

Abstract
With the increasing number and variations of genome sequences available control of gene expression with synthetic, cell permeable molecules is within reach. The variety of sequence-specific-binding agents is, however, still quite limited. Many minor groove binding agents selectivity recognize AT over GC sequences but have less ability to distinguish among different AT sequences. The goal with this paper is to develop compounds that can bind selectively to different AT sequences. A number of studies indicate that AATT and TTAA sequences have significantly different physical and interaction properties and different requirements for minor groove recognition. Although it has been difficult to get minor groove binding at TTAA, DB293, a phenyl-furan-benzimidazole-diamidine, was found to bind as a strong, cooperative dimer at TTAA but with no selectivity over AATT. In order to improve selectivity, modifications were made to each unit of DB293. Binding affinities and stoichiometries obtained from biosensor-surface plasmon resonance experiments show that DB1003, a furan-furan-benzimidazole diamidine binds strongly to TTAA as a dimer and has selectivity (KTTAA/KAATT = 6). CD and DNAse I footprinting studies confirmed the preference of this compound for TTAA. In summary (i) a favorable stacking surface provided by the pi system, (ii) H-bond donors to interact with TA base pairs at the floor of the groove provided by a benzimidazole (or indole) –NH and amidines, and (iii) appropriate curvature of the dimer complex to match the curvature of the minor groove play important roles in differentiating the TTAA and AATT minor grooves.
Keywords: TTAA, Sequence-recognition, DNA, Biosensor-SPR, Cooperative dimer
Introduction
Specific recognition of DNA sequences is essential for many biotechnology applications, for external control of gene expression, and for therapeutic targeting of DNA. Specific recognition of a unique sequence in human chromosomal DNA requires approximately 15 bp if the entire DNA sequence is considered.1 For the design of clinically useful drugs to selectively target cellular DNA, however, the preparation and use of high molecular weight agents that can recognize such a long sequence of DNA is limited by factors such as preparation costs, sample stability, solubility and cell uptake. In exploring methods to selectively target DNA, however, it is clear that the entire length of DNA need not be considered in calculating the requirements for specific recognition since much of biologically relevant DNA is complexed with a variety of tightly bound proteins. It is also clear that reversible binding of a small molecule to an isolated DNA duplex site is generally not sufficient to generate a significant biological response.
An alternative approach, which has recently emerged as a method to increase selective binding to DNA and synergistically generate a significant biological response, is to target locally similar clusters of DNA sequences or structures. Proof of principle of the effectiveness of this approach has come from studies of targeting clustered DNA AT sequences in the AT rich mitochondrial kinetoplast DNA of some parasitic, disease-causing microorganisms2–4, extended triplet repeat sequences that are implicated in a number of diseases,5–7 repeated G-rich sequences of telomeres and ribosomal DNA.8–11 The key feature of this approach is that one or more small molecules can be used to selectively target a clustered set of binding sites in DNA and generate a biological response that is not observed for binding to single sites.
In the model for this synergistic approach small molecules selectively recognize units of a DNA sequence or motif such that a region of DNA is effectively targeted by multiple bound compounds. A fundamental aspect of the model is that the desired biological response occurs only when the locally bound molecules of a compound reach a critical limit. Compounds bound in a more dispersed manner do not reach the critical level to initiate any significant response. With the synergistic interactions, highly selective DNA recognition and associated biological effects can be generated by compounds that each bind selectively to only 4–6 base pair sites. With this design scheme the problems of preparation expense, solubility, stability and uptake can be dealt with in an effective manner. The requirement for multiple bound compounds in a local region for a biological response also helps reduce nonselective toxicity relative to the desired effect. Individual compounds that bind at a nonselective site generate either no response or a repairable response at a low rate.
An additional attractive compound motif for development of sequence-selective DNA binding agents of 4–6 base pairs that can recognize repeated DNA sequences is based on linked-heterocyclic cations that bind in the DNA minor groove. Even with limited development efforts, diamidine derivatives of this class have demonstrated effective cell uptake as well as effective biological activity in animals and humans.12–15 The diamidines, for example, have proven activity against a number of human diseases that are caused by parasitic microorganisms.12,13 A key goal of development of these compounds is to increase the disease spectrum that they can target while taking advantage of their favorable biological and cell uptake properties. As with the natural product dication, netropsin, the diamidines have a general AT binding preference through DNA minor groove complexes. 2,12, 16–18 Selective recognition among different AT base pair sequences, however, has been more difficult to obtain with most minor groove binding compounds and the basis of any selectivity is poorly understood. Research to increase selectivity for different AT sequences by these compounds has been limited, and increased efforts in this direction are necessary to develop the compounds for new applications. Increased recognition selectivity is an essential requirement for a library of compounds for general use in targeting specific, local sequences of DNA to generate a desired biological response.
