

Abstract
HIV protease inhibitors (HIV PI) reduce morbidity and mortality of HIV infection but cause multiple untoward effects. Because certain HIV PI evoke production of reactive oxygen species (ROS) and volume-sensitive Cl− current (ICl,swell) is activated by ROS, we tested whether HIV PI stimulate ICl,swell in ventricular myocytes. Ritonavir and lopinavir elicited outwardly-rectifying Cl− currents under isosmotic conditions that were abolished by the selective ICl,swell-blocker DCPIB. In contrast, amprenavir, nelfinavir, and raltegravir, an integrase inhibitor, did not modulate ICl,swell acutely. Ritonavir also reduced action potential duration, but amprenavir did not. ICl,swell activation was attributed to ROS because ebselen, an H2O2, scavenger, suppressed ritonavir- and lopinavir-induced ICl,swell. Major ROS sources in cardiomyocytes are sarcolemmal NADPH oxidase and mitochondria. The specific NADPH oxidase inhibitor apocynin failed to block ritonavir- or lopinavir-induced currents, although it blocks ICl,swell elicited by osmotic swelling or stretch. In contrast, rotenone, a mitochondrial e− transport inhibitor, suppressed both ritonavir- and lopinavir-induced ICl,swell. ROS production was measured in HL-1 cardiomyocytes with C-H2DCFDA-AM and mitochondrial membrane potential (ΔΨm) with JC-1. Flow cytometry confirmed that ritonavir and lopinavir but not amprenavir, nelfinavir, or raltegravir augmented ROS production, and HIV PI-induced ROS production was suppressed by rotenone but not NADPH oxidase blockade. Moreover, ritonavir, but not amprenavir, depolarized ΔΨm. These data suggest ritonavir and lopinavir activated ICl,swell via mitochondrial ROS production that was independent of NADPH oxidase. ROS-dependent modulation of ICl,swell and other ion channels by HIV PI may contribute to some of their actions in heart and perhaps other tissues.
Keywords: ICl,swell; VRAC; HIV protease inhibitors; reactive oxygen species; mitochondria; mitochondrial membrane potential; NADPH oxidase; swelling; cell volume
1. Introduction
Inclusion of HIV protease inhibitors (HIV PI)1 in highly active anti-retroviral therapy substantially reduces the morbidity and mortality of HIV infections. A significant challenge is their adverse effects, which include hyperlipidemia, lipodystrophy, insulin resistance, and increased risks of atherosclerosis and myocardial infarction (for review, see [1]). Arrhythmogenic effects also are reported. HIV PI cause PR and QTc prolongation and torsade de pointes in patients [2-4] and suppress heterologously expressed hERG currents and IKr in ventricular myocytes [2]. Moreover, HIV PI cause endothelial cell damage [5] and decompensated heart failure [6] in experimental settings, and dilated cardiomyopathy and heart block in human neonates [7].
Recent studies indicate that HIV PI augment production of reactive oxygen species (ROS) in the vasculature [8-12], adipocytes [13,14], macrophage-derived foam cells [15], fibroblast [16], and pancreatic β cells [17], suggesting that oxidative stress may play a role in the adverse effects of HIV PI. HIV PI reduce mitochondrial membrane potential, which triggers mitochondrial ROS production [11,15], and the expression of mitochondrial respiratory chain proteins is diminished [16,18]. Moreover, expression of NADPH oxidase subunit mRNAs is augmented [15].
We hypothesize that HIV PI-induced ROS production modulates ion channel function and, thereby, may contribute to untoward effects in heart and perhaps other tissues. One potential target is the outwardly rectifying, volume- and stretch-sensitive Cl− current, ICl,swell. We previously demonstrated that activation of ROS production by osmotic swelling, stretch, and downstream signaling proteins, as well as exogenous H2O2, elicit ICl,swell in ventricular myocytes [19-21]. ROS-induced ICl,swell activation also was identified in HeLa and HTC hematoma cell lines [22,23] and primary astrocytes [24]. These studies concluded that ROS production by NADPH oxidase is a required intermediate that elicits ICl,swell, and we recently found mitochondrial ROS also is necessary [25].
We tested whether HIV PI elicit ICl,swell in cardiomyocytes and identified the source of ROS. Under isosmotic conditions, clinically relevant concentrations of the HIV PI ritonavir (RTV) and lopinavir (LPV) rapidly induced an outwardly-rectifying Cl− current, and RTV decreased action potential duration (APD) by 60 ms, effects that were fully blocked by the ICl,swell inhibitor DCPIB. HIV PI-induced ICl,swell also was blocked by the ROS scavenger ebselen and by rotenone, which inhibits mitochondrial e− transport. In contrast, the NADPH oxidase inhibitor apocynin had no effect. Furthermore, the HIV PI inhibitors amprenavir (APV) and nelfinavir (NFV) and the HIV integrase inhibitor raltegravir (MK-0518) failed to acutely modulate ICl,swell, and APV failed to alter APD. Flow cytometry confirmed that RTV and LPV, but not APV, NFV or MK-0518, augmented ROS production measured in HL-1 myocytes. As found for ICl,swell, HIV PI-induced ROS production was inhibited by rotenone but not by suppressing NADPH oxidase activity. Additionally, RTV, but not APV, depolarized mitochondrial membrane potential, ΔΨm. These data suggest that RTV and LPV acutely activated ICl,swell via mitochondrial ROS production, most likely at complex III, and that ROS arising from NADPH oxidase were not required.
2. Materials and methods
Detailed methods are given in the Online Supplement. Animal studies were approved by the Institutional Animal Care and Use Committee and were conducted in accordance with the Declaration of Helsinki, and the Guide for the Care and Use of Animals (National Academy Press, Washington, D.C., 1996).
Ventricular myocytes for electrophysiological studies were freshly isolated from anesthetized New Zealand white rabbits (∼3 kg) by enzymatic digestion. HL-1 cardiomyocytes were used for flow cytometry because they provide a more homogeneous population than freshly isolated myocytes, which unavoidably included damaged cells.
2.1. Electrophysiology
Recordings were made at room temperature with fire-polished pipettes (2–3 MΩ). Myocytes were suprafused at ∼2 ml/min, junction potentials were corrected, and a 3-M KCl-agar bridge served as ground. Seal resistances were 5–30 GΩ, and myocytes were dialyzed for 10 min before data collection. Currents were recorded with an Axoclamp 200B and Digidata 1322A under pClamp 9 and were low-pass filtered. Successive 500-ms steps were made at 1-s intervals from −60 mV, and current-voltage (I–V) relationships were plotted from the late current. APD at 50 and 90% repolarization (APD50, APD90) was determined in myocytes stimulated at 1 Hz. At least 30 action potentials were recorded to verify stability, and the last 10 were averaged. Bath and pipette solutions for measuring ICl,swell were designed to isolate anion currents, and those for action potentials contained physiologic anions and cations. Solution composition, preparation of reagents, and the structure of HIV PI and integrase inhibitors are given in the Online Supplement.
