This solution was deposited onto a cryo-EM grid and rapidly frozen into vitreous ice by plunging into liquid ethane.[12] As the molecules freely move about in solution prior to freezing, snapshots of the RNA solution structure were captured.
Figure 1. Cryo-EM and SPR of the S-domain intermediate structure.
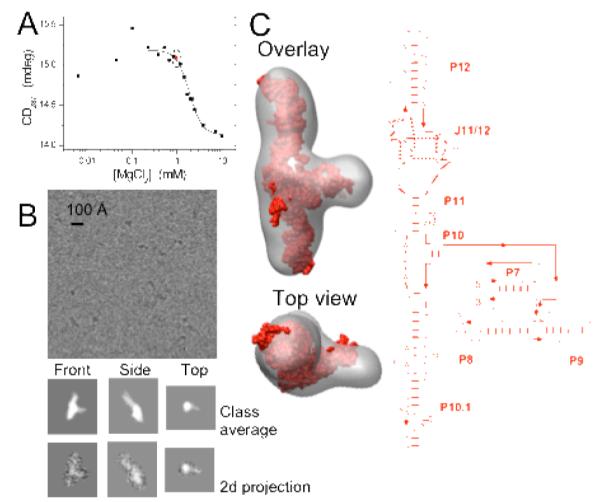
(A) Folding monitered by CD spectroscopy under conditions similar to cryo-EM studies. (B) Selected class averages of the intermediate compared with 2D projections from the 3D cryo-EM reconstruction. (C) SPR of the intermediate overlayed to an atomic model and secondary strucure. The final 3D map was generated using 60 class averages from ~11,600 particles.
During the freezing process, molecules in the intermediate state were unlikely to convert to either the unfolded state or the native state. S-domain becomes more stable at lower temperature which precluded the intermediate reverting to the unfolded state. Forward folding to the native state was precluded as the folding time constant is over ten seconds at 22°C.[8] This was much longer than the freezing time, estimated to be less than 100 msec.
After cryo-EM visualization at 60,000x magnification, single particle reconstruction (SPR) was performed in three stages including particle selection, particle image classification and iterative 3-D refinement using EMAN software.[13] Prior to 3D reconstruction, the particles were subjected to reference-free 2D classification and averaging (Fig. 1B, the black spots are individual RNA molecules). These class-averages exhibited characteristic shapes similar to the expected structure (see below), implying that we successfully selected RNA particles from the raw images. The resulting class averages contained less noise than individual particles and their orientations were determined using a cross-common lines approach. An initial 3D model was generated from the class averages and iteratively refined using standard methods of projection matching to produce a final 3D reconstruction (Fig. 1C).
The cryo-EM density map of the intermediate is elongated with a distinct protrusion near the center. The map contains many features found in a previously generated model of this intermediate using solution data [10, 11] (Fig. 1C). The salient feature of this model is the co-linear arrangement of the P12 and P10.1 helices in the intermediate structure; these regions have a side-by-side arrangement in the native fold. The cryo-EM density map of the intermediate recapitulates this principal feature along with the absolute dimensions of the original model.
The major difference between our previously generated model and the cryo-EM density map is at the tip of the P12 region (Fig. S1A). Because we lacked chemical mapping data for this region, we had left it in its native arrangement in the original, SAXS-derived model. The new cryo-EM density map contains additional density above the P12 helix, and therefore warrants an adjustment of the orientation of the P12 helix. Using the program Assemble,[14] we rotate P12 into a more vertical, extended conformation to fit the EM density map (Fig. S1B). The resulting model also results in better agreement with previous SAXS experiments [10, 11] (data not shown).
The size of the S-domain is below the commonly pursued limit of SPR methods.[15] The size, however, is within the range predicted of being possible to align for SPR[16] and it is further enhanced by the relatively higher scattering of RNA as compared to protein. Because S-domain is close to the limit of theoretical feasibility, we conducted additional analyses and experiments to verify our findings. To test the robustness of the final reconstruction, we initiated a 3D reconstruction starting from a simplified Gaussian ellipsoid (Fig. S1C). Although this reconstruction was devoid of any initial 2D class information, the result agreed well with the cryo-EM density map generated using the 2D class information (Fig. S1D).
To further validate our cryo-EM results, we constructed an S-domain variant with a 16 base pair extension added to the P9 helix as was done previously for EM studies of a group I intron ribozyme.[17] The P9 hairpin does not participate in any non-local interactions in either the native or the intermediate state. Lengthening the P9 stem by one-and-a-half helical turns (~5 nm) did not alter the folding behavior of the S-domain (Fig. 2A). In cryo-EM measurements, the P9 extension significantly helped particle selection and 2D classification because of the additional mass and more identifiable shape. These data were processed similarly as the wild-type data and without any reference to the original intermediate model. The cryo-EM density map of the P9ext-intermediate (Fig. 2B) closely resembled the wild-type intermediate. Importantly, it clearly exhibited the expected density of the P9 extension (shown in green), allowing unambiguous assignment of the P9 helical arm in the intermediate structure.
Figure 2. Cryo-EM of S-domain intermediate with the helical extension on helix P9 (P9ext, green).
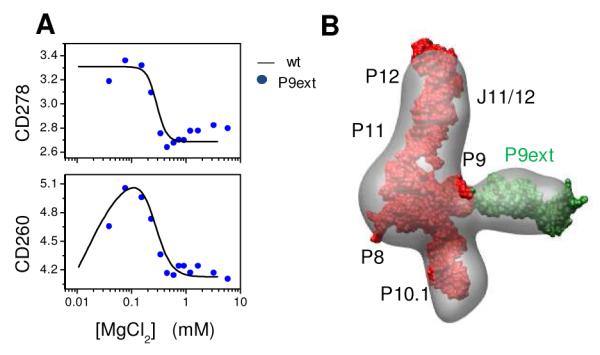
(A) P9ext (blue circles) has the same folding behavior as the wild-type S-domain (black line). (B) Cryo-EM reconstruction of the P9ext intermediate. The final 3D map was generated using 60 class averages from ~19,800 particles.
