

Abstract
Escherichia coli RecX (RecXEc) is a negative regulator of RecA activities both in the bacterial cell and in vitro. In contrast, the Neisseria gonorrhoeae RecX protein (RecXNg) enhances all RecA-related processes in N. gonorrhoeae. Surprisingly, the RecXNg protein is not a RecA protein activator in vitro. Instead, RecXNg is a much more potent inhibitor of all RecANg and RecAEc activities than is the E. coli RecX ortholog. A series of RecXNg mutant proteins representing a gradient of functional deficiencies provide a direct correlation between RecANg inhibition in vitro and the enhancement of RecANg function in N. gonorrhoeae. Unlike RecXEc, RecXNg does not simply cap the growing ends of RecA filaments, but it directly facilitates a more rapid RecA filament disassembly. Thus, in N. gonorrhoeae, recombinational processes are facilitated by RecXNg protein-mediated limitations on RecANg filament presence and/or length to achieve maximal function.
Keywords: DNA, DNA Enzymes, DNA Recombination, DNA Repair, Nucleic Acid Enzymology, Escherichia coli, Neisseria gonorrhoeae, RecA, RecX
Introduction
Cellular recombination systems have a range of functions in all cells. In bacteria, the central function is the repair of stalled or collapsed replication forks (1–5). When a replication fork encounters breaks or lesions in DNA, the fork stalls, and cell death can occur if replication is not restarted. Bacterial replication fork stalling may occur as often as every replication cycle even under normal growth conditions (5, 6). In addition, recombination is used to promote horizontal genetic transfer of alleles between organisms and can be used to promote specialized recombination programs.
The 37.8-kDa, 352-amino acid Escherichia coli RecA (RecAEc)2 protein is essential for the recombinational DNA repair needed to regenerate a functional replication fork. RecAEc protein functions primarily as a dynamic, extended helical filament formed on DNA in the presence of an adenine nucleotide triphosphate co-factor (7, 8). RecAEc filament formation on single-stranded DNA (ssDNA) begins with a slow nucleation step, followed by a faster 5′ to 3′ extension phase (6, 9–11). Once formed, the RecAEc filaments predominantly assemble at the 3′ end and disassemble at the 5′ end with ATP hydrolysis occurring throughout the filament (12–14).
In the human-specific pathogen Neisseria gonorrhoeae (Gc), RecA is not only necessary for DNA repair but is also necessary for DNA transformation and pilus antigenic variation (15–17). Gc pili mediate attachment of the pathogen to the host epithelium, and antigenic variation of these pili is postulated to be necessary for immune avoidance (18). Antigenic variation occurs when silent copies of the pilS gene are transferred unidirectionally into the expressed pilE gene via RecA-mediated recombination (19, 20). Change of the amino acid sequence of PilE, the main component of Gc pili (21) can result in changes in pilus-dependent colony morphology (phase variation) (22, 23), providing a convenient way to estimate the frequency of pilin antigenic variation (23, 24). RecANg and RecAEc share 65% identity and 81% similarity. When expressed in N. gonorrhoeae, RecAEc complements for pilin antigenic variation, partially complements for DNA transformation, but does not complement for DNA damage survival (25). Similarly, RecANg can complement a recA null mutant in E. coli for homologous recombination but not for survival to DNA damage. In vitro characterization of RecANg reveals a protein functionally similar to RecAEc, although ATP hydrolysis by RecANg is somewhat faster.3
Regulation of recombination is critical for DNA repair and genome stability, and RecA represents the primary target for such regulation. RecAEc is regulated on three levels: transcriptional regulation of the recA gene within the SOS regulon (27), autoregulation by the C terminus of the RecA protein (28), and regulation by other proteins. Most allosteric RecA regulators characterized to date are involved in controlling the formation or the disassembly of RecA filaments as reviewed in Ref. 29). Not as much is known about RecA regulation in Gc. The two species both possess orthologs of most genes involved in recombinational DNA repair (19). Until recently, no classical SOS response had been identified in Gc (30). However, a small regulon controlled by a LexA homolog was recently discovered in N. gonorrhoeae that does not regulate RecANg.4 Thus, the regulation of RecA in N. gonorrhoeae and E. coli might differ because RecANg is involved in a process absent in E. coli (pilus antigenic variation), and RecAEc is involved in a process not well conserved in N. gonorrhoeae (SOS response).
One well conserved RecA regulating protein in bacteria is RecX. The 19.4-kDa, 166-amino acid E. coli RecX (RecXEc) protein is encoded by the recX gene (also called oraA) located downstream of the recA gene. Cellular levels of RecXEc, produced by transcriptional read-through from the recA promoter, are less than 5% of RecAEc levels with numbers of less than 50 RecXEc molecules/cell (31) and 15,000 RecAEc molecules/cell (31–33). The only phenotype of an E. coli ΔrecX strain is a 5-fold decrease in UV resistance (31). Overexpression of recX inhibits SOS induction and leads to a decrease in UV resistance and P1 transduction frequency (31). These data indicate that in E. coli, high levels of RecX inhibit the RecA-mediated response to DNA damage and suggest that a low level of RecX is beneficial for conferring UV resistance.
Three-dimensional EM reconstruction shows that RecXEc binds within the major helical groove of a RecAEc filament, spanning the monomer-monomer interface (34). RecXEc inhibits RecAEc-mediated homologous recombination, co-protease, and ATPase activities in vitro at substoichiometric concentrations (31, 35). Kinetic and electron microscopic analysis of the effects of RecXEc on RecAEc provided evidence for a filament capping mechanism (35). Net RecAEc filament disassembly occurs because of RecXEc blocking assembly at the 3′-proximal filament end, whereas disassembly continues at the 5′-proximal end. The crystal structure of RecXEc reveals three bundles of three helices making up a crescent-shaped protein (36).
The 17.7-kDa, 153-amino acid N. gonorrhoeae RecX (RecXNg) protein is encoded by a recX gene that is not located downstream of recA as it is in many other bacteria (37–40). The RecXEc and RecXNg proteins share 25% identity and 54% similarity. A Gc recX loss of function mutant shows a 6-fold decrease in pilus phase variation, a 5-fold decrease in DNA transformation, and a 4-fold decrease in DNA repair ability upon nalidixic acid exposure, whereas the levels of RecANg protein are not affected (41). This demonstrates that the Gc recX gene aids all RecANg-related activities in N. gonorrhoeae and indicates a positive effect of RecXNg on RecANg function (41). It is not clear what role RecXNg plays in these RecA-mediated processes, and no biochemical studies have been carried out.
In the present work, we have examined the effects of RecXNg protein in light of its positive effects on RecANg function in N. gonorrhoeae. Surprisingly, we find that RecXNg is not only an inhibitor of both RecANg and RecAEc in vitro, but it is a much more potent inhibitor than RecXEc. Our results offer important insight into the regulation of RecA-mediated processes by negative regulators and show that inhibition of RecA activities in vitro can result in stimulation of RecA-dependent processes in the bacterial cell.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Growth Conditions
E. coli strains were grown in LB broth or agar at 37 °C. Gc strains were grown on Gc medium base plus Kellogg supplements (22.2 mm glucose, 0.68 glutamine, 0.45 mm cocarboxylase, 1.23 mm Fe(NO3)3) (42) at 37 °C in 5% CO2 or in Gc liquid medium (1.5% proteose peptone no. 3, 0.4% K2HPO4, 0.1% KH2PO4, 0.1% NaCl) with Kellogg supplements. Chloramphenicol was added at 30 mg/liter for E. coli and 0.75 mg/liter for N. gonorrhoeae; kanamycin was added at 50 mg/liter for E. coli.