2.2. ROS and ΔΨm measurement by flow cytometry
ROS production was detected in HL-1 myocytes loaded with C-H2DCFDA-AM (Invitrogen), which is hydrolyzed by intracellular esterases and oxidized to fluorescent carboxy-DCF (excitation 488 nm; emission 525 nm), primarily by H2O2. Confluent cells were incubated with C-H2DCFDA-AM (2.5 or 5 μM) for 30 min (37°C), washed with DPBS, and isolated by brief trypsinization. JC-1 (Invitrogen) was used to detect ΔΨm. HL-1 myocytes were incubate with 1 μM JC-1 for 90 min (37°C) and washed. ΔΨm depolarization increases monomer (green) and decreases J-aggregate (orange-red) fluorescence (excitation: 488 nm; emission 525 and 575 nm). C-H2DCFDA-AM and JC-1-loaded cells were assayed separately (EPICS XL; Beckman Coulter).
2.3. Statistics
Data are reported as mean ± SEM; n denotes number of cells (electrophysiology) or experiments (flow cytometry). One-way Repeated Measures ANOVA and the Holm-Sidak method were used (P <0.05; SigmaStat 3.11). For clarity, percent block (± SEM) is sometimes reported and was calculated using each myocyte as its own control. Geometric means of fluorescence data were not normally distributed and were analyzed using Kruskal-Wallis One-way ANOVA on Ranks and Dunn's method. Non-linear fitting was done in SigmaPlot 10.1.
3. Results
3.1. Ritonavir activates ICl,swell
Exposure of ventricular myocytes to 15 μM RTV for 20 min activated a Cl− current that outwardly rectified. Families of membrane currents (Fig. 1A) and corresponding I–V relationships (Fig. 1B) are shown. This outwardly rectifying current is likely to be ICl,swell because it was abolished by DCPIB, a selective ICl,swell blocker [26], in the presence of RTV. RTV increased the current at +60 mV by 1.70 ± 0.16 pA/pF, from 0.42 ± 0.04 pA/pF under control conditions to 2.12 ± 0.15 pA/pF (n = 19, P <0.001) (Fig. 1C). Adding DCPIB (10 μM; 6–12 min) with RTV inhibited 95 ± 3% (n = 4, P <0.002) of RTV-induced current, and current after block was not different than control (n = 4, P = 0.71).
Fig. 1.
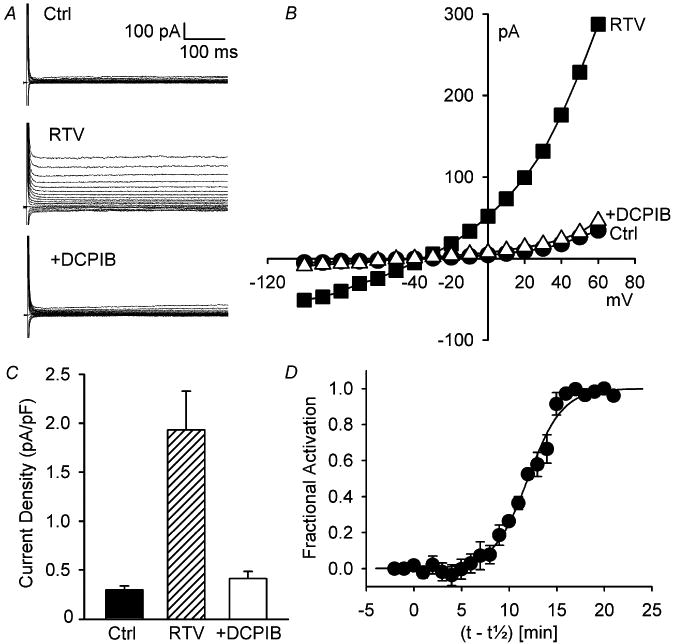
Ritonavir (RTV) activated volume-sensitive Cl− current (ICl,swell) under isosmotic conditions; RTV-induced currents outwardly rectified and were blocked by DCPIB. (A) Families of currents (-100 to + 60 mV, 500 ms) in control (Ctrl), after RTV exposure (15 μM, 20 min), and after addition of DCPIB (10 μM, 6 min) in presence of RTV (+DCPIB). (B) I-V relationships from A. (C) Current densities at +60 mV (n = 4; Ctrl vs. RTV, P <0.002; RTV vs. +DCPIB, P <0.002; Ctrl vs. +DCPIB, P = 0.71). (D) Kinetics of activation of ICl,swell by RTV (n = 4).
Activation of RTV-induced current at +60 mV was sigmoidal; latency for activation was ∼3–7 min, and steady state was reached in 15–20 min. The time course for fractional activation, Y, was well-described by: Y = 1/[1 + exp(-(t-t1/2)/τ)], where t1/2, the time for half-activation, was 12.0 ± 0.9 min, and τ, the time constant, was 1.8 ± 0.2 min (n = 4) (Fig. 1D). Thus, despite variable latency, activation occurred with a consistent time constant once it began.
Although RTV-induced ICl,swell was readily blocked by DCPIB, stimulation of current was not easily reversed. The RTV-induced current was 1.16 ± 0.09 pA/pF and remained at 1.20 ± 0.02 pA/pF despite ∼25 min of washout in control bath solution in 2 cells.
3.2. Mitochondrial ROS elicit RTV-induced ICl,swell
NADPH oxidase and mitochondria both generate O2−• that is readily dismutated to H2O2 spontaneously and by superoxide dismutase. Previous studies found that NADPH oxidase- dependent and exogenous H2O2 activate ICl,swell in ventricular myocytes [19-21,25] and other cells [22-24]. The role of H2O2 in the RTV-induced activation of ICl,swell was tested with ebselen, a membrane-permeant, glutathione peroxidase mimetic that scavenges H2O2. After RTV elicited ICl,swell, adding ebselen (15 μM, 12–20 min) in the presence of RTV blocked 105 ± 3% (n = 5, P <0.001) of RTV-induced current (Fig. 2A-C).
Fig. 2.