The presence of identifiable features in the cryo-EM density map indicated that the intermediate was dominated by conformations having limited global variability. The power of cryo-EM lies in its ability to classify and align thousands of individual snapshots, and thereby reconstruct a 3D map. When interpreting the density map, three possible sample compositions were considered: (i) a significant population of native and unfolded molecules, (ii) a mixture of structures with widely varying angles between the P10.1 and P12 helices, and (iii) a dominant discrete structure. The first two possibilities would result in only the central core intensity being seen in the 2D classification and 3D reconstruction, because the protruding helical arms of the different structures would not be repetitively aligned. Hence, the convergence of both the wild-type and P9ext reconstructions to the observed structural envelopes demonstrates that the intermediate has one dominant conformation.
This study indicates that RNA folding pathways can proceed through discrete and specific global architectures. A sequential folding process may aid in efficient folding by reducing conformational variability and the natural propensity of RNA to misfold. The presence of a distinct energy well for the intermediate would also increase the degree of folding cooperativity because the I-to-N transition occurs between two well-defined structural states. In the case of the S-domain, the discrete structure of the intermediate is a result of both native and non-native interactions.[11]
The application of cryo-EM to determine structural features of biomolecules has been limited to ~200 kD.[15] The present results demonstrate that cryo-EM methods can generate density maps of metastable RNA structures as small as ~150 residues or ~50 kD, which is within the theoretical expectation.[16] The feasibility of visualizing such a “small” molecule in our study can be explained partly by the high scattering of the RNA phosphate backbone and the extended helical features that assist in particle alignment during data processing. The ability to obtain molecular envelopes of “small” RNA opens the possibility of structural determination for metastable RNAs, assuming they maintain a single dominant conformation.
Supplementary Material
Acknowledgment
This work was supported by NIH grants (GM57880 to TP and TRS, P41RR02250 to SL and WC).
Footnotes
Supporting Information Available. Cryo-EM methods and a figure describing modeling of the intermediate structure.
REFERENCES
- 1.Brion P, Westhof E. Hierarchy and dynamics of RNA folding. Ann. Rev. Biophys. Biomol. Struct. 1997;26:113–37. doi: 10.1146/annurev.biophys.26.1.113. [DOI] [PubMed] [Google Scholar]
- 2.Tinoco I, Jr., Bustamante C. How RNA folds. J. Mol. Biol. 1999;293(2):271–81. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- 3.Treiber DK, Williamson JR. Beyond kinetic traps in RNA folding. Curr. Opin. Struct. Biol. 2001;11(3):309–14. doi: 10.1016/s0959-440x(00)00206-2. [DOI] [PubMed] [Google Scholar]
- 4.Thirumalai D, et al. Early events in RNA folding. Annu. Rev. Phys. Chem. 2001;52:751–62. doi: 10.1146/annurev.physchem.52.1.751. [DOI] [PubMed] [Google Scholar]
- 5.Woodson SA. Folding mechanisms of group I ribozymes: role of stability and contact order. Biochem Soc Trans. 2002;30(Pt 6):1166–9. doi: 10.1042/bst0301166. [DOI] [PubMed] [Google Scholar]
- 6.Russell R, et al. Exploring the folding landscape of a structured RNA. Proc. Natl. Acad. Sci. USA. 2002;99(1):155–60. doi: 10.1073/pnas.221593598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su LJ, Waldsich C, Pyle AM. An obligate intermediate along the slow folding pathway of a group II intron ribozyme. Nucleic Acids Res. 2005;33(21):6674–87. doi: 10.1093/nar/gki973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baird NJ, et al. Folding of a universal ribozyme: the ribonuclease P RNA. Q. Rev. Biophys. 2007;40(2):113–61. doi: 10.1017/S0033583507004623. [DOI] [PubMed] [Google Scholar]
- 9.Krasilnikov AS, et al. Crystal structure of the specificity domain of ribonuclease P. Nature. 2003;421(6924):760–4. doi: 10.1038/nature01386. [DOI] [PubMed] [Google Scholar]
- 10.Baird NJ, et al. Structure of a folding intermediate reveals the interplay between core and peripheral elements in RNA folding. J. Mol. Biol. 2005;352(3):712–22. doi: 10.1016/j.jmb.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 11.Baird NJ, et al. Extended structures in RNA folding intermediates are due to nonnative interactions rather than electrostatic repulsion. J Mol Biol. 2010;397(5):1298–306. doi: 10.1016/j.jmb.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubochet J, et al. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 1988;21(2):129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- 13.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 1999;128(1):82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 14.Jossinet F, Ludwig TE, Westhof E. RNA structure: bioinformatic analysis. Curr. Opin. Microbiol. 2007;10(3):279–85. doi: 10.1016/j.mib.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Chiu W, et al. Electron cryomicroscopy of biological machines at subnanometer resolution. Structure. 2005;13(3):363–72. doi: 10.1016/j.str.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 16.Henderson R. The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules. Q. Rev. Biophys. 1995;28(2):171–93. doi: 10.1017/s003358350000305x. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura TM, et al. Relative orientation of RNA helices in a group 1 ribozyme determined by helix extension electron microscopy. Embo J. 1995;14(19):4849–59. doi: 10.1002/j.1460-2075.1995.tb00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.