Enzymes, Biochemicals, and Buffers
The native E. coli wild-type RecA and single-stranded DNA binding (SSB) proteins were purified as described previously (44). The concentration of the purified RecA and SSB proteins was determined from the absorbance at 280 nm using the extinction coefficients of 2.23 × 104 m−1 cm−1 (45) and 2.83 × 104 m−1 cm−1, respectively (46).
The N. gonorrhoeae RecA and SSB proteins were purified as described (26). The E. coli RecX protein was purified as described (47) with modifications described in the supplemental materials. The concentration of E. coli RecX was determined from the absorbance at 280 nm using the native extinction coefficient 2.57 × 104 m−1 cm−1 (47).
Phosphate buffer contained the indicated concentration of potassium phosphate (pH 6.8), 1 mm DTT, 0.1 mm EDTA, 10% (w/v) glycerol. R buffer contained 20 mm Tris-HCl, 80% cation (pH 7.5), 1 mm DTT, 0.1 mm EDTA, 10% (w/v) glycerol. RecA buffer contained 25 mm Tris-OAc (80% cation, pH 7.4), 1 mm DTT, 5% (w/v) glycerol, 3 mm potassium glutamate, and 10 mm Mg(OAc)2.
Cloning, Overexpression, and Purification of the Native N. gonorrhoeae Wild-type RecX (RecXNg) Protein
The Gc recX gene was amplified from the FA1090 genome by PCR using primers GCRXPETFOR and GCRXPETREV (supplemental Table S3). The resulting 470-bp DNA fragment was cloned into vector pBLUNT (Invitrogen), sequenced, and subcloned into pET21a yielding construct pET21a-RecXNg.
Competent cells of E. coli strain BL21(DE3) were transformed with pET21a-RecXNg. Ten liters of culture were grown at 37 °C in LB medium to an A600 of 0.52. RecXNg protein expression was induced by the addition of isopropyl-1-thio-β-d-galactopyranoside to 0.4 mm. Following a 3.75-h incubation at 37 °C, ∼20 g of cells were harvested by centrifugation, flash frozen in liquid N2, and stored at −80 °C. The RecXNg protein was purified as described in the supplemental materials. The concentration of RecXNg was determined from the absorbance at 280 nm using the native extinction coefficient 1.857 × 104 m−1 cm−1. The RecXNg extinction coefficient was determined during the course of the present work using procedures published elsewhere (48, 49).
Cloning, Overexpression, and Purification of the N. gonorrhoeae HA-tagged RecX (RecXNgHA) and the RecX Point Mutants (HisRecXNgHA)
Genes encoding the parent and mutant RecXNgHA proteins were amplified by PCR using Pfu polymerase from clones pGCC6/GCRXHA, pGCC6/G24E, pGCC6/D54A, pGCC6/Y57A, pGCC6/Q81K, and pGCC6/RF130AA using primers GCRXPETFOR and GCRXREVHA (supplemental Table S3). DNAs were cloned into pBLUNT, sequenced, and subcloned into pET28a. Doubly-tagged HisRecXNgHA proteins were purified from strain BL21(DE3).
Two liters (4 liters for R130A/F132A and D54A) of culture were grown, induced, harvested, and stored as described for wild-type RecXNg. The HisRecXNgHA point mutants were purified as described in the supplemental materials. The concentration of each point mutant was calculated from the absorbance at 280 nm using the calculated extinction coefficient 2.54 × 104 m−1 cm−1.
DNA Substrates
The cssDNA from bacteriophage M13mp18 (7249 nucleotides) was prepared as described (50, 51) with the following modifications. Chemically competent E. coli JM101 cells were transfected with gel-purified RFI M13mp18 DNA from New England Biolabs. The CsCl banding was done twice in a Beckman Ti60 rotor at 37,000 rpm for 15–20 h. The concentration of cssDNA was determined by absorbance at 260 nm, using 36 μg/ml A260−1 as the conversion factor. All of the DNA concentrations are given in μm nucleotides with the exception of the fluorescence anisotropy experiments, where the DNA concentrations are given in μm molecules. The oligonucleotides used in the fluorescence anisotropy experiments were purchased from Integrated DNA Technologies and resuspended and diluted in 10 mm Tris-acetate (80% cation) and 1 mm EDTA.
ATPase Assay
A coupled spectrophotometric enzyme assay (52, 53) as described in Ref. 54 was used to measure the DNA-dependent ATPase activities of the RecAEc and RecANg proteins. The reaction mixtures (100 μl) contained 4 μm M13mp18 cssDNA, 2.4 μm RecA, and a coupling system of 1.5 mm NADH and 10 units/ml lactate dehydrogenase in RecA buffer. An ATP regeneration system consisting of 10 units/ml pyruvate kinase and 3.5 mm phosphoenolpyruvate was included. The reactions containing 3 mm dATP instead of ATP contained 60 units/ml pyruvate kinase and 25 units/ml lactate dehydrogenase. The reactions were started after a 10-min incubation of the reaction mixtures by the addition of a mixture of (d)ATP and SSB at 3 mm and 0.4 μm final concentrations, respectively. The RecX protein was added after a 15-min incubation as indicated. The figures and figure legends indicate which RecA, RecX, and SSB proteins were used in each reaction. All of the experiments were carried out at least three times with reproducible results.
Electron Microscopy
A modified Alcian method was used to visualize RecA filaments. Activated grids were prepared as described previously (28).
Samples for electron microscopy analysis were prepared as follows. All of the incubations were carried out at 37 °C. RecAEc or RecANg (3 μm) was preincubated with 6 μm M13mp18 cssDNA in RecA buffer for 10 min. An ATP regeneration system of 10 units/ml creatine phosphokinase and 12 mm phosphocreatine was included in the incubation. ATP and SSB were added to 3 mm and 0.6 μm, respectively, and the reaction was incubated for another 15 min. RecXNg or RecXEc was added to concentrations of 0.11 and 0.1 μm, respectively, and incubated for various times as indicated. ATPγS was then added to 3 mm to stabilize the filaments, followed by another 3-min incubation. The sample was prepared for analysis as described previously (54).
To determine the proportion of molecules that were either fully or partially coated by RecA or bound only by SSB at different times of incubation with RecX, a minimum of 500 molecules from two independent experiments and two separate regions of each grid were counted at an identical magnification. A RecA filament was considered gapped if the ssDNA molecule to which it was bound had a detectable region of SSB-coated DNA of any size. The relative length of the remaining filament and the length of SSB-coated DNA were used to classify the gaps into three categories: small gaps, medium gaps, and big gaps (see supplemental Fig. S3 for examples and results section for descriptions). Linearized DNA molecules originating likely from shearing force during pipetting were also counted. With the total number of filaments counted as 100%, the percentage of each type of nucleoprotein filament was calculated.
Imaging and photography were carried out as described previously (54). Images were obtained of the two types of nucleoprotein filaments showing the highest percentage of all of the filaments counted. Where possible, both of the filament types were captured in the same image. Where this was impossible, the type of filament occurring at the highest percentage was chosen for photography.
A cytochrome c method was used to visualize M13mp18 cssDNA to check the purity of the DNA. The samples were prepared as described previously (54, 55).