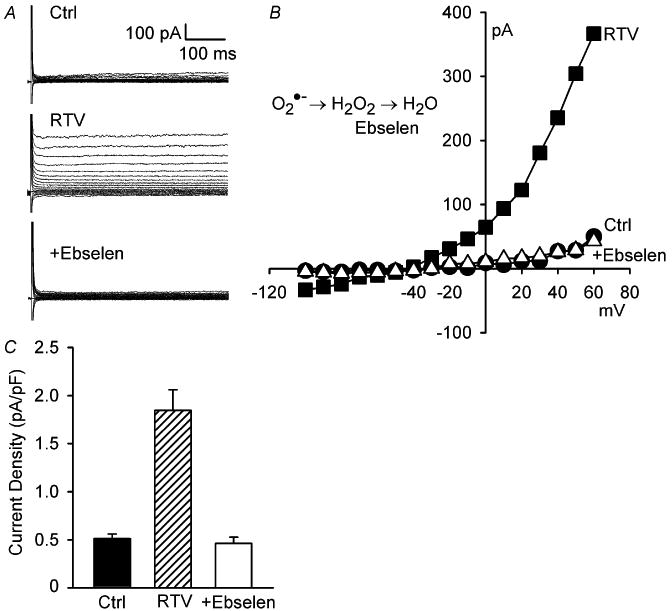
RTV-induced activation of ICl,swell was reversed by ebselen, a H2O2 scavenger. (A) Currents in control (Ctrl), after RTV exposure (15 μM, 20 min), and after addition of ebselen (15 μM, 15 min) in presence of RTV. (B) I-V relationships from A. (C) Current densities at +60 mV (n = 5; Ctrl vs. RTV, P <0.001; RTV vs. +Ebselen, P <0.001; Ctrl vs. +Ebselen, P = 0.81).
Next we considered the source of ROS. NADPH oxidase is a required for stretch and swelling to elicit ICl,swell [19-21,25]. The role of NADPH oxidase in RTV-induced ICl,swell activation was evaluated with apocynin, a selective inhibitor that prevents translocation of p47phox to the membrane and assembly of NADPH oxidase [27]. Apocynin (500 μM, 20 min) failed to inhibit RTV-induced current (n = 4, P = 0.98) (Fig. 3A, B). In contrast, we previously showed that apocynin effectively suppresses ICl,swell elicited by swelling of cardiomyocytes [21].
Fig. 3.
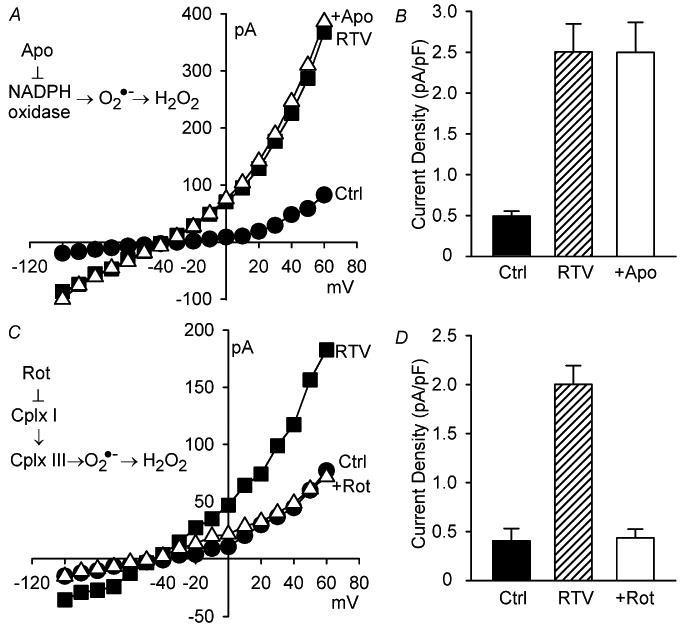
RTV elicited ICl,swell via ROS from mitochondria but not NADPH oxidase. (A, B) Apocynin (Apo; 500 μM, 20 min), a specific NADPH oxidase inhibitor, failed to suppress RTV (15 μM, 20 min)-induced ICl,swell (n = 4; Ctrl vs. RTV, P <0.001; RTV vs. +Apo, P = 0.98; Ctrl vs. +Apo, P <0.001). (C, D) In contrast, rotenone (Rot; 10 μM, 30 min), which suppresses mitochondrial ROS production, fully inhibited RTV-induced ICl,swell (n = 5; Ctrl vs. RTV, P <0.001; RTV vs. +Rot, P <0.001; Ctrl vs. +Rot, P = 0.89).
Besides NADPH oxidase, mitochondria are an important source of ROS in cardiomyocytes. Complex III leaks O2−• to the intermembrane space from which ROS accesses the cytoplasm [28,29]. To test the role of mitochondrial ROS production, we used rotenone, a highly selective complex I inhibitor that prevents e− transport to and ROS production by complex III. Rotenone (10 μM, 26–32 min) inhibited 100 ± 3% (n = 5, P <0.001) of RTV-induced ICl,swell-activation (Fig. 3C, D). Rotenone also suppresses the activation of ICl,swell by antimycin A [25], which directly stimulates complex III ROS production.
3.3. Lopinavir activates ICl,swell via mitochondrial ROS
RTV was not the only HIV PI that activated ICl,swell. As shown in Fig. 4A and B, LPV (15 μM, 20 min) increased current at +60 mV by 1.69 ± 0.14 pA/pF, from 0.81 ± 0.06 to 2.50 ± 0.16 pA/pF (n = 18, P <0.001). The magnitude of current evoked by LPV and RTV, 1.70 pA/pF (Fig. 1), were indistinguishable.
Fig. 4.
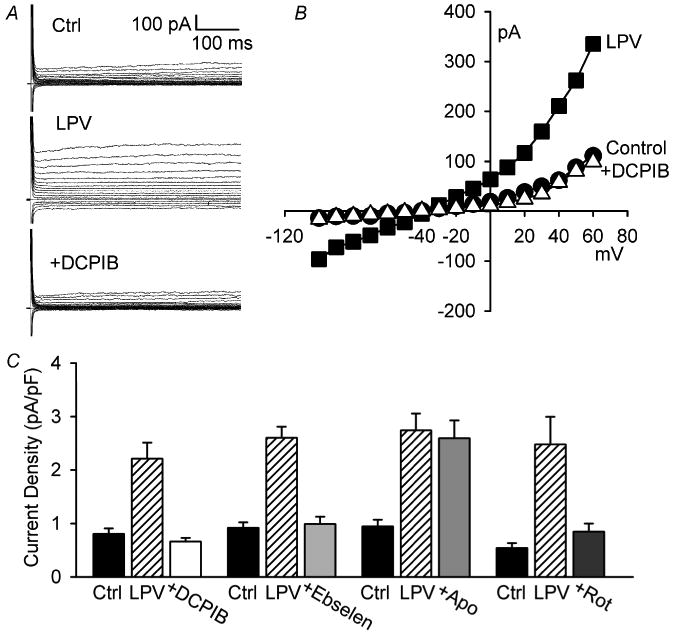
Lopinavir (LPV) activated ICl,swell via mitochondrial ROS production but not NADPH oxidase. (A, B) LPV (15 μM, 25 min) elicited outwardly rectifying Cl− current that was completely blocked by DCPIB. (C) Current densities at +60 mV showing block of LPV-induced current by DCPIB (n = 5, Ctrl vs. LPV, P <0.001; LPV vs. +DCPIB, P <0.001; Ctrl vs. +DCPIB, P = 0.55), ebselen (n = 5, Ctrl vs. LPV, P <0.001; LPV vs. +Ebselen, P <0.001; Ctrl vs. +Ebselen, P = 0.72), and rotenone (n = 4, Ctrl vs. LPV, P <0.003; LPV vs. +Rot, P <0.006; Ctrl vs. +Rot, P = 0.45). In contrast, LPV-induced current was unaffected by apocynin (n = 4, Ctrl vs. LPV, P <0.001; LPV vs. +Apo, P = 0.79; Ctrl vs. +Apo, P <0.001).
