

Abstract
Purpose
To estimate the maximum tolerated dose (MTD); study the pharmacology of escalating doses of gefitinib combined with radiation therapy in patients ≤21 years with newly diagnosed intrinsic brainstem gliomas (BSG) and incompletely resected supratentorial malignant gliomas (STMG); and to investigate epidermal growth factor receptor (EGFR) amplification and expression in STMG.
Patients and methods
Three strata were identified: Stratum 1A - BSG; Stratum IB - incompletely resected STMG not receiving enzyme inducing anti-convulsant drugs (EIACD); and Stratum II - incompletely resected STMG receiving EIACD. Dose escalation using a modified 3 + 3 cohort design was performed in strata IA & II. The initial gefitinib dosage was 100mg/m2/day commencing with radiation therapy and the dose-finding period extended until 2 weeks post-radiation. Pharmacokinetics (PK) and biology studies were performed in consenting patients.
Results
Of 23 eligible patients, 20 were evaluable for dose-finding. MTDs for strata IA and II were not established as accrual was halted due to four patients experiencing symptomatic intratumoral hemorrhage (ITH); 2 during and 2 post dose-finding. ITH was observed in 0 of 11 patients treated at 100mg/m2/day, 1 of 10 at 250mg/m2/day, and 3 of 12 at 375mg/m2/day. Subsequently a second patient at 250mg/m2/day experienced ITH. PK analysis showed the median gefitinib systemic exposure increased with dosage (p=0.04). EGFR was overexpressed in 5 of 11 STMG and amplified in 4 (36%) samples.
Conclusion
This trial provides clear evidence of EGFR amplification in a significant proportion of paediatric STMG and 250mg/m2/day was selected for the Phase II trial.
Keywords: epidermal growth factor receptor, gefitinib, radiotherapy, brain stem neoplasms, supratentorial neoplasms, glioma
Introduction
Diffusely infiltrating brainstem gliomas (BSG) have been amongst the most therapeutically resistant paediatric brain tumors; 1- and 5-year progression-free survival (PFS) rates for these patients are less than 25% and 10%, respectively.1, 2 A paucity of data exist to document the histology and biology of these tumors; in the limited instances where tissue has been obtained, histology has shown high-grade or infiltrating glioma.3–7 More recent experience confirms the similarity of BSG to more accessible supratentorial malignant gliomas, a tumor setting where prognosis for children following incomplete resection is also quite poor with 1- and 5-year PFS rates of less than 50% and 20%, respectively.8–10
In view of these discouraging results, new therapeutic approaches are needed. Recent data demonstrate that a wide variety of tumors, including malignant gliomas, are driven to proliferate by aberrant activation of growth factor receptor-mediated signal transduction pathways.11,12 Constitutive activation of these pathways also contributes to tumor resistance to conventional therapeutic agents.
Cell signaling via the EGFR (also known as ERBB1) has been implicated in the development of adult and paediatric high-grade gliomas.13,14 EGFR amplification and over-expression affect 30%–50% of adult glioblastoma multiformes.13–16 In paediatric high-grade gliomas, available data suggest that while EGFR amplification occurs with low frequency,17,18 EGFR receptor over-expression is relatively common. Bredel et al reported elevated expression of this receptor in 81% of paediatric STMG, with over half demonstrating over-expression in >90% of tumor cells.18 PBTC earlier showed that, EGFR protein is expressed to high levels and amplified in samples of childhood BSG.19 These data suggest that the EGFR constitutes a promising therapeutic target for paediatric STMG and BSG.
Gefitinib (ZD1839, Iressa™, AstraZeneca), a low molecular weight synthetic molecule, is a potent and selective inhibitor of the EGFR tyrosine kinase that works by competing with adenosine triphosphate for its binding site, and blocking signal transduction pathways implicated in cancer cell proliferation, survival and other host-dependent processes thought to promote cancer growth.20 At the time this clinical trial (PBTC-007) was initiated, gefitinib had demonstrated preclinical evidence of antitumor activity alone and in combination with irradiation and had shown good antitumor activity in a wide range of human tumor xenografts after oral administration.
In both BSG and incompletely resected STMG, radiation therapy has demonstrated benefit.21 Preclinical studies have demonstrated radiosensitization with concurrent exposure to EGFR specific inhibitory agents, providing further rationale for trials of upfront combinations of EGFR inhibitors and concurrent irradiation.22
In adult phase I studies, gefitinib was well tolerated after either intermittent or continuous dosing.23–26 In these trials, dose-related toxicity was confined to the skin and gastrointestinal system; rarely, hepatic enzyme elevation occurred. Increasing intolerability was noted at daily doses of ≥600 mg. The combination of preclinical antitumor activity, known over-expression of the target pathway, and acceptable toxicity profile led us to study the agent in paediatric malignant gliomas.