Fluorescence Anisotropy
The experiments were carried out at 25 °C with a Beacon fluorescence polarization system (Invitrogen). Serial dilutions of either RecXNg or RecXEc were incubated with 1 nm fluorescein-labeled oligonucleotide den7 (supplemental Table S3). The final reaction volume was 100 μl, containing RecA buffer, 3 mm ATP, and compensating amounts of RecXEc or RecXNg storage buffer. The reactions were incubated for 30 min shielded from light before measurements were done. The Kd values were calculated using Prism software fitting a nonlinear regression and one site total binding. The data are reported in triplicate.
Kinetic Pilus-dependent Colony Morphology Change Assay
This assay measures the number of visible pilus-dependent colony morphology changes that occur over time and is a very reliable method to report pilin antigenic variation frequencies (56). Briefly, gonococcal strains were revived onto Gc medium base from frozen freezer stocks. After ∼20 h of growth, five individual colonies were passaged onto a plate. Colony variation of 20 colonies of each strain was scored after 18, 20, 22, 24, and 26 h by observing the number of P regions arising from each colony. The scores for the individual colonies at each time point were averaged.
RESULTS
Experimental Design
An array of biochemical approaches, including kinetic assays utilizing RecA-mediated ATP hydrolysis, electron microscopy, and fluorescence anisotropy, were used to analyze the effects of RecXNg and its mutant variants on RecANg and RecAEc function in vitro. This work was complemented by an analysis of RecXNg and its mutant variants on pilin variation in N. gonorrhoeae. Most in vitro experiments described below were carried out using RecXNg and its cognate RecA and SSB proteins and also RecXEc with its cognate RecA and SSB. The SSB protein used is always cognate to the RecA protein used. We also tested the effect of RecXNg on RecAEc (with SSBEc) for three reasons: RecAEc is very well characterized, many mutants are available to use as tools, and we could directly compare RecXNg with RecXEc.
The RecXNg Protein Is a Negative Modulator in Vitro, Inhibiting the ATPase Activity of RecANg and RecAEc More Potently than RecXEc
We first examined the effect of RecXNg or RecXEc on the ATPase activity of preformed RecA filaments on circular ssDNA (cssDNA). Enough RecA (1 RecA/1.67 nucleotides) was present to more than saturate all of the RecA-binding sites on the DNA. Under these conditions, it was previously estimated that two or three disassembling filament ends would be present per M13mp18 DNA molecule coated with RecAEc (57). A 1:24 ratio of RecXEc (100 nm) to RecA (2.4 μm) inhibits the ATPase activity of RecAEc in 15–20 min (Fig. 1A), consistent with previous results (35). RecXEc has almost no effect on the ATPase activity of RecANg at this same 1:24 ratio (Fig. 1C). In contrast, RecXNg completely abolishes the ATPase activity of either RecANg or RecAEc on cssDNA within 3–5 min at a 1:24 ratio of RecX (110 nm) to RecA (2.4 μm) (Fig. 1, B and D). The percentage of ATPase activity remaining 9 min after RecX addition, averaged over three independent experiments, is detailed in supplemental Table S1. The potent inhibition of RecANg by RecXNg is not due to an intrinsically faster disassembly of RecANg because RecXNg has a similar if not a stronger effect on RecAEc. The filament capping mechanism is insufficient to explain this rapid inhibitory effect.
FIGURE 1.

RecXNg inhibits the ATPase activity of RecA more potently than RecXEc. The reaction scheme at the top of the figure indicates the order of addition for this experiment. The reactions were carried out as described under “Experimental Procedures” and contained 2.4 μm RecA protein, 4 μm M13mp18 cssDNA, 0.4 μm SSB, and 3 mm ATP. ATP and SSB were added at t = 0. Each panel (A–D) contains a different combination of RecA and RecX proteins from either N. gonorrhoeae or E. coli as indicated. RecA filaments hydrolyze ATP at a steady rate when assembled on cssDNA. At t = 15 RecX was added at the concentrations indicated in the figure. RecXNg leads to a more rapid decrease of ATP hydrolysis by either RecA protein than does RecXEc.
RecX Point Mutants Affect RecX Function in Gc as Well as in Vitro Functions
Given the positive effect of RecXNg on RecANg function in the bacterial cell (41), we further explored the unexpectedly inhibitory effects of RecXNg in vitro. An amino acid alignment of RecX homologs from a variety of Gram-negative and Gram-positive bacteria and from the plant model organism Arabidopsis thaliana was carried out to guide the design of mutant proteins (supplemental Fig. S1). We noted residues that appeared to be conserved across species (Asp-54, Tyr-57, Arg-130, and Phe-132), and residues that appeared to differ specifically between Gc and the other species (Gly-24 and Gln-81). To determine the importance of these residues to RecXNg function, the conserved residues were mutated to alanines (D54A, Y57A, and R130A/F132A), and the residues that differed in Gc were mutated to the prevailing amino acid in the alignment (G24E and Q81K). RecXNg point mutant constructs were subsequently recombined into the FA1090ΔrecX chromosome (see “Experimental Procedures”).
We tested the effect of each RecX point mutant using the well established pilus-dependent colony morphology change assay, which measures observable changes in colony morphology resulting from the appearance of nonpiliated blebs over time (56) and is a widely used method to measure frequencies of Gc pilin antigenic variation (23, 24) (Fig. 2A). We found that strains R130A/F132A and D54A showed statistically the same amount of variation as the recX null strain. Strains Y57A and G24E showed an intermediate level of variation, statistically distinct from both the recX null mutant and the ΔrecX/recXHA complement strain. Strain Q81K showed a statistically higher amount of antigenic variation than strain ΔrecX/recXHA (Fig. 2A). The RecX protein expression levels in the parent (ΔrecX/recXHA) and mutant strains were measured by Western blot (data not shown) and varied from levels equal to that of the parent RecXHA protein to as low as 3% of the parent protein. However, the differences in protein expression levels of the mutant proteins do not appear to be responsible for the observed differences in phase variation. Constructs created to drive the expression of the parent RecXHA protein from two different promoters resulted in a 252-fold difference in RecXHA protein levels, as measured by Western blot, and spanned the various point mutant expression levels, yet both promoter constructs mediated identical levels of phase variation in Gc (data not shown). Therefore, we conclude that these amino acid residues all influence RecX activity in Gc, with the relative importance of the residues being: R130A/F132A > D54A > Y57A > G24E > Q81K.
FIGURE 2.

RecXNg point mutants that behave like ΔrecX strains are deficient at inhibiting RecA ATPase activity in vitro. A, colony variation analysis of RecXNg point mutants using the kinetic pilus-dependent colony morphology change assay. The variation score is given based on the average number of P-blebs that appear on a colony at each time point. FA1090 is the parental strain; ΔrecX is the recX mutant strain; ΔrecX/recXHA contains the C-terminal HA tagged recX complement recombined into the chromosome of the ΔrecX strain; the remaining strains are HA-tagged recX point mutants recombined into the ΔrecX strain (see “Experimental Procedures”). The error bars represent the standard error of the mean of two or three experiments, with 20 colonies analyzed for each experiment. At the 26-h time point, the asterisk indicates a statistically significant difference (p < 0.05) relative to recXHA; † indicates a statistically significant difference (p < 0.05) relative to ΔrecX; ‡ indicates no significant difference (p > 0.05) relative to FA1090 using the Student's t test. B, ATPase assay with RecXNg point mutants with the same order of addition as in Fig. 1. The reactions contained 2.4 μm RecANg protein, 4 μm M13mp18 cssDNA, 0.4 μm GcSSB, and 3 mm ATP. ATP and SSB were added at t = 0. RecA filaments hydrolyze ATP at a steady rate when assembled on cssDNA. At t = 15, 110 nm of a RecXNg point mutant or compensating storage buffer was added as indicated in the figure. The graph shown is a representative of three independent reproducible experiments. C, quantification of percentage of RecA-dependent ATPase activity remaining after RecX addition. The percentage of ATPase activity after RecX addition was calculated at t = 27 by taking the tangent to the best fit line at t = 27 from three independent experiments.