Experiments identifying the LPV-induced current as ICl,swell and demonstrating that mitochondrial ROS production, rather than NADPH oxidase, was responsible are summarized in Fig. 4C. First, the ICl,swell inhibitor DCPIB (10 μM, 6–12 min) blocked the LPV-induced current at +60 mV (115 ± 10%; n = 5, P <0.001) in the presence of LPV, suggesting the current was ICl,swell. Second, the H2O2 scavenger ebselen (15 μM, 12–20 min) suppressed (95 ± 5%; n = 5, P <0.001) LPV-induced current, demonstrating the involvement of ROS. Third, the NADPH oxidase inhibitor apocynin (500 μM, 20 min) failed to significant suppress LPV-induced ICl,swell (8 ± 5%; n = 4, P = 0.46), implying that ROS production by NADPH oxidase was not required. Fourth, blocking mitochondrial e− transport with rotenone (10 μM, 26–32 min) decreased LPV-induced ICl,swell by 82 ± 4% (n = 4, P <0.01). These data recapitulate the results with RTV (Fig. 1–3) and suggest that LPV and RTV modulate ICl,swell by the same ROS-dependent mechanism.
3.4. Nelfinavir, amprenavir and raltegravir, an integrase inhibitor, did not block ICl,swell
Although both RTV and LPV activated ICl,swell, neither NFV (15 μM, 30 min; n = 5, P = 0.55) nor APV (15 μM, 30 min; n = 6, P = 0.11), two other HIV PI, significantly altered ICl,swell within the period tested (Fig. 5). This failure to activate ICl,swell was not due to the absence of the channels in the particular cells examined; hypoosmotic swelling in 0.7T elicited ICl,swell in the same cells. In the NFV experiments, current at +60 mV increased from 0.89 ± 0.11 pA/pF in control to 1.44 ± 0.08 pA/pF in 0.7T (n = 5, P <0.001), and activation by 0.7T was confirmed in 2 of 2 cells challenged with APV and 0.7T (P <0.001). The HIV integrase inhibitor raltegravir (MK; 15 μM, 30 min; n = 5, P = 0.86) also failed to stimulate ICl,swell, whereas 0.7T elicited ICl,swell in 2 of 2 cells exposed to MK and 0.7T (P <0.001).
Fig. 5.
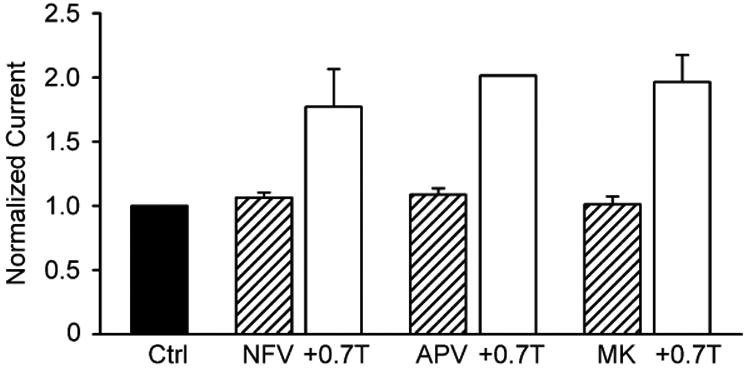
The HIV PI nelfinavir (NFV; 5 μM, 30 min; n = 5) and amprenavir (APV; 15 μM, 30 min, n = 6) and the HIV integrase inhibitor raltegravir (MK; 15 μM, 30 min, n = 5) failed to activate ICl,swell acutely. By contrast, ICl,swell was elicited by osmotic swelling in 0.7T bath solution in the continued presence of HIV protease or integrase inhibitors (for drug + 0.7T, n = 5, 2 and 2, respectively). Currents were normalized to Ctrl for each experimental group.
3.5. APD
If activation of outwardly rectifying ICl,swell by HIV PI is functionally significant, these agents should decrease APD in physiological solutions (Fig. 6). RTV significantly decreased APD50 and APD90 by 37 ± 7 and 60 ± 9 ms, respectively (15 μM, 15 min; n = 4), whereas APV did not alter APD (15 μM, 15 min; n = 3). Moreover, the RTV-induced reduction of APD was reversed by the ICl,swell blocker DCPIB (10 μM, 10 min) in the presence of RTV. These responses are consistent with the effects of RTV, APV, and DCPIB on ICl,swell and illustrate the importance of ICl,swell in modulating APD. Nevertheless, RTV also reduced overshoot and plateau voltage, actions insensitive to suppression of ICl,swell by DCPIB. This suggests ICl,swell was not the only channel modulated by RTV, but the details were not further studied.
Fig. 6.
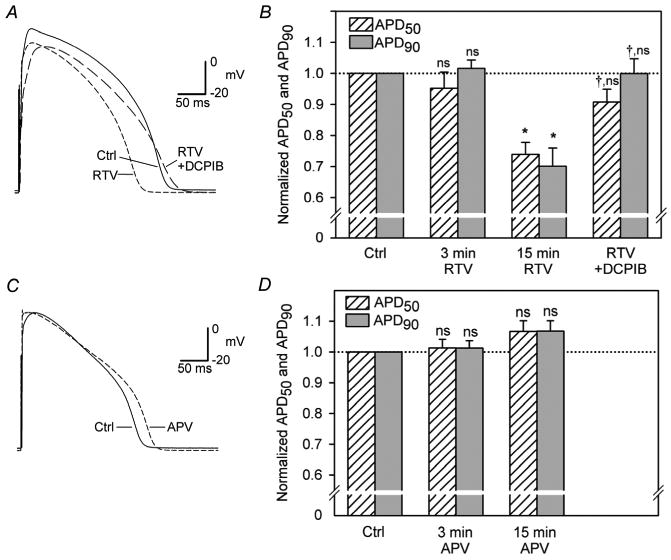
Effects of RTV and APV on ventricular action potentials and normalized APD50 and APD90. (A,B) RTV (15 μM, 15 min; n = 4) significantly decreased APD50 and APD90, and abbreviation of APD was reversed by DCPIB (10 μM, 10 min). *, P < 0.05 vs Ctrl; †, vs 15 min RTV; ns, vs Ctrl. (C,D) APV (15 μM, 15 min; n = 3) did not significantly alter APD; ns, vs Ctrl.