Our knowledge base for specific targeting of DNA by the heterocyclic diamidines is limited. DNase footprinting has indicated that the diamidine berenil has strong interactions with sequences of four or more AT base pairs with a preference for binding to AATT or ATAT over TTAA or TATA. DB75 and many heterocyclic dications follow the same trend.19 In a study of the bisbenzimidazole monocation, DB183, and the dication, DB185 (Figure S1), binding to different AT containing DNA sequences, we found that DB183 could bind to TTAA as a dimer while the dication bound as a monomer.20 This is a clear example of both DNA and compound molecular reorganization for structural complementary in complex formation. The isomeric sequences AATT and TTAA have quite different properties and the results with DB183 suggest that compounds could be designed to selectively recognize one or the other of these sequences (AATT vs TTAA). The generally reduced compound affinity at TTAA and TATA is probably due primarily to their wider minor groove relative to other AT sequences.21–22 AATT, for example, provides an essentially prearranged site for linked aromatic rings but a wider groove is not optimum for binding, even with AT base pairs.17 The DB183 dimer is, thus, one way to take advantage of the wider TTAA groove with monocations that have less electrostatic repulsion in a stacked dimer than with dications.
A large number of compounds, thus, bind to the AATT minor groove in preference to TTAA, but there are currently no small molecules that recognize TTAA more favorably than AATT. DB183 binds quite well to TTAA (K > 108 M−1) in the dimer mode but it still binds better to AATT as a monomer (K > 109 M−1). We have found that the benzimidazole diamidine DB293 can form a dimer to cooperatively bind to the sequence ATGA23 and, as described above, the bisbenzimidazole, DB183, binds cooperatively to TTAA. These results indicate that the benzimidazole-amidine group is especially useful for stacking and binding to wider minor groove sequences with base functional groups that are complementary to the bound compound. To test this hypothesis a set of derivatives related to DB293 (Fig. 1) have been designed, prepared and evaluated for binding to a TTAA site. The present goal is to explore conformational and substituent space around DB293 to find motifs that are more favorable for binding to that sequence than to other AT sequences of similar length. Such initial models will allow rational design of agents with high specificity for different AT binding sequences. To be sure of true selectivity, results with TTAA have been compared to similar experiments with an AATT binding sequence that is generally an especially favorable site for minor groove complexes. We report here the first minor groove compound that can bind strongly to the TTAA sequence with almost 6 fold selectivity over binding to AATT. This complete reversal of affinity preference over most minor groove binding agents clearly shows that, with a combination of focused synthesis, based on experimental and/or theoretical results, followed by biophysical analysis, it is possible to design compounds to selectively recognize a variety of AT minor groove binding sites.
Fig. 1.

(a) Representative structures of compounds classified by their functional group modifications. Structures for all compounds are tabulated in Table 1. (b) DNA sequences used in this study.