The PBTC conducted a phase I trial of gefitinib in combination with radiation therapy in children with newly diagnosed BSG and incompletely resected STMG. The primary objectives were to define the safety of gefitinib administered orally once daily in combination with radiation therapy and to describe dose-limiting toxicities. Secondary objectives included characterizing the pharmacokinetic and pharmacogenetics of gefitinib in this patient population and to investigate EGFR expression and amplification in STMG.
Patients and Methods
Patient Eligibility
Patients ≥3 and ≤21 years of age with a newly diagnosed non-disseminated BSG or incompletely resected STMG were eligible. Other eligibility criteria included Karnofsky or Lansky performance score ≥50%, no prior chemotherapy (except corticosteroids) or radiotherapy, adequate bone marrow, renal, and hepatic function. Patients could not be pregnant, have an uncontrolled infection, or a history of deep venous or arterial thrombosis.
The institutional review boards (IRBs) of each participating PBTC institution approved the protocol before initial patient enrollment, and continuing approval was maintained throughout the study. Patients or their legal guardians gave written informed consent, and assent was obtained as appropriate at the time of enrollment.
Studies Before and During Treatment
A complete history, physical exam including detailed neurological exam and laboratory studies were obtained before treatment and periodically thereafter. Pretreatment evaluation included: CBC, electrolytes including magnesium, renal function tests (serum creatinine and BUN), liver function tests, fibrinogen, anticonvulsant levels in patients receiving enzyme-inducing anticonvulsant drugs (EIACD), and β-HCG in females of childbearing potential. MRI was obtained prior to therapy and at 8 week intervals during therapy.
Dosage, Drug Administration, and Treatment Plan
Gefitinib was provided in tablets that could be dissolved in water, as necessary. Patients received gefitinib once daily; a course was defined as 4 weeks of therapy. In the absence of disease progression or dose-limiting toxicity, treatment was continued for 13 courses (1 year).
Patients received local irradiation using conventional or conformal techniques; imaging and treatment plan were centrally reviewed (Quality Assurance Review Center, QARC). A total dose of 55.8 Gy was given in 180 cGy daily fractions. Treatment was initiated within 4 weeks of diagnosis (BSG) or surgery (STMG).
Dose Escalation, Dose-Limiting Toxicity, and Maximum Tolerated Dose
The initial gefitinib dosage was 100mg/m2/day commencing with radiation therapy. Dose escalation for patients in Strata IA and II were performed as shown in Table 1; patients registered in Stratum IB were assigned to the current dose level in Stratum IA.
Table 1.
Dose-Limiting Toxicities and Intratumoral Hemorrhage Summaries
| Stratum | Dose | No. of Patients Entered | No. of Evaluable Patients | No. of Patients with DLT | Observed DLTs Courses 1 and 2 | Observed CNS Hemorrhage Courses 3+ |
|---|---|---|---|---|---|---|
| IA* (BSG) | 100 | 6 | 6 | 2^ | Gr-2 Fibrinogen Gr-3 Lymphopenia |
|
| 250 | 7 | 6 | 0 | Gr-4 | ||
| 375 | 7 | 5 | 1 | Gr-3 Rash/desquamation | Gr-4 | |
| IB§ (STMG) | 100 | 2 | 2 | 0 | ||
| 250 | 3 | 3 | 0 | Gr-4 | ||
| 375 | 5 | 5 | 3 | Gr-3 CNS hemorrhage Gr-5 CNS hemorrhage Gr-3 Dehydration Gr-3 Fibrinogen Gr-3 Vomiting |
||
| II (STMG + EIACD) | 100 | 3 | 3 | 0 |
No patients in this cohort received enzyme-inducing anticonvulsants.
Upon observing these two DLTs, the protocol was amended and the DLT definition was changed.
Dose-escalation was not based on DLTs observed in stratum IB
Abbreviations: DLT=Dose-limiting Toxicity, BSG=Brain stem gliomas, STMG=Supertentorial Malignant Gliomas, EIACD=Enzyme Inducing Anti-convulsant Drugs, Gr=grade
The dose-finding interval extended from initiation through 2 weeks following completion of radiation therapy. A traditional 3+3 dose-finding algorithm was used to empirically estimate MTDs for strata IA and II. Cohorts of up to six patients could be enrolled at a dosage; the decision to escalate the dosage for the next cohort was made after three patients had completed the dose-finding interval. The MTD was defined as the highest dosage at which at least five of six patients did not experience DLT and the next higher dosage was unacceptably toxic.