3.6. Mitochondrial ROS production and ΔΨm
To more directly assess the proposed mitochondrial ROS-dependent mechanisms of ICl,swell activation, the effect of HIV PI on ROS production in HL-1 cardiomyocytes was determined by flow cytometry with C-H2DCFDA-AM, which primarily detects H2O2. Fig. 7A,B display log fluorescence histograms from a single experiment. Geometric means of histograms normalized by background fluorescence are depicted in Fig. 7C. Measurements of ROS production were fully consistent with the notion that ICl,swell is a ROS sensor. Both RTV and LPV (15 μM, 30 min) significantly increased ROS production 4.2-fold from background levels. This was due to mitochondrial ROS production and did not depend on NADPH oxidase. RTV- and LPV-induced ROS production was suppressed by pretreatment (30 min) with rotenone (10 μM) but was unaffected by pretreatment with the NADPH oxidase blocker gp91ds-tat (500 nM). Moreover, the HIV PI inhibitors NFV and APV and the integrase inhibitor MK failed to elicit ROS production (15 μM, 60 min). In comparison, fluorescence elicited by H2O2 (100 μM, 15 min) was increased 5.6-fold above background (Fig. 7A,C). Negligible responses were obtained from myocytes in the absence of fluorophore (−F) with or without addition of LPV or RTV (Fig. 7A), indicating neither myocytes nor HIV PI themselves contributed to the signal attributed to ROS. We previously demonstrated that gp91ds-tat (500 nM) fully inhibits ROS production elicited by endothelin-1, which stimulates NADPH oxidase [25].
Fig. 7.
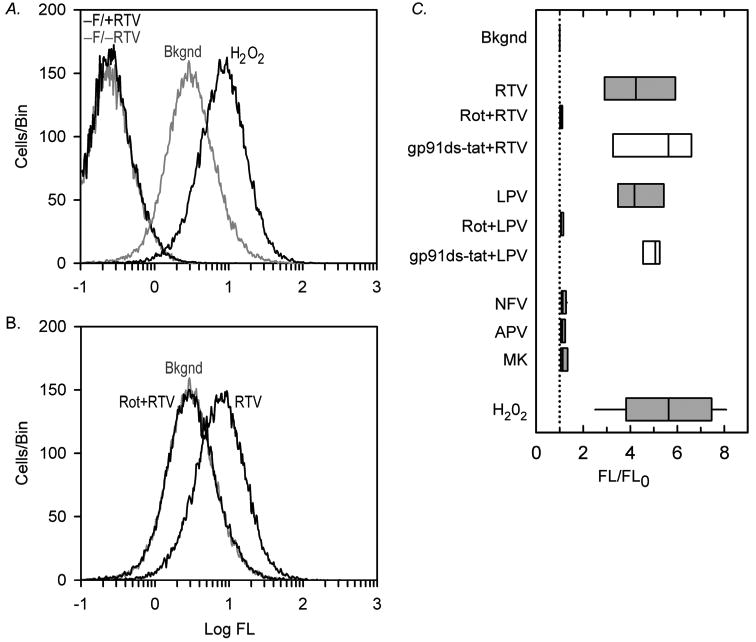
RTV- and LPV-induced mitochondrial ROS production in HL-1 myocytes. (A) Log fluorescence (FL) histograms by flow cytometry. Negative controls in absence of probe (−F) without (−RTV) or with (+RTV) RTV (15 μM, 30 min). In fluorophore-loaded myocytes, background fluorescence (Bkgnd) and response to H2O2 (100 μM, 15 min) reflect ROS. (B) RTV elicited ROS production, and pretreatment with rotenone (10 μM, 30 min; Rot+RTV) suppressed ROS to background levels; A and B, from same experiment. (C) Geometric means of histograms normalized by background fluorescence (FL/FLO). Both RTV and LPV (15 μM, 30 min; n = 5, for each) significantly increased ROS production, which was fully suppressed by pretreatment with Rot (10 μM, 30 min; n = 5, for each) but not by pretreatment with gp91ds-tat (500 nM, 30 min; n = 3, for each). In contrast, NFV (n = 4), APV (n = 3), and MK (n = 3; 15 μM, 60 min, for each) failed to augment ROS production. H2O2 (100 μM, 15 min; n = 9) was positive control. Box plots show 25, 50 and 75%ile, and whiskers show 5 and 95%ile; dotted line, background fluorescence.
ΔΨm also was assessed in HL-1 myocytes with the potential-sensitive dye JC-1. Fig. 8 shows that RTV (15 μM, 60 min, n = 5) significantly depolarized ΔΨm, as reflected by the increased green:orange-red fluorescence ratio over time. After 30 min, a time when ROS production and ICl,swell were augmented, the JC-1 fluorescence ratio was 2.1-fold (median) greater than control. On the other hand, APV (15 μM, 60 min, n = 4) elicited only a 20% increase at 30 min when ROS production and ICl,swell were unaffected. This suggests ICl,swell activation was triggered by significant ΔΨm depolarization. As a positive control, mitochondrial respiration was uncoupled with DNP (0.3 mM, 30 min, n = 3); this evoked a 4.5-fold increase in JC-1 fluorescence, whereas ΔΨm was unchanged over 60 min in control media (n = 3).
Fig. 8.
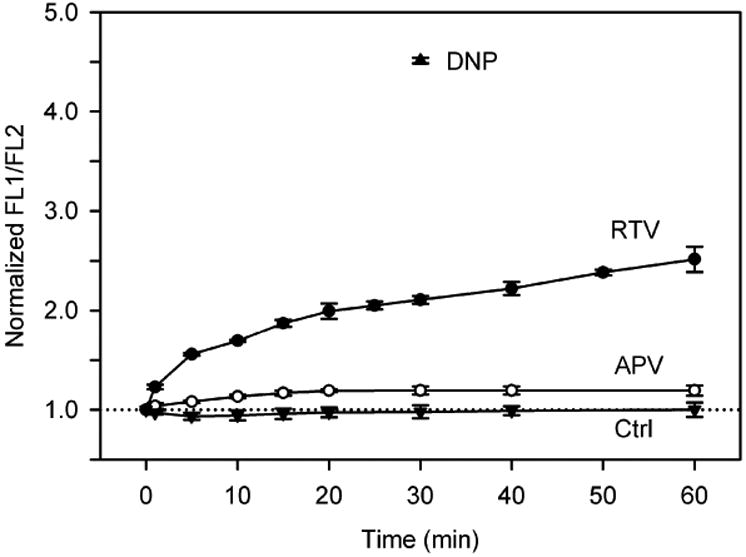
Time-dependence of RTV-induced ΔΨm depolarization in HL-1 myocytes. Ratios of JC-1 fluorescence (FL1/FL2) normalized by control fluorescence ratio are plotted for RTV (15 μM; n = 5), APV (15 μM; n = 4), and a time control (Ctrl; n = 3). Treatment with DNP (0.3 mM, 20 min) served as positive control. Increased FL1/FL2 corresponds to depolarization of ΔΨm.