Results
Specificity and binding mode of DB75 and DB293 for AT sequences
We have evaluated the binding of a range of dicationic heterocyclic compounds to TTAA and AATT hairpin duplexes by using a biosensor surface with immobilized DNAs and SPR (surface plasmon resonance) detection of compound binding. The biosensor-SPR method has an advantage in comparing binding to different DNAs since the same compound solution flows rapidly over the set of immobilized DNAs and, therefore, comparisons are quite accurate.24 To obtain the equilibrium association constants the RU (instrument response in resonance units24) values in the plateau region, where any mass transport effects are eliminated, were determined and plotted versus the free compound concentration, Cf (Fig. 2). In the SPR method the free compound concentration is simply the concentration in the solution that flows over the biosensor surface.24 For these plots the RU in the plateau region was converted to r = moles bound ligand/moles DNA and r = RU/RUpred-max. RU is the observed (experimental) response in the plateau region and RUpred-max is the predicted maximum response for a single small molecule binding to a nucleic acid site. Dividing the observed steady-state response RU by the calculated response RUpred-max yields a stoichiometry normalized binding isotherm24. This is a particularly nice feature of the SPR detection method where each bound molecule gives an identical response that is based on its mass and can be predicted by the amount of immobilized DNA. The binding affinities were determined by fitting the r-Cf plots as described in the Methods Section and results for the TTAA and AATT binding sites are compared over the same compound concentration range in Table 1. In non-linear least fitting of individual SPR binding curves, such as in fig. 2e, we find quite small predicted errors. More important is how reproducible a K value is among several repeat experiments. For our conditions the variability for binding constants between 5×106 and 5×108 M−1 is less than +/− 20%. Higher concentrations in the flow solution are required to maintain this accuracy in binding constants below 1×106 M−1 but such weak binding is of less interest. The errors for binding constants under our conditions for K values between 2×106 and 2×105 M−1 are +/− 35%.
Fig. 2.

Equilibrium binding studies. SPR sensorgrams for binding of (a) DB293, (b) DB75, (c) DB828, and (d) DB270 to the TTAA at 25 °C. The unbound compound concentrations in the flow solutions range from 0.0001 μM in the lowest curve to 1 μM in the top curve. The RU values from the steady-state region of SPR sensorgrams were converted to r by r = RU/RUmax and are plotted versus the unbound compound concentration for DB293, DB75, DB809, and DB270 binding to (e) TTAA; Solid lines represent a one site fit for DB75, DB828 and a two site fit for DB293 and DB270 and (f) AATT; Solid lines represent a one site equation fit. Experiments were conducted in cacodylic acid buffer with 0.1 M NaCl added at 25 °C.
Table 1.
Equilibrium binding constants for DB293 and its analogues in TTAA and AATT using SPR.
| Structure | Code No. | TTAAb K(mol−1) | Kcoop (TTAA) | AATT K (mol−1) | KTTAA/KAATT |
|---|---|---|---|---|---|
| DB75 | K = 0.02 × 107 | a | 1.5×107 | 0.01 | |
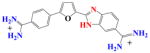 |
DB293 | K1 = 1.1×106 K2 = 1.1×108 K = 1.0×107 |
400 | 2.3×107 | 0.5 |
|
Benzimidazole Modification | |||||
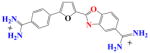 |
DB828 | K ~ 104 | a | K ~104 | NA |
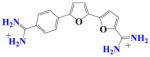 |
DB832 | K ~ 105 | a | 1.7×106 | NA |
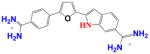 |
DB1878 | K1 = 1.6×106 K2 = 1.5×107 K = 4.9×106 |
38 | 9.0×106 | 0.5 |
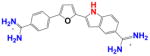 |
DB1478 | K1 = 1.0×106 K2 = 4.4×106 K = 2.0×106 |
18 | 8.0×106 | 0.2 |
|
Phenyl Modification | |||||
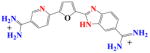 |
DB915 | K1 = 2.3×106 K2 = 3.5×107 K = 0.9×107 |
61 | 5.4×106 | 1.6 |
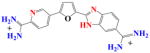 |
DB995 | K1 = 3.8×106 K2 = 2.1×107 K = 0.9×107 |
22 | 4.6×106 | 1.9 |
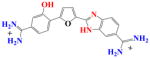 |
DB928 | K1 = 3.9×106 K2 = 1.9×107 K = 0.9×107 |
20 | 1.2×107 | 0.75 |
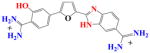 |
DB992 | K1 = 1.8×106 K2 = 6.2×106 K = 0.2×107 |
14 | 5.2×106 | 0.4 |
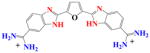 |
DB270 | K1 = 2.5×106 K2 = 3.6×106 K = 0.3×107 |
6 | 1.1×107 | 0.27 |
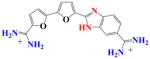 |
DB1003 | K1 = 2.7×106 K2 = 1.2×108 K = 1.8×107 |
178 | 3.1×106 | 5.8 |
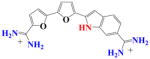 |
DB1871 | K1 = 1.4×106 K2 = 1.5×108 K = 1.5×107 |
428 | 2.8×106 | 5.3 |
|
Furan Modification | |||||
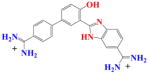 |
DB837 | K1 = 3.6×105 K2 = 6.0×106 K = 0.1×107 |
66 | 5.3×106 | 0.2 |
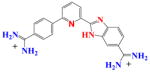 |
DB809 | K1 = 1.5×106 K2 = 3.3×106 K = 0.2×107 |
8 | 1.5×105 | NA |
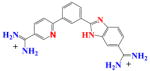 |
DB853 | K = 0.16×107 | a | 6.2×106 | 0.25 |
|
Linear Molecule with Dimer Recognition Mode | |||||
| DB1242 | K < 105 | a | 3.1×105 | NA | |
|
Amidine Modification | |||||
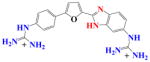 |
DB704 | K ~ 105 | a | 3.7×106 | NA |
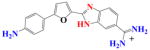 |
DB501 | K < 105 | a | 7.0×105 | NA |
Not applicable
K = √K1K2
Kcoop(Cooperativity index) = K1/K2 × 4; Kcoop = K1/K2 is 0.25 for completely noncooperative interaction.
