

Abstract
Pancreatic cancer is the fourth leading cause of cancer-related deaths in the U.S. and is nearly always fatal. While early detection offers the most promising approach for reducing the mortality of this disease, there is still a need to develop effective drugs for the prevention and treatment of pancreatic cancer. We tested two promising classes of non-cytotoxic drugs, synthetic oleanane triterpenoids and rexinoids, for the prevention of carcinogenesis in the highly relevant LSL-KrasG12D/+;LSL-Trp53R127H/+;Pdx-1-Cre (KPC) mouse model of pancreatic cancer. KPC transgenic mice closely recapitulate the genetic mutations, clinical symptoms, and histopathology found in human pancreatic cancer. Beginning at 4 wks of age, mice were fed powdered control diet or a diet containing the triterpenoids CDDO-methyl ester (CDDO-Me) or CDDO-ethyl amide (CDDO-EA), the rexinoid LG100268 (LG268), or the combination, until the mice displayed overt symptoms of pancreatic cancer. CDDO-Me, LG268, the combination of CDDO-Me and LG268, and the combination of CDDO-EA and LG268 all significantly (P < 0.05) increased survival in the KPC mice by 3–4 wks. Recent studies have shown that gemcitabine, the current standard of care for human pancreatic cancer, does not extend survival in KPC mice. In cell lines developed from the KPC mice, the triterpenoids directly interact with both STAT3 and IKK to decrease constitutive IL-6 secretion, inhibit constitutive STAT3 phosphorylation, and block the degradation of IKBα when challenged with TNFα. These results suggest that oleanane triterpenoids and rexinoids have the potential to prevent pancreatic cancer.
Keywords: Triterpenoid, CDDO-Me, CDDO-EA, rexinoid, LG100268, combination therapy, pancreatic cancer, prevention, KPC mice
Introduction
Over 43,000 new cases of pancreatic cancer are expected to be diagnosed in the United States in 2010, and this disease is almost uniformly fatal, with a 5-year survival rate of just 6% (1). More than 80% of patients present with unresectable or metastatic disease, and pancreatic cancer is resistant to conventional cytotoxic chemotherapy and radiotherapy. Gemcitabine has been the standard palliative therapy for patients with pancreatic cancer for decades. Only recently have combination therapies of gemcitabine, with erlotinib or capecitabine, been shown to prolong survival, but this increased survival is usually measured in weeks (2). Although prevention of pancreatic cancer will ultimately be the most effective strategy for reducing the unacceptable mortality rates for this disease (3;4), the practical realities of the cancer clinic are undeniable evidence that new drugs and drug combinations are needed for both prevention and treatment of pancreatic cancer.
Genomic analyses of human cancers are revealing the complexity of carcinogenesis (5;6). In pancreatic cancer, an average of 63 genetic mutations per cancer involving 12 distinct signaling pathways were altered in 67–100% of the 24 human pancreatic tumors sequenced (7). The high number of genetic mutations and the resulting defects in cellular signaling are thought to contribute to the aggressiveness and chemoresistance of pancreatic cancer (8). Kinzler et al. concluded that monofunctional drugs that target a single mutated gene product will likely not work in pancreatic cancer; instead, drugs or drug combinations that broadly target key regulatory nodes or multiple pathways will be needed in order to reduce the number of deaths from this devastating disease (7).
Synthetic oleanane triterpenoids and rexinoids are two promising classes of multifunctional drugs that are highly effective for preventing and/or treating cancer in a wide variety of preclinical animal models (9;10), and they have also been shown to inhibit proliferation and/or induce apoptosis in human pancreatic cancer cells (11–13). These compounds are neither conventional cytotoxic drugs nor monofunctional agents that target single proteins in a signal transduction pathway. Instead, these multifunctional drugs target either receptors that act as transcription factors themselves (rexinoids) or proteins that regulate transcription factors (triterpenoids) (9). Thus, these oleanane triterpenoids and rexinoids are able to impact a number of biological processes thought to be critical for tumorigenesis in the pancreas (14–16), including inflammation, cell proliferation, angiogenesis, and apoptosis.
Triterpenoids are synthesized endogenously in many different types of plants through the cyclization of squalene, and over 300 new synthetic oleanane triterpenoids have been synthesized and screened (9). Many of these new compounds are over 10,000 fold more potent than the starting material, oleanolic acid, and potently inhibit inflammation, regulate the redox status of cells, block cell proliferation, and induce differentiation and apoptosis of a variety of cancer cells. Moreover, cancer cells are more sensitive than normal cells to apoptosis by the triterpenoids, as concentrations of triterpenoids that induce apoptosis in cancer cells are not toxic to normal lymphocytes or normal epithelial cells (9). The triterpenoid CDDO-Methyl ester (CDDO-Me) has been tested in early clinical trials for the treatment of a variety of cancers, including pancreatic cancer, but is now being evaluated in patients with chronic kidney disease.
Rexinoids are selective ligands for members of the nuclear receptor superfamily known as retinoid X receptors (RXRs). Because RXRs form functional heterodimers with several other nuclear receptors, including RARs, VDRs, TRs, and LXRs, they modulate the activity of many other transcription factors that control development, metabolism and the physiology of cell growth and survival (9;10). Although bexarotene is currently the only rexinoid that is approved for clinical use, both LG100268 (268) and NRX 194204 (4204) are significantly more potent and more selective for RXRs than bexarotene. We have also shown that the combination of a triterpenoid and rexinoid synergizes in vitro and in vivo (9). The use of two synergistic classes of drugs that target different signaling pathways will allow lower doses of drugs to be used to improve drug tolerance in the patient without sacrificing drug efficacy (17).