Although DCPIB distinguishes between ICl,swell and a number of plasmalemmal anion and cation channels [26], its mechanism and site of action are unknown. One possibility is that DCPIB blocks mitochondrial inner membrane Cl− channel, thereby restoring ΔΨm and decreasing ROS production. Rather than enhancing ΔΨm, DCPIB (10 μM, 10 min) increased JC-1 fluorescence in HL-1 myocytes 8.7 ± 0.9-fold (n = 6), indicating it strongly depolarized ΔΨm. In contrast, a low DCPIB concentration (0.1 μM, 10 min) that fails to suppress ICl,swell increased JC-1 fluorescence by only ∼20% (n = 2). These data suggest mitochondrial actions of DCPIB may, at least in part, explain its block ICl,swell.
4. Discussion
We demonstrated that certain HIV PI activate the volume-sensitive Cl− current, ICl,swell, in ventricular myocytes under isosmotic conditions. At clinically relevant concentrations, RTV and LPV acutely induced an outwardly-rectifying Cl− current, RTV decreased APD90 by 60 ms, and both effects were fully abolished by the ICl,swell-blocker DCPIB. Two other HIV PI, APV and NFV, and the HIV integrase inhibitor raltegravir (MK-0518) did not effect ICl,swell, and APV also did not alter APD.
Activation of ICl,swell by RTV and LPV was ROS-dependent. H2O2 is a required intermediate in the signaling cascade regulating ICl,swell, and exogenous H2O2 activates ICl,swell in rabbit ventricular myocytes [19-21,25], adenocarcinoma HeLa cells, hepatoma HTC cells, and cultured primary astrocytes [22-24]. We found RTV- and LPV-induced ICl,swell was fully suppressed by the H2O2 scavenger ebselen. RTV and LPV augmented ROS production, as detected by C-H2DCFDA-AM, in HL-1 myocytes, whereas APV and NFV failed to generate significant ROS after 1 h. Dichlorofluoresceins, such as C-H2DCFDA-AM, are nominally H2O2-selective, but ONOO−•, HOCl, and peroxides also can contribute to its fluorescence [30]. Measurements of ΔΨm with JC-1 in HL-1 myocytes suggest that RTV elicited mitochondrial ROS production, at least in part, by depolarizing ΔΨm. ROS and ΔΨm measurements were made in HL-1 myocytes rather than acutely dissociated adult myocytes to avoid inclusion of cells damaged during isolation. Other studies confirm that ICl,swell is activated by a number of the same mechanisms in HL-1 cells as in adult myocytes [31].
The two major sources of H2O2 in cardiomyocytes are sarcolemmal NADPH oxidase and mitochondria. These systems generate O2−•, which is readily dismutated to H2O2 spontaneously and by superoxide dismutase. Previously we showed that NADPH oxidase is essential for the activation of ICl,swell by β1-integrin stretch and osmotic swelling and that ICl,swell can be blocked by selectively inhibiting NADPH oxidase with apocynin or gp91ds-tat [19-21,25]. However, apocynin failed to suppress the RTV-and LPV-induced currents, and pretreatment with gp91ds-tat failed to suppress C-H2DCFDA-AM fluorescence. These data are consistent with the notion that activation of ICl,swell by HIV PI was independent of NADPH oxidase.
Complex I and Complex III are the major mitochondrial redox centers that leak single e− to molecular O2 to generate O2−• [28,29]. Normally O2−• generated by complex I is exclusively released into the matrix of intact mitochondria and degraded by the matrix anti-oxidant system [32]. By contrast, Complex III releases O2−• to both sides of the mitochondrial inner membrane and accounts for extra-mitochondrial release of O2−• [28,32]. We propose that HIV PI elicit ICl,swell via mitochondrial ROS production by Complex III. RTV- and LPV-induced ICl,swell were completely abolished by rotenone, a selective Complex I inhibitor that limits downstream e− flux to Complex III and, therefore, inhibits mitochondrial ROS generation [28]. In addition, a 1-hr pretreatment with rotenone fully abrogated RTV- and LPV-induced H2O2 production measured directly by flow cytometry. Although we favor H2O2 as the active species, we cannot rule out the possibility that the activation of ICl,swell by HIV PI involves other ROS derived from O2−• or H2O2.
A mitochondrial ROS-dependent mechanism for the activation of ICl,swell is consistent with previous work showing that HIV PI reduce mitochondrial membrane potential and increases leak of e− from the transport chain to O2 to produce O2−• [11,15]. It also is consistent with our previous studies showing that blocking mitochondrial ROS production inhibits activation of ICl,swell by endothelin-1 [31]. We did not evaluate whether mitochondrial ROS-induced ROS release [33] contributed to the response to HIV PI.
Despite the failure of APV and NFV to modulate ICl,swell in 30 min or elicit ROS production after 1 h, it remains possible that these HIV PI also would activate cardiac ICl,swell with chronic administration. APV and NFV have been shown to elicit ROS production in endothelial cells [9,34,35], vascular smooth muscle cells [10], macrophages [14], adipocytes [13,36], fibroblasts [16], and pancreatic β cells [17]. Prolonged exposures to HIV PI often employed to detect oxidative stress cannot be replicated in patch clamp studies using each myocyte as its own control.
DCPIB is regarded as a highly selective ICl,swell blocker because it suppresses ICl,swell without affecting several other plasmalemma Cl−, Na+, Ca2+ and K+ channels [26], but evidence that it binds directly to ICl,swell channels is lacking. Studies with JC-1 indicated that DCPIB very strongly depolarized ΔΨm. This raises the possibility that DCPIB uncouples mitochondrial e− transport and blocks ROS production, which may in part explain how DCPIB inhibits ICl,swell. Nevertheless, DCPIB also suppresses ICl,swell evoked by 100-μM exogenous H2O2 [32], suggesting a mitochondria-independent effect. Although DCPIB is useful to distinguish between ion channels, fully understanding its actions on mitochondrial function and the mechanism of ICl,swell blockade requires additional investigation.