The phenyl-furan-phenyl diamidine, DB75, binds very strongly to AATT and other A-track type DNA sequences25–26 with a binding constant K of ~ 2 × 107 M−1, and our results in Figure 2f and Table 1 are in good agreement with that value. When tested against the TTAA hairpin DNA, however, as shown in sensorgrams in Figure 2b, DB75 has a significantly lower response level compared to AATT (Fig. 2e, f). The flow cells for AATT and TTAA have essentially the same amount of DNA immobilized so that these sensorgram saturation levels can be compared directly for kinetics and stoichiometry differences.24 The results for TTAA-DB75 binding indicate weak, non-specific binding with K approximately 2×105 M−1 compared to the 100 times greater K for the AATT site.
Figure 2a shows sensorgrams for DB293, which has one phenyl of DB75 converted to a benzimidazole, binding to TTAA. As can be seen by the response in Figure 2a, DB293 binds to TTAA much more strongly than DB75 and approaches saturation of the TTAA binding sites at 0.3 μM concentration. The kinetics of DB293 association and dissociation are such that a steady state plateau was reached (Fig. 2a) and plateau RU values were used directly in the r versus Cf plot (Fig. 2e). Binding of DB293 to the AATT DNA sequence is similar to DB75 and binding plots for TTAA and AATT are compared in Figures 2e and 2f, respectively. As can be seen in this comparison, DB293 reaches saturation RU values that are approximately twice as large with TTAA as with the AATT sequence. The data for DB293 and AATT were fitted to a single binding site model which resulted in a strong binding affinity; K= 2.3 × 107 M−1. The data for DB293 and TTAA, however, required fitting to a two site model using equation 1 (Method Section), which resulted in a strong cooperative dimer binding mode (K1= 1.1×106, K2 = 1.1×108). A quantitative definition of non-cooperative binding for a two-identical-site complex is that the ratio of macroscopic equilibrium constants, K2/K1, is equal to 0.25, a purely statistical value. We define Kcoop = (K2/K1) × 4 and this is 1.0 for a non-cooperative 2-site interaction. The interaction occurs with positive cooperativity if the ratio of K2/K1 is significantly larger than the statistical value and with negative cooperativity if the ratio is less than 0.25. Note that the cooperativity in this model arises naturally from the best fit values to the results in Fig. 2e. The cooperativity factor for the DB293-TTAA complex was determined to be Kcoop= 400 (Table 1) indicating strong positive cooperativity. Note that the cooperativity in this model arises naturally from the best fit values to the results in Fig. 2e. For a two site fit to TTAA the reproducibility errors in the individual K values are twice as large as listed as above due to correlation of the Ks but the root means square values, also reported in Table 1, are as above and these constants are comparable to the binding constants for monomer binding to AATT. The root means square values also make for easier total binding comparisons across all single and two-site binding interactions in Table 1.