Activating mutations in the Kras protoncogene are found in over 90% of human pancreatic cancers (7). In mice, conditionally targeting an activating point mutation in Kras (G12D) with a Pdx-1 pancreas-specific promoter results in pancreatic lesions that display a full spectrum of pancreatic intraepithelial neoplasias (mPanINs), precursors to invasive pancreatic cancer (18). These lesions progress to fully invasive and metastatic adenocarcinomas, and thus this transgenic mouse model (LSL-KrasG12D/+;Pdx-1-Cre) mimics both the genetic and histological changes observed in human pancreatic cancer. When a point mutation (R172H) in the p53 tumor suppressor gene, which is mutated in 75% of human pancreatic tumors, is also introduced into the pancreas of mice with a KrasG12D/+ mutation, these triple transgenic KPC mice (LSL-KrasG12D/+;LSL-Trp53R127H/+;Pdx-1-Cre) develop pancreatic cancer more rapidly than do mice with just the Kras mutation (19). Because the carcinomas in the KPC mice do not typically appear until 10 weeks of age, additional genetic alterations may be needed for disease progression.
The median survival time of the KPC triple transgenic mice is only 5 months, and they develop large pancreatic carcinomas that metastasize to the liver, lungs, and diaphragm. The clinical symptoms including cachexia and abdominal distension, the defined histopathological progression, and the genomic instability found in human pancreatic cancer are all replicated in the KPC transgenic mice, making it one of the most relevant animal models for preclinical evaluation of new drugs for pancreatic cancer (20). We hypothesized that the triterpenoids and rexinoids, alone and in combination, could significantly delay tumor development in KPC mice. Based on our studies in 177 mice, we report effective chemoprevention or chemotherapy in this KPC model by the individual drugs but did not find a synergistic effect.
Materials and Methods
Drugs
CDDO-ME and CDDO-EA were synthesized as previously described (21–23) by Reata Pharmaceuticals. LG 100268 (24) was generously provided by William Lamph, Ligand Pharmaceuticals.
Tissue culture and in vitro assays
PDA 4964 pancreatic cancer cells (provided by Anirban Maitra) were grown in DMEM and 10% FBS. To generate new pancreatic cancer cell lines, pancreatic tumors resected from a KPC mouse (19) were minced with a scalpel and digested in 0.25% trypsin/0.02% EDTA (Invitrogen) for 15 min at 37°C with gentle agitation from a stir bar. The cell suspension was filtered through a 40 μm Cell Strainer (BD Bioscience), centrifuged at 220 g for 10 min and plated in DMEM + 10% FBS. Additionally, abdominal ascites from different KPC mice were centrifuged at 220 g for 10 min and then plated in the same media. After passing the cells more than 10 times, they were used for mechanistic studies (PanAsc cells from ascites and Pan cells from tumors). To determine targets of the triterpenoids, cells were treated with 3 μmol/L of a biotinylated triterpenoid (compound 6 in ref. 25) for 1 hr. Cell lysates were incubated with 50 μl DynaBeads MyOne Strepavidin (Invitrogen) as described (26), and then analyzed by Western blotting using antibodies against STAT3, pSTAT3, CREB and EGFR (Cell Signaling), IKK, IKBα, PCNA and RXR (Santa Cruz), tubulin (Calbiochem), and ErbB2 (Lab Vision). Cells were treated with triterpenoids for 3 days, and the amount of IL-6 released into the medium was measured using a Quantikine ELISA kit (R&D Systems). For proliferation, cells were treated with triterpenoids for 2 days, and the amount of 3H-thymidine incorporation was measured after a 2 hr incubation. To measure ROS, cells were treated with triterpenoids for 1 hr and 10 μmol/L of H2DCFDA was added for the last 30 minutes. The mean fluorescence intensity of 10,000 cells was analyzed by flow cytometry using a 480-nm excitation wavelength and a 525-nm emission wavelength. Apoptotic cells were stained using a TACSAnnexin V-FITC apoptosis detection kit (R&D Systems) and counted by flow cytometry.
Prevention of pancreatic cancer in vivo
All animal studies were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Dartmouth Medical School. LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre triple mutant (KPC) mice (19) were obtained by interbreeding LSL-KrasG12D/+;LSL-Trp53R172H/+ and LSL-Trp53R172H/+;Pdx-1-Cre mice. Genomic DNA was extracted from tail snips using the Extract-N-Amp Tissue PCR kit (Sigma) and genotyped as previously described (18;19). Four week-old KPC mice were randomized and fed powdered 5002 rodent chow (PMI Feeds) or the same powdered diet containing drugs ad libitum. The doses selected for these studies were based on previous chemoprevention studies with these drugs and are known to be well-tolerated (9). The drugs are stable in diet for over a month, and food was replaced in the cages twice a week. All mice were carefully observed daily and were weighed weekly. Because of IACUC regulations, death was not used as an endpoint, but instead mice were sacrificed when severe symptoms were evident. These symptoms included cachexia, abdominal distension, rapid weight loss or labored breathing. Death of the mice predictably occurs within 24–72 hrs after such symptoms appear (19).
Statistical analysis
Results are described as mean ± SEM and were analyzed by one-way ANOVA and a Tukey test or by one-way ANOVA on ranks (Wilcoxon signed rank test; SigmaStat3.5). All p values are two-sided.
Results and Discussion
Triterpenoids interact with important regulatory proteins in pancreatic cancer cells
The biological activity of the synthetic triterpenoids depends on their ability to interact with cellular nucleophiles such as the – SH groups of cysteines on target proteins (9). Therefore, the triterpenoids interact with multiple molecular targets, accounting for their numerous biological activities. However, the triterpenoids do not interact non-specifically with cysteine residues in all proteins. Instead, the nucleophilicity of the – SH group of any specific cysteine residue is markedly influenced by its neighboring amino acids as well as the redox potential of the cell. Furthermore, steric factors also determine the accessibility of a drug to such potential nucleophilic sites.