Toxicities were graded according to the NCI Common Toxicity Criteria Version 2.0. DLTs were defined as follows: Grade 3 or 4 thrombocytopenia, grade 4 neutropenia, any grade 3 or 4 non-hematologic toxicity, any grade 2 non-hematologic toxicity persisting for more than 7 days and considered sufficiently significant or intolerable by patients to warrant treatment interruption and/or dose reduction, and any toxicity requiring interruption of radiation therapy for >5 consecutive days or 10 days total.
Patients who came off therapy during the DLT observation period for reasons other than toxicity or missed >7 days of gefitinib for reasons other than toxicity were replaced for purposes of estimating the MTD.
Pharmacokinetic Studies
Pharmacokinetic studies were performed in consenting patients. Serial blood samples for pharmacokinetic studies were collected on days 10, 11, or 12 of course 1 before gefitinib administration, and at 1, 2, 4, 6, 8, 12, and 24 hours after administration. Gefitinib concentrations were analyzed by isocratic reversed-phase high performance liquid chromatography with electrospray ionization mass spectrometric detection.27 A one-compartment model was fitted to the gefitinib plasma concentrations using maximum likelihood estimation as implemented in ADAPT II.28 The model parameters for each patient were used to simulate the plasma concentration-time profile, from which the area under the plasma concentration-time curve (AUC0-24) was calculated using the log-linear trapezoidal method.
EGFR amplification and expression
Diagnostic fixed tumour samples were obtained from consenting patients and analyzed by immunohistochemistry (IHC) to determine the expression of total EGFR, phospho-ERK1/2, and phospho-S6S235/236. To remove observer bias, IHC staining of tumours was scored blind to treatment response using ImageJ software analysis that provides a measure of the mean percentage of immunopositivity detected in each ×200 field.33 Dual-probe fluorescence in situ hybridization (FISH) was performed on paraffin-embedded sections with locus-specific probes for EGFR (RP11-148P17) and the 7q control probe (7q31.2, RP11-460J21 + CTB-133K23).
Results
Three of 23 patients enrolled in stratum IA and II were not evaluable for dose-finding: all withdrew without toxicity 14, 15, and 46 days after the start of gefitinib. Ten patients were enrolled in stratum IB. Patient characteristics are summarized in Table 2.
TABLE 2.
Patient demographics
| Numbers of Patients | ||||
|---|---|---|---|---|
| Stratum IA (BSG) | Stratum IB (STMG) | Stratum II (STMG + EIACDs) | ||
| Gender | Male | 6 | 8 | 1 |
| Female | 14 | 2 | 2 | |
| Ethnicity | Unknown | . | 1 | . |
| Hispanic or Latino | 5 | 1 | . | |
| Non-Hispanic | 15 | 8 | 3 | |
| Diagnosis | AA | 2* | . | 1 |
| BSG | 18 | . | . | |
| GBM | . | 9 | 1 | |
| MG | . | . | 1 | |
| MOA | . | 1 | . | |
|
Age at Study Entry: Median (min, max) |
7.4 (3.4, 15.1) |
13.5 (3.1, 20.9) |
12.8 (10.4, 15.2) |
|
Two BSG patients were biopsied
Abbreviations: AA=Anaplastic Astrocytoma, BSG=Brain Stem Gliomas, GBM=Glioblastoma Multiforme, MG=Malignant Gliomas, MOA=Mixed Oligoastrocytoma, STMG=Supertentorial Malignant Gliomas, EIACD=Enzyme Inducing Anti-Convulsant Drugs
Estimation of MTD
Twenty patients were evaluable for dose-finding, including 17 in stratum IA, and 3 in stratum II (Table 1). Of the first six patients treated at 100mg/m2/day in Stratum IA, one experienced grade 2 fibrinogen (hypofibrinogenemia) and another grade 3 lymphopenia; both DLTs, prompting a protocol amendment to exclude non-clinically significant hematologic toxicities as DLTs. Subsequently only one of five evaluable patients in stratum IA treated at the highest dosage investigated (375mg/m2/day) experienced a DLT (grade 3 rash/desquamation). None of the three patients in stratum II treated at the highest dosage (100mg/m2/day) evaluated experienced a DLT. While dose escalation was not based on Stratum IB, three of five patients at 375mg/m2/day experienced DLTs (one grade 5 ITH, one grade 3 ITH & one grade 3 hypofibrinogenemia, dehydration and vomiting) during courses 1 and 2 (Table 1).
Although systemic toxicities were generally mild-moderate and reversible (Tables 2 and 3), there were four instances of symptomatic ITH noted among the 33 eligible patients prompting closure of the trial without having estimated MTDs in strata IA or II. Three patients experienced ITH among 12 treated at the 375mg/m2/day dosage, one among 10 at the 250mg/m2/day dosage and none among 11 at 100mg/m2/day (Table 3). Subsequent to closure another patient treated at 250mg/m2/day was retrospectively noted to have experienced an ITH post dose-finding. The difference in cumulative incidence functions of ITH among the three dosages investigated was not statistically significant (p=0.20). No relationship to concomitant dexamethasone administration or evidence of thrombocytopenia or coagulation abnormality was noted. Three instances of ITH occurred in patients with STMG and two in patients with BSG. In one patient with a BSG, ITH was associated with progressive tumour, and the death of a patient with a GBM was attributed to ITH.