These comparative results illustrate how the small structural differences between DB75 and DB293 affect their DNA recognition pattern. DB75, like DB293 maintains the expected ability to bind strongly to AATT but has weak binding affinity to TTAA with no dimer formation. We have previously shown that DB185, the bisbenzimidazole dication analog of the monocation, DB183 (Supplemental, Fig. S1), does not bind to TTAA as a dimer. 20 The strong binding of the dication, DB293, to this sequence is thus a consequence of the ability of DB293 to form a stacked dimer that achieves satisfactory separation of the four charges when bound in the anionic environment of the DNA minor groove.
CD studies were performed to evaluate the mode of DB293 binding with TTAA. CD results in Figure 3a for DB293 with the TTAA hairpin are characterized by large, positive induced CD signals in the compound absorption wavelength range above 300 nm. The induced CD signals monitor the asymmetric environment of compounds when bound to DNA and, therefore, can be used to obtain information on the binding mode.27 As shown in Figure 3a, ten additions of 1 μM each of DB293 to the 5 μM TTAA sequence induced a CD curve with a maximum peak at 367 nm. The significant positive induced CD and DNA saturation at 2:1 compound to DNA ratio indicate that DB293 binds as a stacked dimer in the minor groove of the TTAA sequence in agreement with SPR experiments. The isoelliptic points in Figure 3 are consistent with only two species present in significant amounts. This is as expected for highly cooperative formation of 2:1 dimer complex where only the free DNA and 2:1 complex present at any time.
Fig. 3.

Study of binding mode. CD titration for TTAA in (a) DB293, (b) DB832, (c) DB1003, and AATT in (d) DB1003. The concentration of duplex hairpin was 5 μM and the compound was added with 1 μM increment with total of 10 increments. In case of DB293 and DB1003 in TTAA, DNA saturates at 2:1 compound to DNA ratio confirming dimer formation for these compounds. DB832 in TTAA and DB1003 in AATT with similar compound to DNA ratio give weaker binding.
To determine the contribution of each part of the DB293 molecule to cooperative dimer formation at TTAA, a number of modified compounds were synthesized with each part of DB293 modified (Fig. 1), and their binding evaluated as for DB293.
Benzimidazole modifications
The benzimidazole ring system was converted to a number of other groups (Table 1 and Fig. 1), and sensorgrams were compared over the same compound concentration range for these compounds. DB828, a benzoxazole derivative, has very weak binding to both TTAA (Fig. 2c and 2e) and to AATT (Fig. 2f). Figures 4b, c, and d for TTAA and AATT illustrate weak and non-cooperative binding of DB832 (benzimidazole → furan) to both of these sequences. These results indicate the importance of the benzimidazole ring in DB293 for strong dimer formation ability in TTAA but it is not clear if both benzimidazole nitrogens are required. To address this question the benzimidazole of DB293 was converted to an indole in DB1478 with the indole -NH pointed in the opposite direction as the amidine (reverse indole) and in DB1878 with the -NH and amidine in the same direction. DB1478 has a fivefold reduction in affinity for TTAA with low cooperativity dimer formation while binding to AATT is reduced by a factor of about three. DB1878 binds more strongly to TTAA as a more cooperative dimer with approximately one-half the affinity of DB293. Binding to AATT is 2–3 times weaker than with DB293. Clearly the benzimidazole is favored for recognition of TTAA sites but the indole with the indole –NH and amidine in the same direction is quite good and is preferred over the –NH and amidine in opposite directions.
Fig. 4.

Equilibrium binding studies. SPR sensorgrams for binding of (a) DB1003 and (b) DB832 to the TTAA at 25 °C. The unbound compound concentrations in the flow solutions range from 0.0001 μM in the lowest curve to 0.8 μM in the top curve. The RU values from the steady-state region of SPR sensorgrams were converted to r by r = RU/RUmax and are plotted versus the unbound compound concentration for DB1003 and DB832 binding to (c) TTAA and (d) AATT.
CD titrations of DB832 with the TTAA hairpin duplex conducted under the same conditions as with DB293 are shown in Figure 3b. DB832 is not optically active when free in solution, however, when titrated into TTAA, a small, positive induced CD signal was observed, which was very small compared to DB293 (Fig. 3a), in agreement with very weak binding of DB832 to TTAA. DB1878 gives an induced CD curve that is very similar to that for DB293 (not shown).