Recent proteomic studies (manuscript in preparation) have revealed several new putative targets for the triterpenoids, so we tested whether these drugs directly interact with regulatory proteins that have been implicated in the pathogenesis of pancreatic cancer (27;28). In PDA 4964 pancreatic cancer cells treated with a biotinylated triterpenoid followed by streptavidin precipitation with DynaBeads (29), the triterpenoids directly interacted with STAT3, IKK, RXRα, CREB, EGFR, ErbB2, and α-tubulin (Fig. 1) but did not interact with PCNA. All of these targets, except RXRα, are known to be important in pancreatic cancer (4;30). For example, proteins in the NF-κB pathway including the p65 subunit of NF-κB (RelA) and IKBα are constitutively activated or overexpressed in pancreatic adenocarcinomas and in pancreatic cancer cell lines but not in normal pancreatic tissue (4;15;31). Constitutive activation of STAT3 drives cellular transformation and tumor progression while protecting against apoptosis in a number of tumors including pancreatic cancer; blocking STAT3 signaling limited invasion and decreased the expression of proangiogenic molecules in pancreatic cancer cells (32;33).
Fig. 1. Triterpenoids directly interact with critical regulatory proteins in pancreatic cancer cells.
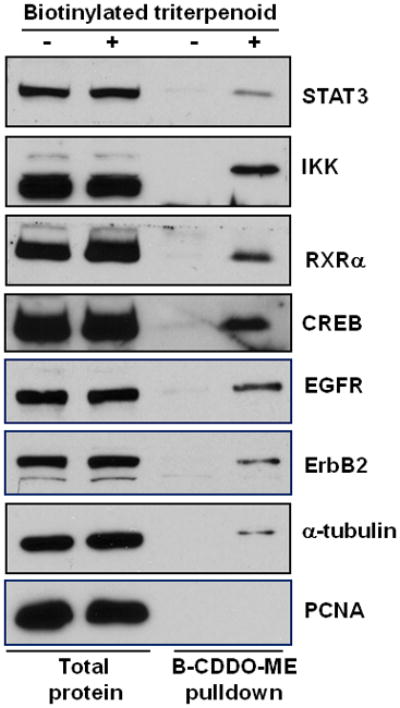
PDA 4964 cells were treated with a biotinylated triterpenoid (B-CDDO-Me, 3 μM) for 1 hr, triterpenoid-protein complexes were precipitated from cell lysates with streptavidin DynaBeads, proteins were separated by SDS-PAGE, and various antibodies were used for immunoblotting.
Furthermore, high levels of ErbB2/Her2 are consistently expressed in KPC mice in mPanIn lesions, in invasive carcinomas and in metastases (19). Overexpression of the EGFR has been described in a subset of human pancreatic cancers, especially at advanced clinical stages (4;34). When the EGFR inhibitor erlotinib was combined with gemcitabine, this combination did provide a small but statistically significant improvement in survival for pancreatic cancer patients (2). Rexinoids bind selectively to retinoid X receptors (RXR) and regulate complex cell survival and differentiation processes by heterodimerizing with other nuclear hormone receptors (9). The discovery that triterpenoids can directly bind to RXRα may help explain the striking synergy between triterpenoids and rexinoids for preventing and treating experimental breast and lung cancer (9;26).
In order to make the in vitro studies as relevant as possible to the in vivo model, we developed several new cell lines from solid pancreatic carcinomas (Pan cells) or abdominal ascites (PanAsc cells) from KPC mice. In all of the new cell lines tested (2 Pan and 3 PanAsc cell lines), the biotinylated triterpenoid directly interacted with STAT3 and IKK (Fig. 2A is a representative blot from PanAsc 2159 cells). These protein interactions are biologically relevant as the triterpenoids CDDO-ME and CDDO-EA reduced constitutive expression of phosphorylated STAT3 (pSTAT3) in a dose-dependent manner in the PanAsc 2159 cells but had no effect on STAT3 levels (Fig. 2B). All of the new pancreatic cancer cells secreted detectable levels of IL-6, with higher levels in the PanAsc cells from ascites than in the Pan cells from solid tumors. IL-6 can induce phosphorylation of STAT3, resulting in constitutive STAT3 phosphorylation (35), and treatment of pancreatic cancer cells with CDDO-ME and CDDO-EA significantly (P < 0.05) reduced IL-6 secretion in a dose-dependent manner in PanAsc 2159 cells (Fig. 2C). The interaction of the triterpenoids with IKK is also highly relevant in these pancreatic cancer cells as pretreatment with triterpenoids for 2 hrs (data not shown) or 24 hrs (Fig. 2D) prevented the degradation of IKBα when challenged with TNFα; the interaction of the triterpenoids with IKK is also known to block nuclear translocation of p65 as has been shown in a variety of cells (29). The same concentrations of triterpenoids that blocked constitutive pSTAT3 expression (Fig. 2B) and prevented the degradation of IKBα (2D) also induced apoptosis in the pancreatic cancer cells (Fig. 3C), possibly through the accumulation of reactive oxygen species (Fig. 3A), as has been previously reported for the induction of apoptosis in human pancreatic cancer cells by triterpenoids (13). The triterpenoids, but not the rexinoid 268, also inhibited proliferation of the pancreatic cancer cells in a dose-dependent manner (Fig. 3B). Because the triterpenoids suppressed constitutive IL-6 secretion and STAT3 phosphorylation as well as inhibited IKBα degradation in pancreatic cancer cells from the KPC mice, we next tested whether triterpenoids could extend survival in vivo.