Table 3.
Non dose-limiting toxicities (≥ Grade 2) attributed to therapy and observed in 4 or more (> 10%) of the 30 evaluable patients
| Grade | Stratum IA (BSG) | Stratum IB (STMG) | Stratum II (STMG + EIACDs) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crs-1 and 2 | Crs-3+ | Crs-1 and 2 | Crs-3+ | Crs-1 and 2 | Crs-3+ | |||||||
| 2 | 3 | 2 | 3 | 4 | 2 | 3 | 2 | 3 | 4 | 2 | 2 | |
| Lymphopenia | 2 | 6 | 3 | 5 | 2 | |||||||
| Diarrhea patients without colostomy | 3 | 3 | 1 | 1 | ||||||||
| Rash/desquamation | 3 | 2 | 2 | 1 | ||||||||
| Leukocytes (total WBC) | 6 | 1 | ||||||||||
| Vomiting | 3 | 1 | 1 | 2 | ||||||||
| Fatigue (lethargy, malaise, asthenia) | 2 | 3 | 1 | 1 | ||||||||
| SGPT (ALT) (serum glutamic pyruvic transminase) | 3 | 1 | 1 | 1 | ||||||||
| Neutrophils/granulocytes (ANC/AGC) | 3 | 1 | 1 | |||||||||
| Headache | 2 | 1 | 1 | |||||||||
| Fibrinogen | 1 | 2 | 1 | |||||||||
| Nausea | 1 | 2 | ||||||||||
| CNS hemorrhage/bleeding | 2 | 1 | ||||||||||
Abbreviations: BSG=Brain stem gliomas, STMG=Supertentorial Malignant Gliomas, EIACD=Enzyme Inducing Anti-convulsant Drugs, Crs=course
For the twenty patients with BSG (Stratum IA), the 1-year survival and PFS rates were 48% (SE 11%) and 16.1% (SE 7.4%), respectively. For the 13 patients with STMG, the 1-year survival and PFS rates were 28.8% (SE 12.2%) and 15.4% (SE 8.2%), respectively.
Pharmacokinetics
Serial samples for gefitinib pharmacokinetic studies were collected from 22 patients during week 2 of course one – none of whom were receiving EIACD. As summarized in Table 4, the median gefitinib AUC value increased with increasing dosage (p=0.04). At the recommended phase II dosage (250mg/m2/day), the median peak gefitinib plasma concentration was 0.83 μg/ml (range 0.45 to 1.36 μg/ml); observed at a median of 4.2 hr (range 2 to 6 hr) after drug administration. The median gefitinib half-life for all patients studied was 11.1 hr (range 1.8 to 41.3 hr). The median apparent oral clearance for all patients studied was 15.7 L/hr/m2 (range 1.8 to 34.0 L/hr/m2). The effect of dexamethasone administration on gefitinib apparent oral clearance was studied in seven patients who had pharmacokinetic studies conducted while on a stable dexamethasone dosage and later after discontinuing dexamethasone: no statistically significant difference was noted (paired t-test; p=0.15). In our limited analyses no evidence of a relationship between gefitinib pharmacokinetics and toxicity was observed.
Table 4.
Summary* of gefitinib pharmacokinetic parameters in relation to dosage
| 100 mg/m2/d | 250 mg/m2/d | 375 mg/m2/d | |
|---|---|---|---|
| No. of patients | 8 | 6 | 8 |
| Actual dosage (mg/m2/d) | 98.1 (86.2–104.2) | 252/6 (244.8–261.6) | 379.9 (367.6–410.9) |
| Cmax† (μg/mL) | 0.44 (0.28–0.71) | 0.83 (0.45–1.36) | 1.89 (0.93–2.42) |
| Tmax† (h) | 4.9 (1.2–6.2) | 4.2 (2.0–6.0) | 4.2 (2.3–6.4) |
| t1/2 (h) | 9.9 (1.8–19.9) | 17.6 (4.8–41.3) | 10.4 (3.8–39.2) |
| Cl/F (L/h/m2) | 12.8 (1.8–21.8) | 20.9 (12.6–33.9) | 15.0 (5.8–25.8) |
| AUC0-24 (μg/mL*h) | 5.3 (3.7–11.8) | 11.8 (4.3–16.7) | 25.3 (18.4–35.3) |
Values are median and range.
Observed Cmax and Tmax.