We purified His-tagged versions of the same point mutants and characterized their effect on RecA using ATPase assays. Circular dichroism spectra indicate that all of the mutants are folded correctly except for R130A/F132A, which was slightly misfolded (supplemental Fig. S2). The His-tagged wild-type RecXNg has the same inhibitory effects on RecAEc and RecANg filaments as the native protein, as seen in the ATPase assay (Fig. 2B). ATPase assays showed that the RecXNg point mutants that lead to decreased pilus phase variation similar to ΔrecX strains (R130A/F132A and D54A) are also deficient in inhibiting RecA-catalyzed ATP hydrolysis (Fig. 2B). The Q81K mutant was proficient in RecA ATPase inhibition, whereas the Y57A and G24E mutants showed intermediate activities in both assays (Fig. 2B). A quantitative measure of the percentage of ATPase activity 12 min after RecX addition compared with a reaction where no RecX was added is shown in Fig. 2C. The RecXNg mutants exhibited deficiencies in RecA ATPase activity inhibition in the order: R130A/F132A > G24E > D54A > Y57A > Q81K. This is the same trend seen for deficiencies in pilus phase variation, with only the G24E mutation exhibiting somewhat greater effects in vitro than in N. gonorrhoeae. This indicates that the inhibitory effect of RecXNg on RecA activities in vitro corresponds to a positive effect on pilus phase and antigenic variation in the bacterial cell.
The Ratio of RecX to RecA Proteins in N. gonorrhoeae Is Higher than in E. coli
RecXNg is able to inhibit the RecANg ATPase activity at far substoichiometric levels in vitro (Fig. 1). To determine the relative levels of RecA and RecX in Gc, we performed a semi-quantitative Western blot analysis of the two proteins. Serial dilutions of purified RecX and RecA were used as standards for comparison against cell extracts made from Gc strain FA1090. We found that there averaged one RecX molecule for every 5–10 RecA molecules (data not shown), a higher RecXNg to RecA ratio than used in all of the assays shown above and a much higher ratio than the ratio of the E. coli proteins in the bacterial cell (approximately one RecX to 300 RecA) (31–33). These data suggest that the strong inhibition of RecA by RecX is important for optimal RecA function in Gc.
RecXNg Addition Leads to Disassembly of RecA Filaments within 3–5 min
The data presented in Fig. 1 and the correlation between RecX activity in N. gonorrhoeae and the in vitro activities of RecXNg shown above prompted us to investigate the mechanism by which RecXNg inhibits RecA activities and compare it with the established filament capping mechanism for RecXEc. We used EM to determine whether the addition of RecXNg leads to RecA filament disassembly or whether RecXNg inhibits RecA ATPase activity, whereas RecA remains bound to DNA. The addition of ATPγS at different time points after RecX addition allows us to fix RecA filaments for visualization of the filament state at that time. In our experiments we observed many different states of RecA filaments and categorized them into five main groups for analysis. Representative images of each category are shown in supplemental Fig. S3. The categories are: full filaments where the cssDNA is coated with RecA (supplemental Fig. S3A), small gaps where a short region of SSB-coated DNA interrupts RecA filaments (supplemental Fig. S3B; generally more than 80% RecA coverage), medium gaps where SSB coats a larger region of the DNA and the RecA filaments are considerably shorter than in a full filament (supplemental Fig. S3C; corresponding roughly to 20–80% coverage), big gaps where the RecA filaments are very short (<20% coverage), the rest of the DNA molecule, which is coated by SSB (supplemental Fig. S3D), and SSB-coated DNA where the whole DNA molecule is coated with SSB (supplemental Fig. S3E).
When no RecX is added, most RecA filaments show full filaments and no regions of SSB-coated DNA (Fig. 3, A–C). When 100 nm RecXEc was added, RecAEc filaments were considerably shorter after 10 min, and after 20 min only big gaps (short RecA-coated DNA segments) remained. The addition of 110 nm RecXNg led to a complete disassembly of RecAEc or RecANg filaments after 3–5 min. A panel with representative images of the two most prominent categories of filaments for each time point is shown in Fig. 3. The rapid disassembly of RecA filaments observed via EM correlates well with the decrease in ATPase activity (Fig. 1), suggesting that the addition of RecXNg causes a substantial increase in RecA disassembly from cssDNA reflected by the sharp decrease in ATPase activity.
FIGURE 3.

Electron microscopy time course of RecA protein filaments on cssDNA shows RecA disassembly upon the addition of RecXEc or RecXNg protein. Electron micrographs show RecANg or RecAEc filaments formed on cssDNA in the absence of the RecX protein (A–C) and after RecX addition (D–S). The reactions are carried out according to the reaction scheme in Fig. 1 and include 3 μm RecA, 6 μm M13mp18 cssDNA, 0.6 μm SSB, and 3 mm ATP. RecX protein to 100 nm for RecXEc and 110 nm for RecXNg or the equivalent volume of RecX storage buffer (A–C) were added 15 min after RecA nucleoprotein filaments were established. After the time indicated in the figure, the filaments were fixed with ATPγS. The reaction mixtures were diluted 8- or 16-fold before adhesion to the electron microscopy grid.
RecXNg Addition Increases the Number of Gaps per RecA-ssDNA Filament
Faster RecA disassembly could be explained by at least two possible mechanisms. RecXNg could actively promote RecA disassembly, or it could create more disassembling ends in a RecA filament by disrupting interactions between RecA monomers. To test whether RecXNg creates new gaps in RecA-DNA filaments, we closely examined the EM time course data. We counted the number of gaps and size of gaps of over 400 DNA molecules/time point and condition (supplemental Table S2). Using a rate of disassembly of 60–70 RecAEc monomers/minute (57), 7–12 gaps would be needed to account for complete disassembly within 3–5 min. The average number of visible gaps per filament for each time point is plotted in Fig. 4. The differences between the number of gaps produced by the two RecX proteins is small at best. At none of the time points do the gaps/filament average more than 2.5. This number is far below the number of gaps that would be expected (7–12) if RecXNg was simply creating more ends from which RecA would disassemble at 60–70 monomers/minute. The small differences evident in Fig. 4 argue that creating more disassembling RecA filament ends accounts at best for only a small part of the increase in inhibition by RecXNg. We thus continued to explore other potential effects of RecX, focusing on whether a direct facilitation of filament disassembly could play a role in the mechanism of RecXNg action.
FIGURE 4.

Gap numbers increase modestly in the presence of RecXNg. Graphical representation of the number of gaps per filament over time. The total number of gaps for each time point was calculated and divided by the number of filaments.