Fig. 2. Direct interaction of the triterpenoids with STAT3 and IKK inhibits these pathways in PanAsc 2159 cells developed from KPC mice.
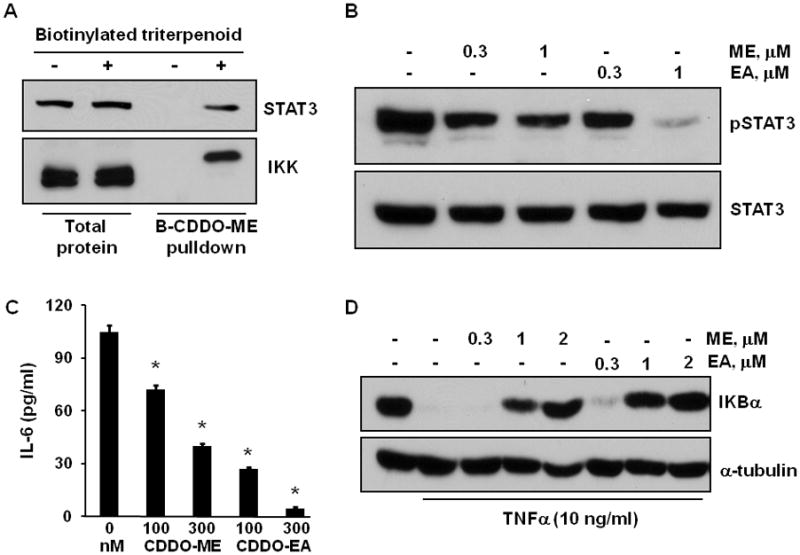
A. PanAsc2159 pancreatic cancer cells were treated with 3 μM of a biotinylated triterpenoid for 1 hr, triterpenoid-protein complexes were precipitated from cell lysates with streptavidin DynaBeads, and STAT3 and IKK were detected by Western blotting. B. Cells were treated with triterpenoids (0.3–1 μM) for 24 h, and lysates were immunoblotted with antibodies against phosphorylated STAT3 (pSTAT3) or STAT3. C. Cells were treated with triterpenoids for 3 days and supernatants were assayed by ELISA for IL-6 secretion. D. Cells were pretreated with triterpenoids for 24 hr then stimulated with TNFα for 15 min; lysates were analyzed by Western blotting. *, P < 0.05 vs. control.
Fig. 3. Triterpenoids inhibit proliferation and induce ROS and apoptosis in pancreatic cancer cells.
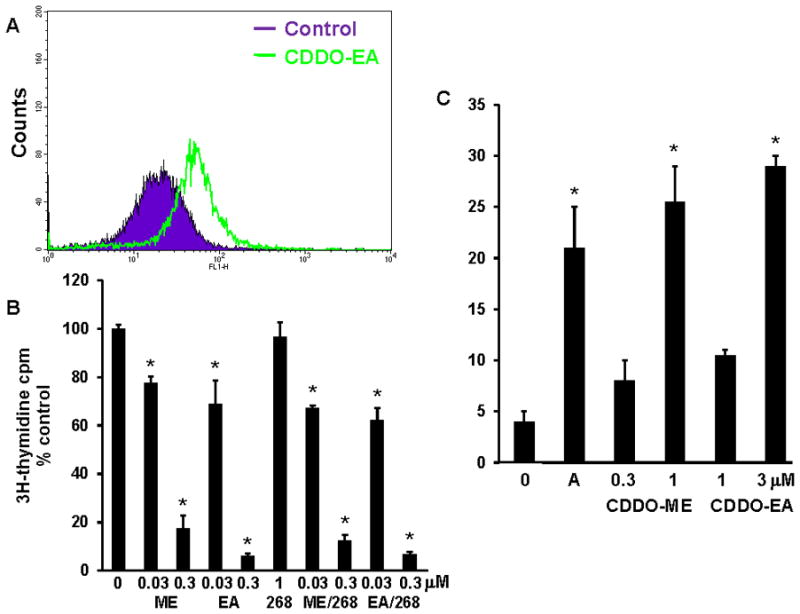
A. PanAsc2159 cells were treated with CDDO-EA (3 μM) for 1 hr, loaded with H2DCFDA for the last 30 min of treatment, and the mean fluorescence intensity analyzed by flow cytometry. B. PanAsc 2159 cells were treated with drugs for 2 days, and proliferation was measured using a 3H-thymidine incorporation assay. C. To measure apoptosis, Pan1343 cells were treated for 24 hr, and Annexin V and propidium iodide staining were measured by flow cytometry. A = anisomycin, 10 μg/ml. *, P < 0.05 vs. control.
Triterpenoids and the rexinoid LG268 extend survival in the KPC mouse model of pancreatic cancer
Beginning at 4 wks of age when pancreata are histologically normal, KPC transgenic mice were started on powdered control diet or diet containing a triterpenoid or rexinoid until the mice developed symptoms found in human pancreatic cancer, such as cachexia or abdominal distension caused by the accumulation of up to 10 ml of hemorrhagic ascites. Other symptoms of the disease in this model include significant and rapid weight loss of up to 20% of body weight caused by intestinal obstruction by a pancreatic tumor or labored breathing from metastases in the lungs or diaphragm. Once any of these symptoms appear, we and others (19) have found that death predictably and consistently occurs within 24–72 hrs, and gross observation of pancreatic cancer was confirmed for all mice included in our studies. Although this KPC model of pancreatic cancer is very aggressive with an average survival time of only 20.5 ± 0.9 wks of age in control mice (n = 83), the mice fed CDDO-Me (60 mg/kg diet or ~ 15 mg/kg body weight; n = 17) lived an average of 24.2 ± 2.7 wks, significantly longer than the controls (Fig. 4A; P < 0.001). In 13 litter-mate matched pairs, 12 of 13 mice fed the triterpenoid CDDO-Me lived an average of 5 wks longer than the controls (P = 0.021 for all 13 pairs; data not shown).