Abbreviations: Cmax,=maximum concentration; Tmax,=time of maximum gefitinib concentration; Cl/F=apparent oral clearance; AUC0-24=area under the concentration-time curve from 0 to 24 hours.
Tumour EGFR amplification and expression
Intense membrane EGFR immunoreactivity was evident in tumour material from 5 of 11 patients with STMG (IB n=8, II n=3). Three of the eleven cases displayed high-level EGFR amplification (Figure 1); one additional case displayed low-level gain in the context of polyploidy. Each of these four cases displayed intense membrane-associated EGFR immunoreactivity in contrast to only one tumour sample that was diploid for the EGFR locus (p=0.01, Fisher’s Exact test).
FIGURE 1.
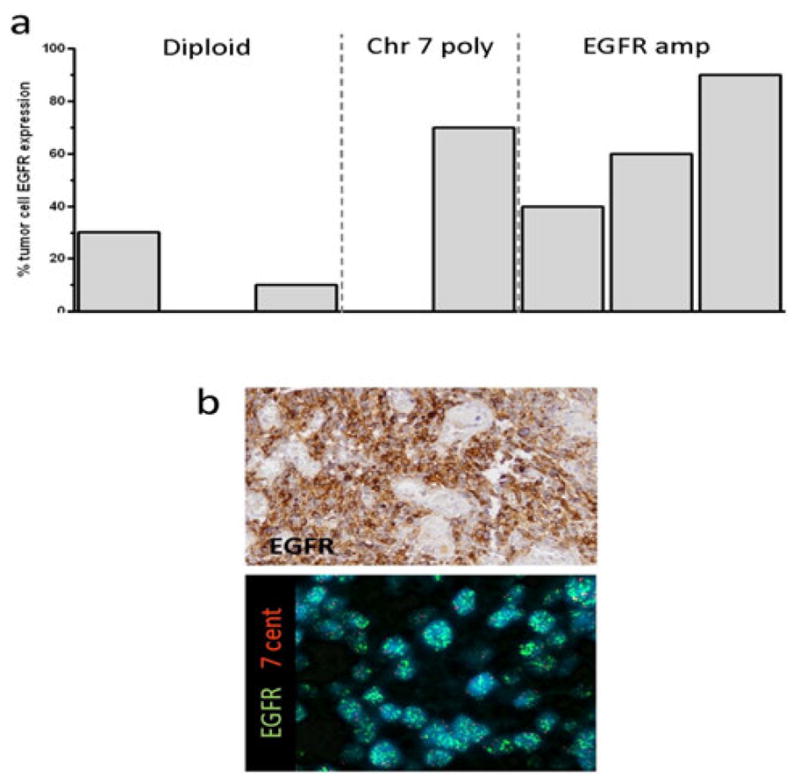
a. Graph summarizing percent of tumour cells expressing epidermal growth factor receptor (EGFR) according to EGFR copy number status. b. Top panel: EGFR immunohistochemistry showing intense membrane staining in the sample in which 90% of cells express EGFR. Bottom panel: FISH analysis confirming high-level EGFR amplification in this same tumour.
Discussion
This study provides clear evidence that EGFR is amplified and highly-expressed in a significant proportion of paediatric STMG. The PBTC reported previously that one-half of high-grade intrinsic BSG express elevated levels of EGFR in the context of gene amplification.19 EGFR overexpression was also reported in paediatric STMG although only a handful of tumours displayed an increase in gene copy number.15–17 Here, we identified intense membrane EGFR immunoreactivity in 5 of 11 (45%) STMG; of which three (27%) and one (9%) contained high-level and low-level gene amplification, respectively. A very recent publication by Zarghoon et al38 based on whole-genome profiling techniques suggest that platelet-derived growth factor receptor α and Poly polymerase as potential therapeutic targets in BSG. They also noted EGFR immunopositivity in 7 of their 11 (64%) cases and low copy number gain in one. Together these data indicate that EGFR is likely to play an important role in the biology of paediatric as well as adult STMG and justifies a phase II evaluation of gefitinib in newly diagnosed paediatric patients with these tumours.