RecXNg and RecXEc Bind to ssDNA with Similar Affinity
We tested whether RecXNg binds ssDNA with higher affinity than RecXEc to find an explanation for the strong RecA-mediated ATPase inhibition by RecXNg and for the possibility that RecXNg creates slightly more disassembling ends. Fluorescence anisotropy experiments with fluorescein-labeled 50-nucleotide ssDNA show an increase in anisotropy as the RecX concentration is increased (Fig. 5). For RecXNg, an apparent dissociation constant (Kd) of 88.6 ± 7.1 nm was extracted from analysis of the binding curve. The apparent Kd for RecXEc using the same DNA substrate is 81.0 ± 6.4 nm. These two values are within standard error from each other, and we can conclude an almost identical affinity for ssDNA for both RecX proteins. Therefore, the stronger inhibitory effect of RecXNg on RecA ATP hydrolysis is not due to RecXNg binding more tightly to ssDNA than RecXEc. We note that this assay reveals a stronger binding of RecXEc to ssDNA than reported previously (35) (see “Discussion”).
FIGURE 5.
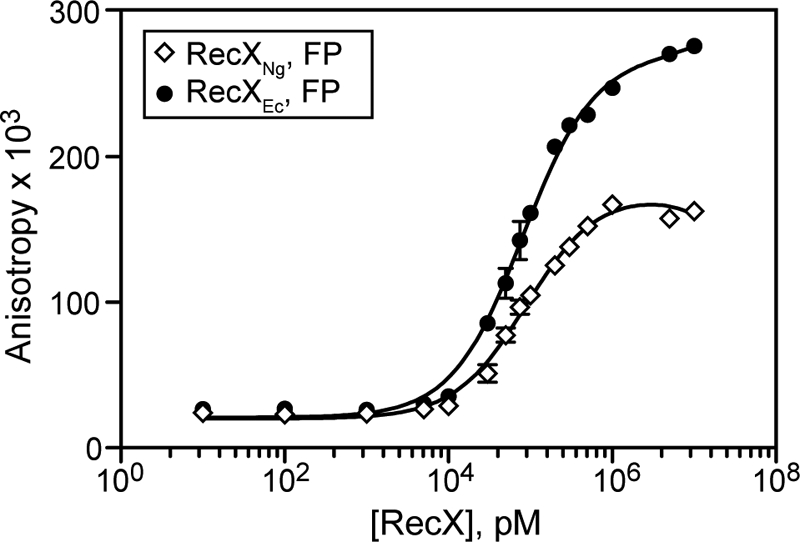
RecXNg does not have stronger ssDNA binding affinity than RecXEc. Serial dilutions of RecXNg and RecXEc were incubated with fluorescein-labeled ssDNA as described under “Experimental Procedures.” The data are reported in triplicate, and error bars are one standard deviation of the mean.
RecXNg and RecXEc Do Not Facilitate Disassembly of the ATPase-deficient RecA Mutant E38K K72R
To further investigate how RecXNg causes such an abrupt RecA inhibition, we used the RecAEc E38K K72R double mutant, and the fact that RecA-mediated ATP hydrolysis is coupled to RecA disassembly, as a tool to study the effect of RecXNg on RecA in the absence of RecA ATP hydrolysis (13, 57). RecA E38K K72R is proficient in forming filaments on cssDNA (54), but it hydrolyzes little or no ATP and is thus deficient in disassembly. Electron micrographs show that preformed RecA E38K K72R filaments on cssDNA are not substantially affected after a 15-min incubation with either RecXNg or RecXEc (Fig. 6). This indicates that neither of the RecX proteins can cause disassembly of a RecA in the absence of ATP hydrolysis. Therefore, ATP hydrolysis by RecA is necessary for RecX to mediate RecA disassembly.
FIGURE 6.

Electron microscopy of RecA E38K K72R filaments after RecX addition. Electron micrographs show RecA E38K K72R filaments formed on cssDNA in the absence of RecX (A) and after EcRecXRecXEc (B) or RecXNg (C) addition. The reactions follow the reaction scheme depicted in Fig. 1 and include 3 μm RecA E38K K72R, 6 μm M13mp18 cssDNA, 0.6 μm SSB, and 3 mm ATP. RecX protein to 100 nm for RecXEc and 110 nm for RecXNg or the equivalent volume of RecX storage buffer (A–C) were added 15 min after RecA nucleoprotein filaments were established. After 15 additional min, the filaments were fixed with ATPγS. The reaction mixtures were diluted 8- or 16-fold before adhesion to the electron microscopy grid.
RecXNg Inhibits the ATPase Activity of RecAEc E38K and the dATPase Activity of Wild-type RecAEc, whereas RecXEc Does Not
Certain conditions are known in which RecAEc is ATPase-proficient, but disassembly is uncoupled from ATP hydrolysis. For example, in the presence of dATP, the wild-type RecAEc does not disassemble significantly from cssDNA (13). A similar observation is made when reactions are carried out with RecAEc E38K, using ATP as a nucleotide co-factor.5 When RecXEc is added to RecAEc in the presence of dATP or RecAEc E38K, it causes no effect on dATP or ATP hydrolysis, respectively (Fig. 7). These results are consistent with the proposed filament capping mechanism for RecXEc, in which RecXEc acts to cap the growing ends of RecA filaments. If no end-dependent filament disassembly occurs, no change in ATPase activity can be observed. Contrary to RecXEc, RecXNg rapidly inhibits both the dATPase activity of wild-type RecAEc (Fig. 7A) and the ATPase activity of RecAEc E38K (Fig. 7B). EM experiments under these conditions show that RecA filaments are disassembled when RecXNg is present (data not shown), strongly suggesting that the inhibition of ATPase activity by RecXNg results from RecA disassembly. This again indicates that the RecXNg protein has an effect on RecA function beyond that of filament capping. We note that the slight increase in the number of gaps per filaments might be related to and may be symptomatic of a facilitated disassembly process.
FIGURE 7.