Phenyl modifications
Figure 6a shows sensorgrams of DB915, which has a phenyl to pyridine change with the nitrogen ortho to the furan ring, with TTAA. Binding plots in Figure 6b indicate that DB915 binds as a monomer to AATT with weaker binding affinity than with DB293 while it binds as a cooperative dimer to TTAA with the same affinity. DB995 also has a phenyl to pyrimidine change but with the pyridine nitrogen meta to the furan ring. The affinity with TTAA is not affected significantly though, the cooperativity factor is reduced from 61 with DB915 to 20 (Table 1). Both compounds are the first in the series that actually show a binding preference to TTAA.
Fig. 6.

Equilibrium binding studies. SPR sensorgrams for binding of (a) DB915 to the TTAA. The unbound compound concentrations in the flow solutions range from 0.0001 μM in the lowest curve to 0.8 μM in the top curve. (b) The binding plot for DB915 in the presence of TTAA and AATT.
With DB270 the phenyl of DB293 is converted to a benzimidazole. The sensorgrams (Fig. 2d) and binding plot for TTAA (Fig. 2e) and for AATT (Fig. 2f) indicate that DB270 binds strongly to AATT (K =1.1×107), as with DB293, but binds ten times more weakly to TTAA than DB293 with two essentially equivalent sites and K= 3×106. The two phenol derivatives, DB928 and DB992 (Table 1), do not show any significant improvement over DB293 in recognition of TTAA.
DB1003 has a phenyl to furan change much like the conversion of DB75 to DB832 described above. DB832 binds very poorly to TTAA (Fig. 4b), but, surprisingly, DB1003 binds as a cooperative dimer with high affinity to TTAA (Fig. 4a) and a cooperativity factor of 178 (Table 1). DB1003 binds as a monomer with significantly weaker binding affinity (Fig. 4d) to AATT. The conversion of the phenyl in DB293 to a furan in DB1003, thus, enhances the TTAA binding affinity, cooperative dimer formation, and specificity for TTAA over the AATT sequence. The strong cooperativity for DB1003 can be seen in the Scatchard binding plot in Fig. 5. The figure shows the pronounced convex curvature that is characteristic of significant positive cooperativity. The line in the figure is constructed from the binding constants in Table 1. With the phenyl of DB293 replaced by the smaller furan group in DB1003, significant selective dimer recognition of TTAA becomes possible for the first time. DB1003 is thus an example of how the functional units of a compound are sensitive to different sequences and can be exploited to modify the specificity in recognition of DNA.
Fig. 5.

Scatchard plots of the results for binding of DB1003 and DB832 to TTAA.
On binding of DB1003 to TTAA and AATT DNAs, CD signals arise between 300–400 nm (Fig. 3c) where the compounds absorb. The induced CD with the AATT sequence has a maximum at 377 nm while with TTAA the maximum is at a lower wavelength, 372 nm. The CD spectra of DB1003 complexes with AATT show one isoelliptic point at around 270 nm. For the TTAA complex three isoelliptic points are observed at 409, 324, and 234 nm. The strong positive induced CD for DB1003 with TTAA indicates minor groove binding of the dimer.
Thermal melting of the AATT and TTAA DNA hairpin duplexes was monitored by UV spectral changes in the absence and presence of DB1003 at pH 6.25 in buffer with no added NaCl, [Na+] = 0.008 M. AATT has a monophasic melting curve in the unbound and DB1003 bound states (Supplemental Fig. S2). With the TTAA sequence DB1003 has a biphasic melting curve at ratios less than 2:1 (compound/hairpin duplex) and a monophasic curve at a 2:1 ratio in agreement with dimer formation. ΔTm was found to be 28.7 °C for the DB1003-TTAA complex which supports strong dimer binding as observed in SPR.
Furan modifications
DB809, a phenyl-pyridine-benzimidazole diamidine was synthesized with the pyridine N in the same relative position as the furan O in DB293. The compound, however, binds weakly as a monomer to AATT and as a low cooperativity dimer to TTAA with a K that is ten times lower than for DB293 (Table 1). Switching the pyridine and phenyl groups gives DB853, which also binds very weakly to AATT and TTAA (Table 1). The phenyl-phenol derivative DB837 is also not as good in recognizing the AT sequences as DB293. Modification of the furan does not appear to be a useful approach for TTAA binding in this series.