Fig. 4. Triterpenoids and rexinoids, alone or in combination, extend survival in the KPC mouse model of pancreatic cancer.
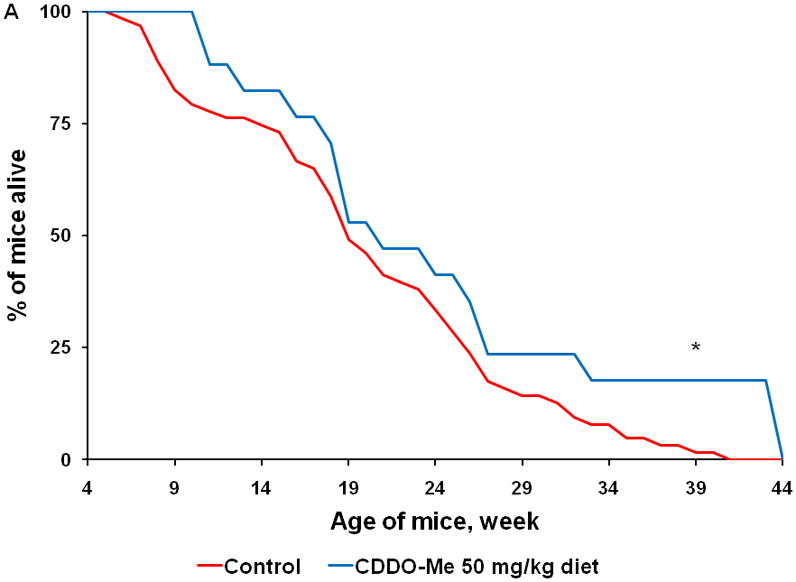
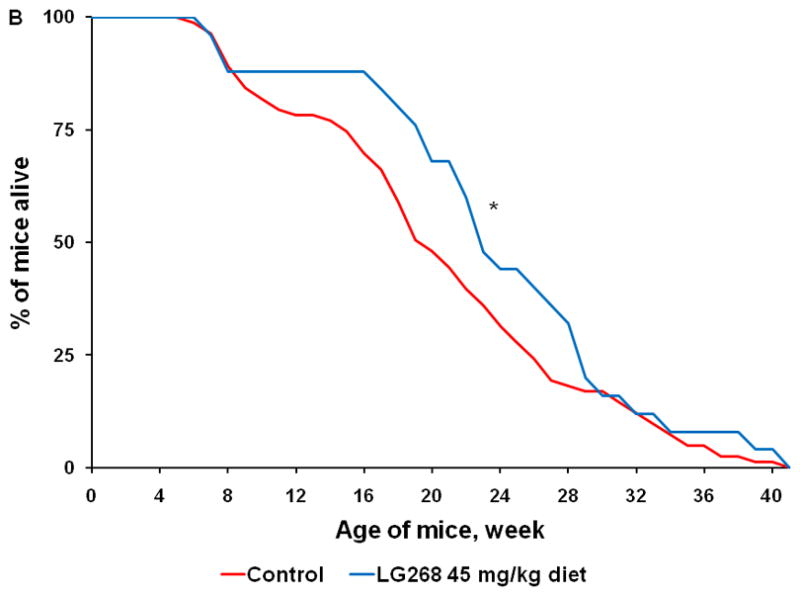
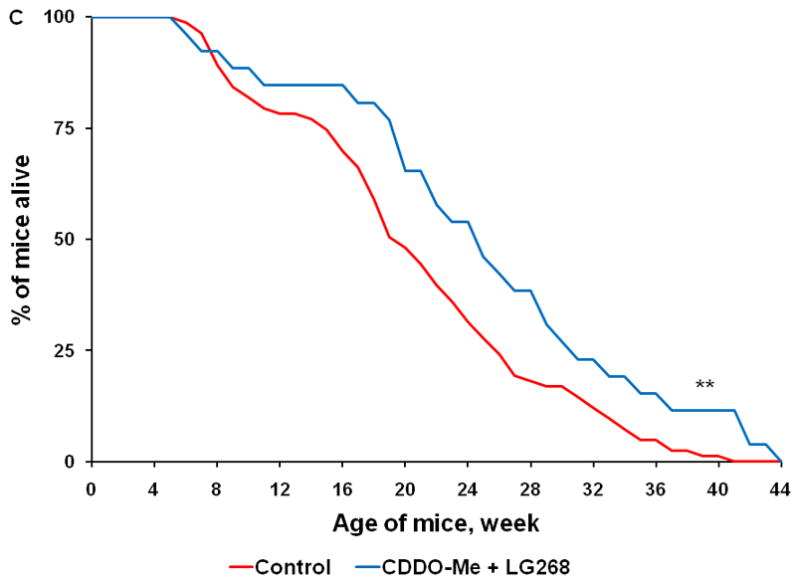
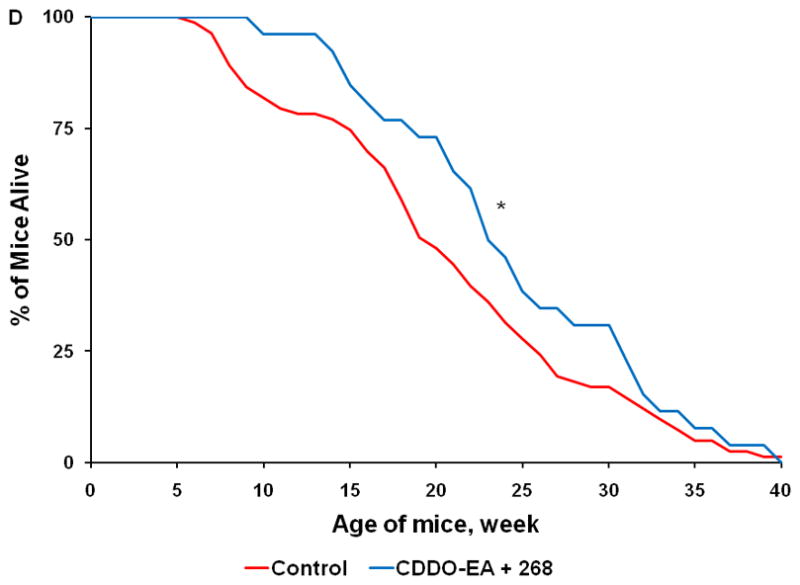
Beginning at 4 weeks of age, KPC transgenic mice were fed powdered control diet, A) CDDO-Me (60 mg/kg diet), B) 268 (45 mg/kg diet), C) the combination of CDDO-Me and LG268, or D) the combination of CDDO-EA (400 mg/kg diet) and LG268. n = 83 for control, 17 for CDDO-Me, 25 for LG268, 26 for CDDO-Me + LG268, 26 for CDDO-EA + LG268; *, P < 0.05 vs. control, **, P < 0.05 vs. control and P < 0.05 vs. LG268 alone.
We also tested the rexinoid LG268 in this model. As has been reported previously (9), the rexinoids have almost no effects on epithelial cells in vitro and thus did not inhibit proliferation of pancreatic cancer cells (Fig. 3B) and did not change the expression or activity of any of the proteins or pathways shown in Fig. 1–3 (data not shown). However, rexinoids are very effective for prevention and treatment of cancer in a number of experimental animal models, as they target inflammatory cells and the tumor microenvironment to inhibit inflammation, angiogenesis, invasion, and metastases (9). When fed in diet (45 mg/kg diet or ~ 11 mg/kg body weight), LG268 also significantly (P < 0.001) prolonged survival of KPC mice (Fig. 4B; n = 25). The average lifespan was 23.7 ± 1.7 wks for the LG268 group vs. 20.5 ± 0.9 wks for the controls. In 14 litter-mate matched pairs, 12 of 14 mice fed the rexinoid LG268 lived an average of 8.4 wks longer than the controls (P = 0.021).
The combination of a triterpenoid and a rexinoid is also effective in this model, but unlike previous studies in experimental breast and lung cancer (26;36), the combination of these two multifunctional drugs was not synergistic. Mice fed the combination of CDDO-Me and LG268 (Fig. 4C; n = 26) lived an average of 24.8 ± 2 wks, which was longer than the average of 20.5 ± 0.9 wks in the controls (P < 0.001). In 18 litter-mate matched pairs, 14 of 18 mice fed the combination of CDDO-Me and