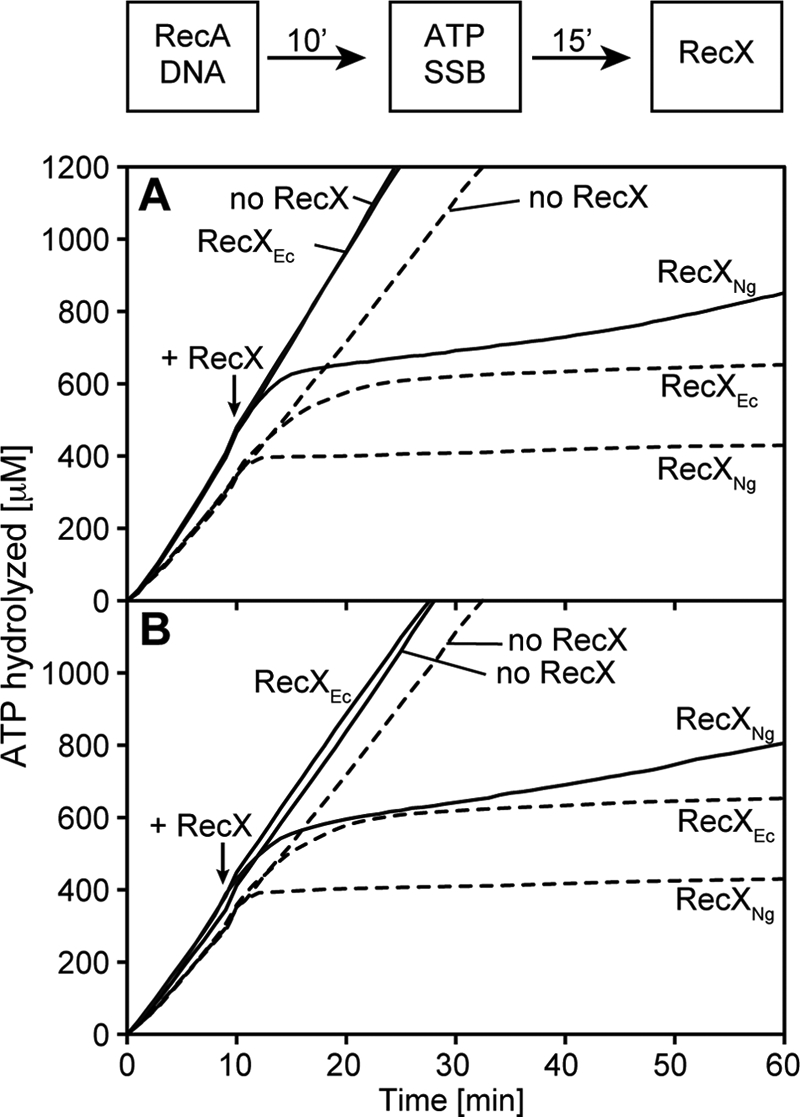
RecXNg inhibits the ATPase activity of RecA E38K and the dATPase activity of wild-type RecA whereas RecXEc does not. The order of addition and the incubation times are detailed in the reaction scheme at the top of the figure. A, reactions in contained 2.4 μm RecAEc protein, 4 μm M13mp18 cssDNA, 0.4 μm SSB, and 3 mm ATP (dashed lines) or dATP (solid lines). (d)ATP and SSB were added at t = 0. RecA filaments hydrolyze (d)ATP at a steady rate when assembled on cssDNA. At t = 10 either RecXEc (100 nm), RecXNg (110 nm) or compensating storage buffer was added as indicated in the figure. RecXNg leads to significant decrease of dATP hydrolysis by RecA, whereas RecXEc has a negligible effect on the dATPase activity of RecA. B, reactions contained 2.4 μm wild-type RecAEc (solid lines) or RecAEc E38K protein (dashed lines), 4 μm M13mp18 cssDNA, 0.4 μm SSB, and 3 mm ATP. ATP and SSB were added at t = 0. RecA filaments hydrolyze (d)ATP at a steady rate when assembled on cssDNA. At t = 10 either RecXEc (100 nm), RecXNg (110 nm), or compensating storage buffer was added as indicated in the figure. RecXNg leads to a significant decrease of ATP hydrolysis by RecA E38K, whereas RecXEc has a negligible effect on the ATPase activity of this RecA mutant.
DISCUSSION
There are two main conclusions to this work. First, RecXNg, a protein with a strong inhibitory effect on RecA protein filaments, has a decidedly positive effect on the function of RecANg protein in the cell. This indicates that optimal RecA function requires the substantial constraint of RecA filament length and/or the length of time the filament exists. For recombinational DNA repair, DNA transformation, and pilus antigenic variation, less can be more. Second, the RecXNg protein is a more potent inhibitor than the previously characterized RecXEc protein and may reveal a broader range of potential RecX functionality. It is likely that RecXNg accelerates end-dependent RecA filament disassembly using mechanisms that are absent or cryptic in the RecXEc protein.
The general result that RecXNg inhibits in vitro while enhancing recombination in N. gonorrhoeae is strengthened by the effects of a series of RecXNg mutant proteins exhibiting a range of functional deficiencies. There is a strong correlation between the capacity of a RecXNg variant to inhibit the RecANg ATPase and the capacity of that same mutant to support cellular functions such as RecANg-mediated pilus phase variation. Two mutations that affect residues conserved among all RecX proteins (R130A/F132A and D54A; see supplemental Fig. S2 for an alignment) are deficient in RecA ATPase inhibition and show the lowest frequency of pilus phase variation. These residues therefore are likely important for RecXNg activity. Mutations that convert residues that are highly conserved in most RecX proteins (but are altered in RecXNg) to the consensus residue generally have intermediate effects. Overall, the trends allow us to conclude that strong inhibition of RecA ATPase activity corresponds to high pilus phase variation frequency.
Mechanistically, the RecXNg protein imposes substantial inhibition on RecA filaments (both RecANg and RecAEc) at rates too rapid to be explained by the filament capping mechanism proposed for RecXEc (35). The decline in ATP hydrolysis in all cases corresponds closely to filament disassembly as seen by EM. Thus, RecXNg is somehow facilitating this more rapid disassembly. This could occur by 1) a displacement mechanism in which RecX actively removes RecA from the DNA (a RecX-based activity that does not rely on any action of RecA); 2) a disruption of subunit-subunit interactions to create and cap more filament ends, the wedge model, leading to more rapid disassembly at the now more numerous disassembling ends; or 3) a facilitation of ATPase- and end-dependent RecA filament disassembly (an active RecX mechanism, but one that relies on RecA-mediated ATP hydrolysis). In mechanism 3, RecXNg could simply enhance the normal RecA disassembly process, perhaps by means of conformation changes brought about by RecXNg binding in the filament groove.
We argue against active removal of RecA (mechanism 1) in a manner that does not rely on any function of RecA, because RecXNg is unable to displace the RecA K72R E38K mutant protein from the DNA. This mutant does not hydrolyze ATP, indicating that RecA-mediated ATP hydrolysis is required for RecA filament disassembly whether RecXNg is present or not. Mechanism 2, the wedge model, is an elaboration of the capping idea, in which filament ends are both created and capped by RecXNg. The small apparent increase in filament gaps observed by EM when RecXNg is present relative to those present with RecXEc supports the idea that RecXNg may have a limited capacity to disrupt subunit-subunit interactions in a RecA filament. However, the observed increase in filament gaps (Fig. 4 and supplemental Fig. S4) is small at best and insufficient to explain the enhancement of activity with RecXNg. Although some RecXNg-generated gaps may have been missed, we do not think the potential underestimation is sufficient to allow for a complete explanation of the effects of RecXNg. If mechanism 2 is insufficient to explain RecXNg activity, then there may be a direct facilitation of RecA dissociation by RecX (mechanism 3), a possibility we currently favor based on these data (Fig. 8). In all cases, the mechanism by which RecXNg promotes RecA filament disassembly requires ATP hydrolysis by RecA.
FIGURE 8.