4.1. Potential Implications
Adverse cardiovascular effects of HIV PI include coronary artery disease and myocardial infarction [37,38], and certain HIV PI are associated with QT interval prolongation and torsade de pointes [2,4]. Anson et al. [2] demonstrated that RTV, LPV, NFV, and saquinavir substantially suppress HERG currents expressed in HEK293 cells and the corresponding endogenous current, IKr, in mouse neonatal myocytes. Block of HERG favors APD prolongation, early afterdepolarizations, and torsade de pointes [39]. Because ICl,swell produces an outward current near the plateau potential, its activation favors reduction of APD. Consequently, HIV PI-induced ICl,swell would antagonize APD prolongation due to block of HERG and thereby reduce the risk of drug-induced long QT syndrome and torsade de pointes. On the other hand, APD abbreviation will favor reentrant arrhythmias, such as atrial fibrillation, but an association between HIV PI and reentrant arrhythmias has not been documented. Although the present acute studies on isolated cardiomyocytes from normal animals raise important several questions, they are not sufficient to establish the role of ICl,swell in HIV-infected patients who are treated with HIV PI as part of multidrug highly active anti-retroviral therapy.
Although several mechanisms underlying the untoward effects of HIV PI have been the focus of research, the potential role of ion channels in these outcomes largely has been ignored. Because ICl,swell is present in a number of tissues affected by HIV PI, such as macrophages, hepatocytes, endothelial cells, and pancreatic β cells, it is reasonable to consider whether ICl,swell activation might contribute to the untoward effects of HIV PI. HIV PI-induced ROS production also may regulate other redox sensitive ion channels and transporters that are widely distributed. Na channels [40], L-type Ca2+ channels, transient outward K+ channels, inwardly rectifying K+ channels, ATP-sensitive K+ channels, ryanodine receptors, inositol-1,4,5- trisphosphate receptors, sarcoplasmic reticulum and plasma membrane Ca2+ ATPases, and the Na+–Ca2+ exchanger [41] are influenced by ROS. Whether HIV PI modulate these channels and transporters and whether they contribute to the adverse effects of HIV PI in heart and other systems remains to be elucidated.
Supplementary Material
Acknowledgments
We thank Frances K. H. White and Julie Farnsworth for training and assistance in flow cytometry. Supported by grants from the American Heart Assoc. (0855044E), NIH (R01AT004148, R21AI068432, R01AI057189), and by the Massey Cancer Center Flow Cytometry & Imaging Shared Resource Facility (P30CA16059).
Footnotes
Abbreviations: APD, action potential duration; DNP, 2,4-dinitrophenol; HIV PI, HIV protease inhibitors; ROS, reactive oxygen species; ICl,swell, volume- sensitive Cl− current; ΔΨm, mitochondrial membrane potential; RTV, ritonavir; LPV, lopinavir; APV, amprenavir; NFV, nelfinavir; and MK-0518, raltegravir.
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Flint OP, Noor MA, Hruz PW, Hylemon PB, Yarasheski K, Kotler DP, et al. The role of protease inhibitors in the pathogenesis of HIV-associated lipodystrophy: cellular mechanisms and clinical implications. Toxicol Pathol. 2009;37:65–77. doi: 10.1177/0192623308327119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anson BD, Weaver JG, Ackerman MJ, Akinsete O, Henry K, January CT, et al. Blockade of HERG channels by HIV protease inhibitors. Lancet. 2005;365:682–6. doi: 10.1016/S0140-6736(05)17950-1. [DOI] [PubMed] [Google Scholar]
- 3.Busti AJ, Tsikouris JP, Peeters MJ, Das SR, Canham RM, Abdullah SM, et al. A prospective evaluation of the effect of atazanavir on the QTc interval and QTc dispersion in HIV-positive patients. HIV Med. 2006;7:317–22. doi: 10.1111/j.1468-1293.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 4.Chinello P, Lisena FP, Angeletti C, Boumis E, Papetti F, Petrosillo N. Role of antiretroviral treatment in prolonging QTc interval in HIV-positive patients. J Infect. 2007;54:597–602. doi: 10.1016/j.jinf.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Fiala M, Murphy T, MacDougall J, Yang W, Luque A, Iruela-Arispe L, et al. HAART drugs induce mitochondrial damage and intercellular gaps and gp120 causes apoptosis. Cardiovasc Toxicol. 2004;4:327–37. doi: 10.1385/ct:4:4:327. [DOI] [PubMed] [Google Scholar]
- 6.Hruz PW, Yan Q, Struthers H, Jay PY. HIV protease inhibitors that block GLUT4 precipitate acute, decompensated heart failure in a mouse model of dilated cardiomyopathy. FASEB J. 2008;22:2161–7. doi: 10.1096/fj.07-102269. [DOI] [PubMed] [Google Scholar]
- 7.McArthur MA, Kalu SU, Foulks AR, Aly AM, Jain SK, Patel JA. Twin preterm neonates with cardiac toxicity related to lopinavir/ritonavir therapy. Pediatr Infect Dis J. 2009;28:1127–9. doi: 10.1097/INF.0b013e3181acd17e. [DOI] [PubMed] [Google Scholar]
- 8.Conklin BS, Fu W, Lin PH, Lumsden AB, Yao Q, Chen C. HIV protease inhibitor ritonavir decreases endothelium-dependent vasorelaxation and increases superoxide in porcine arteries. Cardiovasc Res. 2004;63:168–75. doi: 10.1016/j.cardiores.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 9.Chai H, Yang H, Yan S, Li M, Lin PH, Lumsden AB, et al. Effects of 5 HIV protease inhibitors on vasomotor function and superoxide anion production in porcine coronary arteries. J Acquir Immune Defic Syndr. 2005;40:12–9. doi: 10.1097/01.qai.0000172368.05327.7b. [DOI] [PubMed] [Google Scholar]
- 10.Rudich A, Ben-Romano R, Etzion S, Bashan N. Cellular mechanisms of insulin resistance, lipodystrophy and atherosclerosis induced by HIV protease inhibitors. Acta Physiol Scand. 2005;183:75–88. doi: 10.1111/j.1365-201X.2004.01383.x. [DOI] [PubMed] [Google Scholar]
- 11.Jiang B, Hebert VY, Li Y, Mathis JM, Alexander JS, Dugas TR. HIV antiretroviral drug combination induces endothelial mitochondrial dysfunction and reactive oxygen species production, but not apoptosis. Toxicol Appl Pharmacol. 2007;224:60–71. doi: 10.1016/j.taap.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Chai H, Lin PH, Yao Q, Chen C. Roles and mechanisms of human immunodeficiency virus protease inhibitor ritonavir and other anti-human immunodeficiency virus drugs in endothelial dysfunction of porcine pulmonary arteries and human pulmonary artery endothelial cells. Am J Pathol. 2009;174:771–81. doi: 10.2353/ajpath.2009.080157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben-Romano R, Rudich A, Etzion S, Potashnik R, Kagan E, Greenbaum U, et al. Nelfinavir induces adipocyte insulin resistance through the induction of oxidative stress: differential protective effect of antioxidant agents. Antivir Ther. 2006;11:1051–60. [PubMed] [Google Scholar]