LG268 lived an average of 7.3 wks longer than the controls (P = 0.023). Although the triterpenoid CDDO-EA alone (data not shown) did not significantly prolong survival in the KPC at the one dose tested (400 mg/kg diet or ~ 100 mg/kg body weight), the combination of CDDO-EA and LG268 was effective (Fig. 4D, n = 26, P = 0.005), with an average lifespan of 24.3 ± 1.5 wks vs. the above controls. These drugs were all well-tolerated at the doses used, and the mice continued to gain weight throughout the experiment, until they were symptomatic with pancreatic cancer. After 15 wks on diet, the controls gained an average of 8.2 g compared to 7.1 g for mice fed CDDO-Me, 8.2 g for mice fed LG268, 10 g for mice fed the combination of CDDO-Me and LG268, and 7.4 g for mice fed the combination of CDDO-EA and LG268. Additional experiments with higher doses of both CDDO-EA and LG268, alone and in combination, are ongoing but appear to be even more effective than the doses of drugs used in the current studies. Comprehensive pharmacokinetic studies with these drugs have not been done in the KPC mice. We have, however, published tissue levels of these drugs in other strains of mice and thus know that oral administration as described here produces pharmacologically useful drug levels (37). Pilot studies in KPC mice found detectable drug levels in the pancreas for both triterpenoids and rexinoids, with an average of 120 ± 60 nM in mice fed CDDO-EA and 285 ± 80 nM in mice fed LG268 (n=2–4 per group); further pharmacokinetic analysis will be performed.
Traditionally, screening of potential new drugs for pancreatic cancer has relied on xenograft models, which can replicate the genetic features of the parental tumors and are rapid, easy, and relatively high-throughput. Although a number of drugs, including gemcitabine, are effective in these models, these findings have not translated into the clinic. The lack of predictive ability, the inability to mount an immune response, and the absence of a physiological microenvironment in xenograft models suggest that transgenic mouse models that better replicate the human disease should be used for drug testing (20;38).