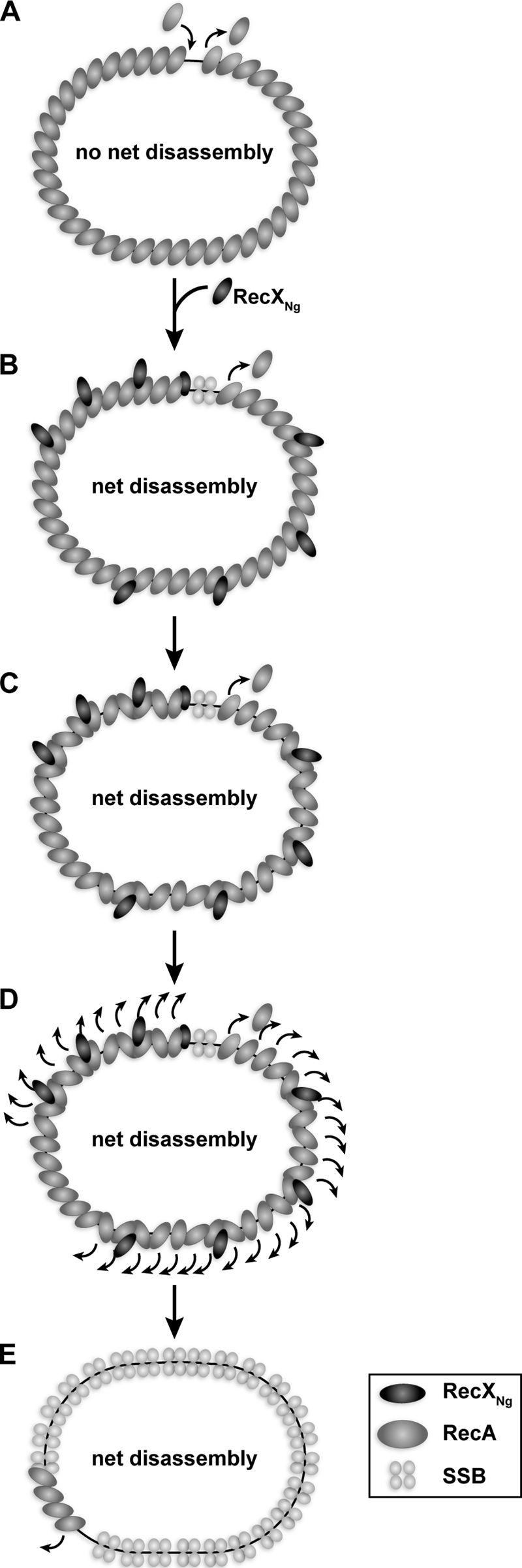
Model of the proposed mechanism of RecA inhibition by RecXNg. A, RecA filament extends at the 3′ end and disassembles at the 5′ end such that on cssDNA no net disassembly occurs. B, upon RecXNg addition at a substoichiometric ratio to RecA, we hypothesize that RecXNg binds along RecA filaments and also blocks assembling filament ends. C, binding of RecXNg could lead to a disruption of the cooperativity between RecA monomers in the vicinity of where RecXNg is bound. D, this then results in faster net disassembly of RecA and overall dissociation of the RecA filament. E, after a few minutes most RecA has disassembled, and SSB coats the cssDNA, preventing RecA from rebinding. Short RecA filaments may remain in regions where no RecXNg was bound.
When disassembly is uncoupled from ATP hydrolysis, using dATP (13) or RecA E38K,5 RecXNg addition leads to RecA disassembly, whereas RecXEc addition does not. This is one of the key differences in the mechanisms between the two RecX proteins and allows us to argue that complex formation of RecXNg with RecANg may result in more rapid but still ATPase-dependent RecA disassembly. In contrast, RecXEc relies on the intrinsic disassembly rate of RecA. A more active facilitation of RecA disassembly by the RecXEc protein, similar to mechanism 3, was previously proposed by Ragone et al. (36) based on surface plasmon resonance measurements of very short RecA filaments. We have found no evidence for an active RecXEc mechanism with our much longer RecA filaments. However, the new results with RecXNg suggest that such an active mechanism may be well within the functional potential of RecX proteins, and the assays we are using for RecXEc studies may simply obscure it. Mechanisms 2 and 3 both have the virtue of providing a role for the RecX protein previously shown to bind along the major groove of a RecA filament (34).
When assayed by fluorescence anisotropy, RecXEc has a more than 10-fold lower Kd for ssDNA than had previously been reported (35). These assays are likely more reflective of the actual dissociation constants than the ones reported previously using electrophoretic mobility shift assays. Other cellular factors may greatly influence the interaction of RecX with ssDNA. For example the presence of a RecA filament on ssDNA could increase the affinity of RecX to the DNA. Alternatively, the presence of SSB could decrease the affinity, which would be consistent with the competition of RecXEc with SSBEc (26) and RecXNg with SSBNg (data not shown). In sum, ssDNA binding by RecX may play a more important role in the mechanisms of either RecX protein than assumed so far. However, the fact that RecXNg has slightly lower ssDNA binding affinity than RecXEc implies that ssDNA binding is not the reason for the much stronger inhibitory effect on RecA by RecXNg.
We acknowledge that the strong inhibition of RecA may not directly affect pilus phase variation. There might be another enzyme or protein not yet discovered that could shed light on this apparently counterintuitive correlation. For example, another protein or proteins could partially counter the effects of RecX and perhaps cooperate with it to create the ideal RecA filament species. However, based on the evidence to date, we suggest that the key function for RecX is to keep RecA filaments at an optimal length. The process of pilus phase variation involves the recombination of very short DNA sequences, which may require shorter RecA filaments. Longer RecA filaments might inhibit the process of pilus phase variation by producing nonproductive products. Regardless, the very different mechanism-of-action of the RecXNg and RecXEc proteins shows how these two Gram-negative bacteria have optimized their recombination processes to respond to different selective pressures.
Acknowledgments
We thank Dr. Darrel McCaslin for analyzing CD spectra of the RecXNg point mutants and the Dr. James Keck laboratory for allowing us to use their fluorescence polarimeter.
This work was supported, in whole or in part, by National Institutes of Health Grants GM32335 (to M. M. C.) and R37 AI033493 and R01 AI044239 (to H. S. S.). This work was also supported by the Dr. James Chieh-Hsia Mao and the William R. & Dorothy E. Sullivan Wisconsin Distinguished Graduate Fellowships (to M. C. G.) and partially supported by American Cancer Society Postdoctoral Fellowship PF-00-016-01-GMC (to E. A. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental text, Tables S1–S3, and Figs. S1–S4.