The pharmacokinetics of gefitinib are similar to those previously reported for children with refractory solid tumours receiving gefitinib as a single agent.34 The median apparent oral gefitinib clearance in that study was 14.8 L/hr/m2 compared with 15.7 L/hr/m2 for this trial. A wide variation was noted in our gefitinib apparent oral clearance, similar to the wide interpatient variability noted in the previous paediatric study (4.8 to 24.8 L/hr/m2).34 Although the maximum gefitinib plasma concentration and area under the concentration-time curve generally increased with gefitinib dosage, marked inter-patient variability within each dosage level was noted. Recent reports have suggested that gefitinib undergoes preferential distribution from the blood into brain tumour tissue.35,36 Thus, at the dosages tolerated and gefitinib exposures achieved in the present study it is likely that tumour concentrations approached or exceeded the concentration (2 μmol/l) necessary for inhibition of wild-type EGFR.35
Due to concern that the observed intratumoural hemorrhages could be related to the study drug, accrual to the Phase I component of the study was halted before MTDs could be estimated. Because of the apparently higher incidence of ITH at 375mg/m2, treating patients at this or higher gefitinib dosages in the efficacy phase of the trial would be unacceptable, leading us to recommend studying the 250mg/m2 dosage level in the phase II trial for patients newly diagnosed with a BSG. This is in contrast to the MTD of 400 mg/m2/day established for paediatric solid tumours.34
A subsequent retrospective review of newly diagnosed BSG treated at a single institution with radiation therapy with or without concurrent cytotoxic agents showed symptomatic ITH in 9 of 48 patients within 12 months of diagnosis.37 Results which are similar to symptomatic ITH occurring in 2 of 20 patients with BSG and 3 of 13 patients with STMG in our trial.
Our current knowledge of the tumour- or radiation-related ITH, as discussed above, suggests the apparent dose-related incidence associated with gefitinib may have been spurious. However given data available at the time, it was reasonable to stop the trial early to ensure patient safety. The pharmacokinetics of gefitinib indicate that the 250mg/m2/day dosing selected for the phase II trial will achieve tumour concentrations necessary for inhibition of wild-type EGFR, which our data suggest will be amplified or highly-expressed in a significant proportion of high-grade gliomas.
Acknowledgments
Role of Funding Source
This study was sponsored by the US National Cancer Institute (NCI) and coordinated by the Pediatric Brain Tumor Consortium. This work was supported by NIH grant U01 CA81457 for the Pediatric Brain Tumor Consortium, NIH grant 5M01RR000188 (General Clinical Research Center for Texas Children’s Cancer Center), AstraZeneca, and the American Lebanese Syrian Associated Charities (ALSAC). The NCI provided input on the design of the trial and reviewed the final manuscript but had no role in the data collection, analysis and interpretation or decision to publish. Astrazeneca provided partial financial support for the conduct of the pharmacokinetic studies but no input into the study design or conduct. The GCRC and ALSAC had no role in the study design, data collection, data analysis, data interpretation, or writing of the report.
The authors and the PBTC acknowledge administrative and statistical support of Ms. Dana Wallace and the clinical research assistant support of Ms. Stacye Richardson.
Footnotes
Presented in part at the American Society of Clinical Oncology meeting held May 31 – June 3, 2003 in Chicago, IL; the American Society for Therapeutic Radiology and Oncology held 3–7 October 2004 in Atlanta, GA; and the American Society of Neuroradiology held April 29 – May 5, 2006 in San Diego, CA.
Conflict of interest statement
Dr. Stewart received research funding from Astrazeneca for the conduct of the pharmacokinetic studies. Dr. Gururangan disclosed a consultant or advisory role with Boehringer Ingelheim Pharma. No other conflicts of interest have been declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pollack IF. Brain tumors in children. N Engl J Med. 1994;331:1500–1507. doi: 10.1056/NEJM199412013312207. [DOI] [PubMed] [Google Scholar]
- 2.Jennings MT, Freeman ML, Murray MJ. Strategies in the treatment of diffuse pontine gliomas: the therapeutic role of hyperfractionated radiotherapy and chemotherapy. J Neurooncol. 1996;28:207–222. doi: 10.1007/BF00250200. [DOI] [PubMed] [Google Scholar]