In pancreatic cancer, the KPC mouse model mimics the genetics, the pathophysiology, and metastatic potential of the human disease (18;19). Although the age at which symptoms first appear in this KPC transgenic mouse model is highly variable, the mice treated with the triterpenoids CDDO-Me or CDDO-EA, the rexinoid LG268, or the combination lived an average of 3–4 weeks longer than the control mice. When compared to littermate matched controls, the average lifespan of mice fed triterpenoid or rexinoid was 5–6 weeks longer than the controls. In comparison, cyclopamine increased median survival by only 6 days in a LSL-KrasG12D;Ink4a/Arflox/lox;Pdx-1-Cre transgenic mouse model of pancreatic cancer (39), and the combination of gemcitabine with IPI-926 increased survival in the KPC model by 14 days (38). Both cyclopamine and IPI-926 are inhibitors of the hedgehog signaling pathway, an important pathway during pancreatic development that can be reactivated in pancreatic carcinogenesis; inhibitors of the hedgehog pathway have gained increasing attention as possible new therapies for pancreatic cancer (30). When tested in the LSL-KrasG12D;Pdx-1-Cre mouse model, the COX-2 inhibitor nimesulide (40) significantly reduced the number of pancreatic intraepithelial neoplasia (mPanIN) lesions, early precursor lesions that proceed to pancreatic ductal adenocarcinoma (PDA). Moreover, both aspirin and enalapril, an angiotensin-I converting enzyme inhibitor, delayed the progression of mPanINs in LSL-KrasG12D;Pdx-1-Cre mice and partially inhibited the development of invasive PDA in KPC mice (41). It would be interesting to compare the efficacy of nimesulide, aspirin, or enalapril to the triterpenoids or rexinoid, but no survival data in the KPC model was reported for these compounds.
In summary, we have shown that the triterpenoid CDDO-Me and the rexinoid LG268, two multifunctional drugs with different mechanisms of action, can significantly extend the lifespan of KPC mice. The KPC transgenic mouse model is an aggressive but relevant animal model that accurately recapitulates the genetics, the histopathology, and the metastatic potential of human pancreatic cancer. We are currently using this model to optimize the doses of triterpenoids and rexinoids and testing these and others drugs in combination with standard therapies, such as gemcitabine and erlotinib. We also will explore the signaling pathways identified in Fig. 1 that are known to be relevant in pancreatic cancer to determine the mechanisms of action of these effective drugs in pancreatic cancer and to determine if these pathways could be used as biomarkers of activity. Furthermore, we are testing whether triterpenoids and rexinoidscan reduce the number of early pancreatic lesions, or mPanINs, in the less aggressive LSL-KrasG12D;Pdx-1-Cre transgenic mice, as has recently been reported for the EGFR inhibitor gefitinib (42).
Acknowledgments
We thank William W. Lamph for supplying the rexinoid LG100268 and David Tuveson for establishing the KPC model and for his advice on using the model.
Grant support: This research was supported by an American Cancer Society Research Grant, #IRG-82-003-23 (KL), a Sidney Kimmel Foundation for Cancer Research Scholar Award (KL), by the National Foundation for Cancer Research (MBS), NIH R01 CA78814 (MBS), and Reata Pharmaceuticals, Inc. (MBS).
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA Cancer J Clin. doi: 10.3322/caac.20073. in press. [DOI] [PubMed] [Google Scholar]
- 2.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–17. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 3.Doucas H, Garcea G, Neal CP, Manson MM, Berry DP. Chemoprevention of pancreatic cancer: a review of the molecular pathways involved, and evidence for the potential for chemoprevention. Pancreatology. 2006;6:429–39. doi: 10.1159/000094560. [DOI] [PubMed] [Google Scholar]
- 4.Sarkar FH, Banerjee S, Li Y. Pancreatic cancer: pathogenesis, prevention and treatment. Toxicol Appl Pharmacol. 2007;224:326–36. doi: 10.1016/j.taap.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 6.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 7.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koorstra JB, Hustinx SR, Offerhaus GJ, Maitra A. Pancreatic carcinogenesis. Pancreatology. 2008;8:110–25. doi: 10.1159/000123838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7:357–69. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]
- 10.Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov. 2007;6:793–810. doi: 10.1038/nrd2397. [DOI] [PubMed] [Google Scholar]
- 11.Suh N, Wang Y, Honda T, et al. A novel synthetic oleanane triterpenoid, CDDO, with potent differentiating, antiproliferative, and anti-inflammatory activity. Cancer Res. 1999;59:336–41. [PubMed] [Google Scholar]
- 12.Balasubramanian S, Chandraratna RA, Eckert RL. Suppression of human pancreatic cancer cell proliferation by AGN194204, an RXR-selective retinoid. Carcinogenesis. 2004;25:1377–85. doi: 10.1093/carcin/bgh122. [DOI] [PubMed] [Google Scholar]
- 13.Samudio I, Konopleva M, Hail N, Jr, et al. CDDO-Im directly targets mitochondrial glutathione to induce apoptosis in pancreatic cancer. J Biol Chem. 2005;280:36273–82. doi: 10.1074/jbc.M507518200. [DOI] [PubMed] [Google Scholar]
- 14.Garcea G, Dennison AR, Steward WP, Berry DP. Role of inflammation in pancreatic carcinogenesis and the implications for future therapy. Pancreatology. 2005;5:514–29. doi: 10.1159/000087493. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Rigas B. NF-kappaB, inflammation and pancreatic carcinogenesis: NF-kappaB as a chemoprevention target. Int J Oncol. 2006;29:185–92. [PubMed] [Google Scholar]