- 14.Lagathu C, Eustace B, Prot M, Frantz D, Gu Y, Bastard JP, et al. Some HIV antiretrovirals increase oxidative stress and alter chemokine, cytokine or adiponectin production in human adipocytes and macrophages. Antivir Ther. 2007;12:489–500. [PubMed] [Google Scholar]
- 15.Wang X, Mu H, Chai H, Liao D, Yao Q, Chen C. Human immunodeficiency virus protease inhibitor ritonavir inhibits cholesterol efflux from human macrophage-derived foam cells. Am J Pathol. 2007;171:304–14. doi: 10.2353/ajpath.2007.060965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caron M, Auclair M, Donadille B, Bereziat V, Guerci B, Laville M, et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007;14:1759–67. doi: 10.1038/sj.cdd.4402197. [DOI] [PubMed] [Google Scholar]
- 17.Chandra S, Mondal D, Agrawal KC. HIV-1 Protease Inhibitor Induced Oxidative Stress Suppresses Glucose Stimulated Insulin Release: Protection with Thymoquinone. Exp Biol Med (Maywood) 2009;234:442–53. doi: 10.3181/0811-RM-317. [DOI] [PubMed] [Google Scholar]
- 18.Viengchareun S, Caron M, Auclair M, Kim MJ, Frachon P, Capeau J, et al. Mitochondrial toxicity of indinavir, stavudine and zidovudine involves multiple cellular targets in white and brown adipocytes. Antivir Ther. 2007;12:919–29. [PubMed] [Google Scholar]
- 19.Browe DM, Baumgarten CM. Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl- current elicited by β1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol. 2004;124:273–87. doi: 10.1085/jgp.200409040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Browe DM, Baumgarten CM. EGFR kinase regulates volume-sensitive chloride current elicited by integrin stretch via PI-3K and NADPH oxidase in ventricular myocytes. J Gen Physiol. 2006;127:237–51. doi: 10.1085/jgp.200509366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ren Z, Raucci FJ, Jr, Browe DM, Baumgarten CM. Regulation of swelling-activated Cl current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovasc Res. 2008;77:73–80. doi: 10.1093/cvr/cvm031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varela D, Simon F, Riveros A, Jorgensen F, Stutzin A. NAD(P)H oxidase-derived H2O2 signals chloride channel activation in cell volume regulation and cell proliferation. J Biol Chem. 2004;279:13301–4. doi: 10.1074/jbc.C400020200. [DOI] [PubMed] [Google Scholar]
- 23.Shimizu T, Numata T, Okada Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl- channel. Proc Natl Acad Sci U S A. 2004;101:6770–3. doi: 10.1073/pnas.0401604101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haskew-Layton RE, Mongin AA, Kimelberg HK. Hydrogen peroxide potentiates volume-sensitive excitatory amino acid release via a mechanism involving Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2005;280:3548–54. doi: 10.1074/jbc.M409803200. [DOI] [PubMed] [Google Scholar]
- 25.Deng W, Baki L, Baumgarten CM. Endothelin signaling regulates volume-sensitive Cl current via NADPH oxidase and mitochondrial reactive oxygen species. Cardiovasc Res. 2010 doi: 10.1093/cvr/cvq125. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Decher N, Lang HJ, Nilius B, Bruggemann A, Busch AE, Steinmeyer K. DCPIB is a novel selective blocker of ICl,swell and prevents swelling-induced shortening of guinea-pig action potential duration. Br J Pharmacol. 2001;134:1467–79. doi: 10.1038/sj.bjp.0704413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ximenes VF, Kanegae MP, Rissato SR, Galhiane MS. The oxidation of apocynin catalyzed by myeloperoxidase: proposal for NADPH oxidase inhibition. Arch Biochem Biophys. 2007;457:134–41. doi: 10.1016/j.abb.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 28.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–31. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 29.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol. 2004;286:R431–R444. doi: 10.1152/ajpregu.00361.2003. [DOI] [PubMed] [Google Scholar]
- 31.Deng W, Raucci FJ, Jr, Baki L, Baumgarten CM. Regulation of volume-sensitive chloride current in cardiac HL-1 myocytes. Biophys J. 2010;98(Suppl 1):338a. [Google Scholar]
- 32.Muller FL, Liu Y, Van RH. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–73. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 33.Aon MA, Cortassa S, Marban E, O'Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. 2003;278:44735–44. doi: 10.1074/jbc.M302673200. [DOI] [PubMed] [Google Scholar]
- 34.Mondal D, Pradhan L, Ali M, Agrawal KC. HAART drugs induce oxidative stress in human endothelial cells and increase endothelial recruitment of mononuclear cells: exacerbation by inflammatory cytokines and amelioration by antioxidants. Cardiovasc Toxicol. 2004;4:287–302. doi: 10.1385/ct:4:3:287. [DOI] [PubMed] [Google Scholar]
- 35.Grigorian A, Hurford R, Chao Y, Patrick C, Langford TD. Alterations in the Notch4 pathway in cerebral endothelial cells by the HIV aspartyl protease inhibitor, nelfinavir. BMC Neurosci. 2008;9:27. doi: 10.1186/1471-2202-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vincent S, Tourniaire F, El Yazidi CM, Compe E, Manches O, Plannels R, et al. Nelfinavir induces necrosis of 3T3F44-2A adipocytes by oxidative stress. J Acquir Immune Defic Syndr. 2004;37:1556–62. doi: 10.1097/00126334-200412150-00003. [DOI] [PubMed] [Google Scholar]
- 37.Lekakis J, Tsiodras S, Ikonomidis I, Palios J, Poulakou G, Rallidis L, et al. HIV-positive patients treated with protease inhibitors have vascular changes resembling those observed in atherosclerotic cardiovascular disease. Clin Sci (Lond) 2008;115:189–96. doi: 10.1042/CS20070353. [DOI] [PubMed] [Google Scholar]
- 38.Vaughn G, Detels R. Protease inhibitors and cardiovascular disease: analysis of the Los Angeles County adult spectrum of disease cohort. AIDS Care. 2007;19:492–9. doi: 10.1080/09540120701203329. [DOI] [PubMed] [Google Scholar]
- 39.Roden DM. Cellular basis of drug-induced torsades de pointes. Br J Pharmacol. 2008;154:1502–7. doi: 10.1038/bjp.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–22. doi: 10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- 41.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–21. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.