- 3.Berger MS, Edwards MS, LaMasters D, Davis RL, Wilson CB. Pediatric brain stem tumors: radiographic, pathological, and clinical correlations. Neurosurgery. 1983;12:298–302. doi: 10.1227/00006123-198303000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Mantravadi RV, Phatak R, Bellur S, Liebner EJ, Haas R. Brain stem gliomas: an autopsy study of 25 cases. Cancer. 1982;49:1294–1296. doi: 10.1002/1097-0142(19820315)49:6<1294::aid-cncr2820490636>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 5.Epstein F, Wisoff JH. Intrinsic brainstem tumors in childhood: surgical indications. J Neurooncol. 1988;6:309–317. doi: 10.1007/BF00177425. [DOI] [PubMed] [Google Scholar]
- 6.Cartmill M, Punt J. Diffuse brain stem glioma. A review of stereotactic biopsies. Childs Nerv Syst. 1999;15:235–237. doi: 10.1007/s003810050379. [DOI] [PubMed] [Google Scholar]
- 7.Albright AL, Packer RJ, Zimmerman R, Rorke LB, Boyett J, Hammond GD. Magnetic resonance scans should replace biopsies for the diagnosis of diffuse brain stem gliomas: a report from the Children’s Cancer Group. Neurosurgery. 1993;33:1026–1029. doi: 10.1227/00006123-199312000-00010. [DOI] [PubMed] [Google Scholar]
- 8.Roujeau T, Machado G, Garnett MR, Miquel C, Puget S, Geoerger B, et al. Stereotactic biopsy of diffuse pontine lesions in children. J Neurosurg. 2007;107:1–4. doi: 10.3171/PED-07/07/001. [DOI] [PubMed] [Google Scholar]
- 9.Pirotte BJ, Lubansu A, Massager N, Wikler D, Goldman S, Levivier M. Results of positron emission tomography guidance and reassessment of the utility of and indications for stereotactic biopsy in children with infiltrative brainstem tumors. J Neurosurg. 2007;107:392–399. doi: 10.3171/PED-07/11/392. [DOI] [PubMed] [Google Scholar]
- 10.Chastagner P, Kalifa C, Doz F, Bouffet E, Gentet JC, Ruchoux MM, et al. Outcome of children treated with preradiation chemotherapy for a high-grade glioma: results of a French Society of Pediatric Oncology (SFOP) Pilot Study. Pediatr Blood Cancer. 2007;49:803–807. doi: 10.1002/pbc.21051. [DOI] [PubMed] [Google Scholar]
- 11.Pollack IF, Bredel M, Erff M. Application of signal transduction inhibition as a therapeutic strategy for central nervous system tumors. Pediatric Neurosurgery. 1998;29:228–244. doi: 10.1159/000028729. [DOI] [PubMed] [Google Scholar]
- 12.Aaronson SA. Growth factors and cancer. Science. 1991;254:1146–1153. doi: 10.1126/science.1659742. [DOI] [PubMed] [Google Scholar]
- 13.Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313:144–147. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 14.Wong AJ, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci U S A. 1987;84:6899–6903. doi: 10.1073/pnas.84.19.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Deimling A, Louis DN, von Ammon K, Petersen I, Hoell T, Chung RY, et al. Association of epidermal growth factor receptor gene amplification with loss of chromosome 10 in human glioblastoma multiforme. J Neurosurg. 1992;77:295–301. doi: 10.3171/jns.1992.77.2.0295. [DOI] [PubMed] [Google Scholar]
- 16.Humphrey PA, Gangarosa LM, Wong AJ, Archer GE, Lund-Johansen M, Bjerkvig R, et al. Deletion-mutant epidermal growth factor receptor in human gliomas: effects of type II mutation on receptor function. Biochem Biophys Res Commun. 1991;178:1413–1420. doi: 10.1016/0006-291x(91)91051-d. [DOI] [PubMed] [Google Scholar]
- 17.Cheng Y, Ng HK, Zhang SF, Ding M, Pang JC, Zheng J, et al. Genetic alterations in pediatric high-grade astrocytomas. Human Pathology. 1999;30:1284–1290. doi: 10.1016/s0046-8177(99)90057-6. [DOI] [PubMed] [Google Scholar]
- 18.Bredel M, Pollack IF, Hamilton RL, James CD. Epidermal growth factor receptor expression and gene amplification in high-grade non-brainstem gliomas of childhood. Clin Cancer Res. 1999;5:1786–1792. [PubMed] [Google Scholar]
- 19.Gilbertson RJ, Hill DA, Hernan R, Kocak M, Geyer R, Olson J, et al. ERBB1 is amplified and overexpressed in high-grade diffusely infiltrative pediatric brain stem glioma. Clin Cancer Res. 2003;9:3620–3624. [PubMed] [Google Scholar]
- 20.Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacology and Therapeutics. 1999;82:241–250. doi: 10.1016/s0163-7258(98)00045-x. [DOI] [PubMed] [Google Scholar]