- 16.Algul H, Treiber M, Lesina M, Schmid RM. Mechanisms of disease: chronic inflammation and cancer in the pancreas--a potential role for pancreatic stellate cells? Nat Clin Pract Gastroenterol Hepatol. 2007;4:454–62. doi: 10.1038/ncpgasthep0881. [DOI] [PubMed] [Google Scholar]
- 17.Sporn MB. Dichotomies in cancer research: some suggestions for a new synthesis. Nat Clin Pract Oncol. 2006;3:364–73. doi: 10.1038/ncponc0536. [DOI] [PubMed] [Google Scholar]
- 18.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 19.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 20.Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J Mol Diagn. 2008;10:111–22. doi: 10.2353/jmoldx.2008.070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Honda T, Rounds BV, Bore L, et al. Novel synthetic oleanane triterpenoids: a series of highly active inhibitors of nitric oxide production in mouse macrophages. Bioorg Med Chem Lett. 1999;9:3429–34. doi: 10.1016/s0960-894x(99)00623-x. [DOI] [PubMed] [Google Scholar]
- 22.Honda T, Honda Y, Favaloro FG, Jr, et al. A novel dicyanotriterpenoid, 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-onitrile, active at picomolar concentrations for inhibition of nitric oxide production. Bioorg Med Chem Lett. 2002;12:1027–30. doi: 10.1016/s0960-894x(02)00105-1. [DOI] [PubMed] [Google Scholar]
- 23.Yates MS, Tauchi M, Katsuoka F, et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther. 2007;6:154–62. doi: 10.1158/1535-7163.MCT-06-0516. [DOI] [PubMed] [Google Scholar]
- 24.Boehm MF, Zhang L, Zhi L, et al. Design and synthesis of potent retinoid X receptor selective ligands that induce apoptosis in leukemia cells. J Med Chem. 1995;38:3146–55. doi: 10.1021/jm00016a018. [DOI] [PubMed] [Google Scholar]
- 25.Honda T, Janosik T, Honda Y, et al. Design, synthesis, and biological evaluation of biotin conjugates of CDDO for the isolation of the protein targets. J Med Chem. 2004;47:4923–32. doi: 10.1021/jm049727e. [DOI] [PubMed] [Google Scholar]
- 26.Liby K, Risingsong R, Royce DB, et al. Prevention and treatment of experimental estrogen receptor-negative mammary carcinogenesis by the synthetic triterpenoid CDDO-methyl ester and the rexinoid LG100268. Clin Cancer Res. 2008;14:4556–63. doi: 10.1158/1078-0432.CCR-08-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6:2969–72. [PubMed] [Google Scholar]
- 28.Maitra A, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20:211–26. doi: 10.1016/j.bpg.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 29.Yore MM, Liby KT, Honda T, Gribble GW, Sporn MB. The synthetic triterpenoid CDDO-imidazole blocks nuclear factor-kappaB activation through direct inhibition of IkappaB kinase beta. Mol Cancer Ther. 2006;5:3232–9. doi: 10.1158/1535-7163.MCT-06-0444. [DOI] [PubMed] [Google Scholar]
- 30.Mihaljevic AL, Michalski CW, Friess H, Kleeff J. Molecular mechanism of pancreatic cancer--understanding proliferation, invasion, and metastasis. Langenbecks Arch Surg. 2010;395:295–308. doi: 10.1007/s00423-010-0622-5. [DOI] [PubMed] [Google Scholar]
- 31.Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–27. [PubMed] [Google Scholar]
- 32.Huang C, Yang G, Jiang T, Huang K, Cao J, Qiu Z. Effects of IL-6 and AG490 on regulation of Stat3 signaling pathway and invasion of human pancreatic cancer cells in vitro. J Exp Clin Cancer Res. 2010;29:51. doi: 10.1186/1756-9966-29-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin L, Hutzen B, Zuo M, et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res. 2010;70:2445–54. doi: 10.1158/0008-5472.CAN-09-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harsha HC, Jimeno A, Molina H, et al. Activated epidermal growth factor receptor as a novel target in pancreatic cancer therapy. J Proteome Res. 2008;7:4651–8. doi: 10.1021/pr800139r. [DOI] [PubMed] [Google Scholar]
- 35.Deng J, Grande F, Neamati N. Small molecule inhibitors of Stat3 signaling pathway. Curr Cancer Drug Targets. 2007;7:91–107. doi: 10.2174/156800907780006922. [DOI] [PubMed] [Google Scholar]
- 36.Liby K, Risingsong R, Royce DB, et al. Triterpenoids CDDO-methyl ester or CDDO-ethyl amide and rexinoids LG100268 or NRX194204 for prevention and treatment of lung cancer in mice. Cancer Prev Res. 2009;2:1050–8. doi: 10.1158/1940-6207.CAPR-09-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liby K, Royce DB, Williams CR, et al. The synthetic triterpenoids CDDO-Methyl ester and CDDO-Ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Can Res. 2007;67:2414–9. doi: 10.1158/0008-5472.CAN-06-4534. [DOI] [PubMed] [Google Scholar]
- 38.Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feldmann G, Habbe N, Dhara S, et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut. 2008;57:1420–30. doi: 10.1136/gut.2007.148189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Funahashi H, Satake M, Dawson D, et al. Delayed progression of pancreatic intraepithelial neoplasia in a conditional Kras(G12D) mouse model by a selective cyclooxygenase-2 inhibitor. Cancer Res. 2007;67:7068–71. doi: 10.1158/0008-5472.CAN-07-0970. [DOI] [PubMed] [Google Scholar]
- 41.Fendrich V, Chen NM, Neef M, et al. The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progression of pancreatic intraepithelial neoplasia and cancer formation in a genetically engineered mouse model of pancreatic cancer. Gut. 2010;59:630–7. doi: 10.1136/gut.2009.188961. [DOI] [PubMed] [Google Scholar]
- 42.Mohammed A, Janakiram NB, Li Q, et al. EGFR inhibitor gefitinib prevents progression of pancreatic lesions to carcinoma in a conditional LSL-KrasG12D/+ transgenic mice model. Cancer Prev Res. 2010;3:XXX. doi: 10.1158/1940-6207.CAPR-10-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]