P. O. P. Schook, E. A. Stohl, A. K. Criss, and H. S. Seifert, submitted for publication.
E. A. Stohl, M. C. Gruenig, M. M. Cox, and H. S. Seifert, submitted for publication.
R. Britt, unpublished results.
- Ec
- E. coli
- ss
- single-stranded
- css
- circular ss
- Gc
- gonococcus
- Ng
- N. gonorrhoeae
- SSB
- single-stranded DNA-binding protein
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
REFERENCES
- 1.Cox M. M., Goodman M. F., Kreuzer K. N., Sherratt D. J., Sandler S. J., Marians K. J. (2000) Nature 404, 37–41 [DOI] [PubMed] [Google Scholar]
- 2.Cox M. M. (2002) Mutat. Res. Fund. Mol. Mech. Mutagen. 510, 107–120 [Google Scholar]
- 3.Kowalczykowski S. C. (2000) Trends Biochem. Sci. 25, 156–165 [DOI] [PubMed] [Google Scholar]
- 4.Kuzminov A. (1999) Microbiol. Mol. Biol. Rev. 63, 751–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michel B., Boubakri H., Baharoglu Z., LeMasson M., Lestini R. (2007) DNA Repair 6, 967–980 [DOI] [PubMed] [Google Scholar]
- 6.Lusetti S. L., Cox M. M. (2002) Annu. Rev. Biochem. 71, 71–100 [DOI] [PubMed] [Google Scholar]
- 7.Yu X., Egelman E. H. (1992) J. Mol. Biol. 227, 334–346 [DOI] [PubMed] [Google Scholar]
- 8.Chen Z., Yang H., Pavletich N. P. (2008) Nature 453, 489–494 [DOI] [PubMed] [Google Scholar]
- 9.Register J. C., 3rd, Griffith J. (1985) J. Biol. Chem. 260, 12308–12312 [PubMed] [Google Scholar]
- 10.van Loenhout M. T., van der Heijden T., Kanaar R., Wyman C., Dekker C. (2009) Nucleic Acids Res. 37, 4089–4099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galletto R., Amitani I., Baskin R. J., Kowalczykowski S. C. (2006) Nature 443, 875–878 [DOI] [PubMed] [Google Scholar]
- 12.Brenner S. L., Mitchell R. S., Morrical S. W., Neuendorf S. K., Schutte B. C., Cox M. M. (1987) J. Biol. Chem. 262, 4011–4016 [PubMed] [Google Scholar]
- 13.Shan Q., Bork J. M., Webb B. L., Inman R. B., Cox M. M. (1997) J. Mol. Biol. 265, 519–540 [DOI] [PubMed] [Google Scholar]
- 14.Schutte B. C., Cox M. M. (1987) Biochemistry 26, 5616–5625 [DOI] [PubMed] [Google Scholar]
- 15.Koomey M., Gotschlich E. C., Robbins K., Bergström S., Swanson J. (1987) Genetics 117, 391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koomey J. M., Falkow S. (1987) J. Bacteriol. 169, 790–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seifert H. S. (1997) Gene 188, 215–220 [DOI] [PubMed] [Google Scholar]
- 18.Boslego J. W., Tramont E. C., Chung R. C., McChesney D. G., Ciak J., Sadoff J. C., Piziak M. V., Brown J. D., Brinton C. C., Jr., Wood S. W., Brian J. R. (1991) Vaccine 9, 154–162 [DOI] [PubMed] [Google Scholar]
- 19.Kline K. A., Sechman E. V., Skaar E. P., Seifert H. S. (2003) Mol. Microbiol. 50, 3–13 [DOI] [PubMed] [Google Scholar]
- 20.Nassif X., Lowy J., Stenberg P., O'Gaora P., Ganji A., So M. (1993) Mol. Microbiol. 8, 719–725 [DOI] [PubMed] [Google Scholar]
- 21.Swanson J., Kraus S. J., Gotschlich E. C. (1971) J. Exp. Med. 134, 886–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagblom P., Segal E., Billyard E., So M. (1985) Nature 315, 156–158 [DOI] [PubMed] [Google Scholar]
- 23.Haas R., Meyer T. F. (1986) Cell 44, 107–115 [DOI] [PubMed] [Google Scholar]
- 24.Swanson J., Bergström S., Robbins K., Barrera O., Corwin D., Koomey J. M. (1986) Cell 47, 267–276 [DOI] [PubMed] [Google Scholar]
- 25.Stohl E. A., Blount L., Seifert H. S. (2002) Microbiology 148, 1821–1831 [DOI] [PubMed] [Google Scholar]
- 26.Baitin D. M., Gruenig M. C., Cox M. M. (2008) J. Biol. Chem. 283, 14198–14204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Little J. W., Mount D. W. (1982) Cell 29, 11–22 [DOI] [PubMed] [Google Scholar]
- 28.Lusetti S. L., Wood E. A., Fleming C. D., Modica M. J., Korth J., Abbott L., Dwyer D. W., Roca A. I., Inman R. B., Cox M. M. (2003) J. Biol. Chem. 278, 16372–16380 [DOI] [PubMed] [Google Scholar]
- 29.Cox M. M. (2007) Crit. Rev. Biochem. Mol. Biol. 42, 41–63 [DOI] [PubMed] [Google Scholar]
- 30.Black C. G., Fyfe J. A., Davies J. K. (1998) Gene 208, 61–66 [DOI] [PubMed] [Google Scholar]
- 31.Stohl E. A., Brockman J. P., Burkle K. L., Morimatsu K., Kowalczykowski S. C., Seifert H. S. (2003) J. Biol. Chem. 278, 2278–2285 [DOI] [PubMed] [Google Scholar]
- 32.Sassanfar M., Roberts J. W. (1990) J. Mol. Biol. 212, 79–96 [DOI] [PubMed] [Google Scholar]
- 33.Moreau P. L. (1987) J. Mol. Biol. 194, 621–634 [DOI] [PubMed] [Google Scholar]
- 34.VanLoock M. S., Yu X., Yang S., Galkin V. E., Huang H., Rajan S. S., Anderson W. F., Stohl E. A., Seifert H. S., Egelman E. H. (2003) J. Mol. Biol. 333, 345–354 [DOI] [PubMed] [Google Scholar]
- 35.Drees J. C., Lusetti S. L., Chitteni-Pattu S., Inman R. B., Cox M. M. (2004) Mol. Cell 15, 789–798 [DOI] [PubMed] [Google Scholar]
- 36.Ragone S., Maman J. D., Furnham N., Pellegrini L. (2008) EMBO J. 27, 2259–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Papavinasasundaram K. G., Movahedzadeh F., Keer J. T., Stoker N. G., Colston M. J., Davis E. O. (1997) Mol. Microbiol. 24, 141–153 [DOI] [PubMed] [Google Scholar]
- 38.Vierling S., Weber T., Wohlleben W., Muth G. (2000) J. Bacteriol. 182, 4005–4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Mot R., Schoofs G., Vanderleyden J. (1994) Nucleic Acids Res. 22, 1313–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pagès V., Koffel-Schwartz N., Fuchs R. P. (2003) DNA Repair 2, 273–284 [DOI] [PubMed] [Google Scholar]
- 41.Stohl E. A., Seifert H. S. (2001) Mol. Microbiol. 40, 1301–1310 [DOI] [PubMed] [Google Scholar]
- 42.Kellogg D. S., Jr., Peacock W. L., Jr., Deacon W. E., Brown L., Pirkle D. I. (1963) J. Bacteriol. 85, 1274–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deleted in proof
- 44.Petrova V., Chitteni-Pattu S., Drees J. C., Inman R. B., Cox M. M. (2009) Mol. Cell 36, 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Craig N. L., Roberts J. W. (1981) J. Biol. Chem. 256, 8039–8044 [PubMed] [Google Scholar]
- 46.Lohman T. M., Overman L. B. (1985) J. Biol. Chem. 260, 3594–3603 [PubMed] [Google Scholar]
- 47.Drees J. C., Lusetti S. L., Cox M. M. (2004) J. Biol. Chem. 279, 52991–52997 [DOI] [PubMed] [Google Scholar]
- 48.Marrione P. E., Cox M. M. (1995) Biochemistry 34, 9809–9818 [DOI] [PubMed] [Google Scholar]
- 49.Robu M. E., Inman R. B., Cox M. M. (2004) J. Biol. Chem. 279, 10973–10981 [DOI] [PubMed] [Google Scholar]
- 50.Messing J. (1983) Methods Enzymol. 101, 20–78 [DOI] [PubMed] [Google Scholar]
- 51.Neuendorf S. K., Cox M. M. (1986) J. Biol. Chem. 261, 8276–8282 [PubMed] [Google Scholar]
- 52.Morrical S. W., Lee J., Cox M. M. (1986) Biochemistry 25, 1482–1494 [DOI] [PubMed] [Google Scholar]
- 53.Lindsley J. E., Cox M. M. (1990) J. Biol. Chem. 265, 9043–9054 [PubMed] [Google Scholar]
- 54.Gruenig M. C., Renzette N., Long E., Chitteni-Pattu S., Inman R. B., Cox M. M., Sandler S. J. (2008) Mol. Microbiol. 69, 1165–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schnos M., Inman R. B. (2000) Mol. Biotech. 16, 77–86 [DOI] [PubMed] [Google Scholar]
- 56.Sechman E. V., Rohrer M. S., Seifert H. S. (2005) Mol. Microbiol. 57, 468–483 [DOI] [PubMed] [Google Scholar]
- 57.Arenson T. A., Tsodikov O. V., Cox M. M. (1999) J. Mol. Biol. 288, 391–401 [DOI] [PubMed] [Google Scholar]