- 21.Packer RJ, Boyett JM, Zimmerman RA, Rorke LB, Kaplan AM, Albright AL, et al. Hyperfractionated radiation therapy (72 Gy) for children with brain stem gliomas. A Childrens Cancer Group Phase I/II Trial. Cancer. 1993;72:1414–1421. doi: 10.1002/1097-0142(19930815)72:4<1414::aid-cncr2820720442>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 22.O’Rourke DM, Kao GD, Singh N, Park BW, Muschel RJ, Wu CJ, et al. Conversion of a radioresistant phenotype to a more sensitive one by disabling erbB receptor signaling in human cancer cells. Proc Natl Acad Sci USA. 1998;95:10842–10847. doi: 10.1073/pnas.95.18.10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ranson M, Hammond LA, Ferry D, Kris M, Tullo A, Murray PI, et al. ZD1839, a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: results of a phase I trial. J Clin Oncol. 2002;20:2240–2250. doi: 10.1200/JCO.2002.10.112. [DOI] [PubMed] [Google Scholar]
- 24.Herbst RS, Maddox AM, Rothenberg ML, Small EJ, Rubin EH, Baselga J, et al. Selective oral epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 is generally well-tolerated and has activity in non-small-cell lung cancer and other solid tumors: results of a phase I trial. J Clin Oncol. 2002;20:3815–3825. doi: 10.1200/JCO.2002.03.038. [DOI] [PubMed] [Google Scholar]
- 25.Baselga J, Rischin D, Ranson M, Calvert H, Raymond E, Kieback DG, et al. Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. J Clin Oncol. 2002;20:4292–4302. doi: 10.1200/JCO.2002.03.100. [DOI] [PubMed] [Google Scholar]
- 26.Nakagawa K, Tamura T, Negoro S, Kudoh S, Yamamoto N, Takeda K, et al. Phase I pharmacokinetic trial of the selective oral epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (‘Iressa’, ZD1839) in Japanese patients with solid malignant tumors. Ann Oncol. 2003;14:922–930. doi: 10.1093/annonc/mdg250. [DOI] [PubMed] [Google Scholar]
- 27.Bai F, Iacono LC, Johnston B, Stewart CF. Determination of gefitinib in plasma by liquid chromatography with a C 12 column and electrospray tandem mass spectrometry detection. J Liquid Chromatography. 2004;27:2743–2758. [Google Scholar]
- 28.D’Argenio DZ, Schumitzky A. ADAPT II User’s Guide. USC, Los Angeles: Biomedical Simulations Resource; 1990. [Google Scholar]
- 29.Kadlubar FF, Berkowitz GS, Delongchamp RR, Wang C, Green BL, Tang G, et al. The CYP3A4*1B variant is related to the onset of puberty, a known risk factor for the development of breast cancer. Cancer Epidemiology, Biomarkers and Prevention. 2003;12:327–331. [PubMed] [Google Scholar]
- 30.Lee SJ, Goldstein JA. Functionally defective or altered CYP3A4 and CYP3A5 single nucleotide polymorphisms and their detection with genotyping tests. Pharmacogenomics. 2005;6:357–371. doi: 10.1517/14622416.6.4.357. [DOI] [PubMed] [Google Scholar]
- 31.Zheng H, Webber S, Zeevi A, Schuetz E, Zhang J, Lamba J, et al. The MDR1 polymorphisms at exons 21 and 26 predict steroid weaning in pediatric heart transplant patients. Human Immunology. 2002;63:765–770. doi: 10.1016/s0198-8859(02)00426-3. [DOI] [PubMed] [Google Scholar]
- 32.Zamber CP, Lamba JK, Yasuda K, Farnum J, Thummel K, Schuetz JD, et al. Natural allelic variants of breast cancer resistance protein (BCRP) and their relationship to BCRP expression in human intestine. Pharmacogenetics. 2003;13:19–28. doi: 10.1097/00008571-200301000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Haley JD, Hsuan JJ, Waterfield MD. Analysis of mammalian fibroblast transformation by normal and mutated human EGF receptors. Oncogene. 1989;4:273–83. [PubMed] [Google Scholar]
- 34.Daw NC, Furman WL, Stewart CF, Iacono LC, Krailo M, Bernstein ML, et al. Phase I and pharmacokinetic study of gefitinib in children with refractory solid tumors: a Children’s Oncology Group Study. J Clin Oncol. 2005;23:6172–6180. doi: 10.1200/JCO.2005.11.429. [DOI] [PubMed] [Google Scholar]
- 35.Prados MD, Yung WK, Wen PY, Junck L, Cloughesy T, Fink K, et al. Phase-1 trial of gefitinib and temozolomide in patients with malignant glioma: a North American brain tumor consortium study. Cancer Chemother Pharmacol. 2008;61:1059–67. doi: 10.1007/s00280-007-0556-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hofer S, Frei K. Gefitinib concentrations in human glioblastoma tissue. J Neurooncol. 2007;82:175–6. doi: 10.1007/s11060-006-9257-3. [DOI] [PubMed] [Google Scholar]
- 37.Broniscer A, Laningham FH, Kocak M, Krasin MJ, Fouladi M, Merchant TE, et al. Intratumoral hemorrhage among children with newly diagnosed, diffuse brainstem glioma. Cancer. 2006;106:1364–71. doi: 10.1002/cncr.21749. [DOI] [PubMed] [Google Scholar]
- 38.Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, Bouffet E, Hawkins C. Whole-Genome Profiling of Pediatric Diffuse Intrinsic Pontine Gliomas Highlights Platelet-Derived Growth Factor Receptor α and Poly (ACP-ribose) Polymerase As Potential Therapeutic Targets. Journal of Clinical Oncology. 2010;28(8):1337–1344. doi: 10.1200/JCO.2009.25.5463. [DOI] [PubMed] [Google Scholar]