Benzimidazole and furan modifications
DB1242 is a linear triaryl derivative that forms a stacked dimer in some GC rich sequences.28 It thus appeared to be a promising dication for dimer binding at TTAA. The compound, however, binds weakly to AATT and binding can not be detected with TTAA. Other similar compounds with linear six-member aromatic systems also do not bind significantly to TTAA (Table 1). Thus linear compounds do not appear promising for TTAA recognition.
Amidine modifications
Converting the amidines of DB293 to guanidinium groups, DB704 resulted in weaker binding to AATT and a complete loss of binding to TTAA (Table 1 and Supplemental Fig. S3). Clearly the guanidinium in the DB293 structure context does not fit well into the minor groove in either sequence. DB501, a monocation, binds much more weakly to AT DNA sequences as compared to all other compounds and does not produce any evidence for dimer formation in TTAA(Table 1).
DNase I Footprinting
The experiments described above were all conducted with small DNA duplexes and the question of whether these results are relevant to higher molecular weight DNAs arises. To address this point DNase I footprinting experiments were conducted with DNAs that contain both TTAA and several other AT binding sites as described in the Methods Section. Footprinting results are shown in Figure 8 and Supplemental Figure S4. DB75, DB1003 and DB1871, the indole analog of DB1003, were evaluated with the MS1-198bp radio-labeled DNA, a sequence that contains TTAA, TATA, AATTA and ATTT sites of four or five AT base pairs (Fig. 8a and corresponding densitrometric analysis on Fig. 8b). The results among the compounds are strikingly different and clearly illustrate the difference between classical minor groove binders and those that bind well to TTAA. DB75 gives a strong footprint at concentrations at 1 μM and above with AATTA and ATTT but it gives no significant protection to the TTAA or TATA sites, classical minor groove binder behavior. DB1003 gives exactly the opposite footprinting pattern, a strong footprint at TTAA but no significant footprint at AATTA or ATTT (Fig. 8a), and the indole, DB1871, gives a similar pattern. Interestingly, none of the compounds give any significant protection to the TATA site.
Fig. 8.

DNase I footprinting assays. (a) The radiolabeled MS1-198 bp (lanes “0”) or (c) TTAA-69 bp DNA fragments were incubated alone (lanes “DNA”) or in the presence of graded concentrations (μM) of the various compounds as indicated on the top of each lanes. Lanes “G” correspond to the G-track ladders that were used as markers for guanines positions to locate the protected sites. The respective densitometric analyses are presented in panels (b) and (d). Black boxes localize the sites that are protected by the compounds from DNase I cleavage.
In Figure 8c and 8d, a DNase I footprinting gel and densitometer trace for DB75, DB293, DB832, DB1003 and DB1871 with the TTAA-69bp DNA fragment that contains TTAA and TATA type sites of four or more base pairs are shown. This sequence contains no other four base pair AT sites. Up to a concentration of 1 μM neither DB75 nor DB832 give significant protection. At 0.5 μM DB293 gives a footprint at the TTAA site and at 0.75 μM it gives a broad cleavage across the TATA site. Below 1 μM DB1003 and DB1871 show significant cleavage only at the TTAA site. The selectivity for the TTAA sequence using DB1003 and its indole derivative DB1871 was also addressed from comparison with the AATT binding of DB75 using a 265 bp DNA fragment containing the long AT-rich sequence 5′-AAATTAA which contains both AATT (underlined) and TTAA (bold) as overlapping sites (Supplemental Fig. 4). The densitometric analysis clearly evidenced a shift of the sequence recognition from the 5′ part of the AT-sequence using DB75 to its 3′ part (TTAA portion) using DB1003 and DB1871. Clearly the footprinting studies are in excellent agreement with results obtained with the oligomer hairpin duplexes.
Discussion
General, minor groove binding compounds, which are heterocyclic cations with a concave shape and an H-bonding donor group on the concave edge of their structure, favor binding to DNA sites that have AT base pair sequences. Although relatively little is known about the binding specificity of such compounds for specific AT sites, the important therapeutic and biotechnology applications of minor groove binding, suggest that the design/discovery of structures with increased binding specificity of AT sequences would open up new uses of these agents. A general finding is that most AT selective compounds bind preferentially at A-tract sequences, such as AATT, with narrow minor grooves that do not contain TA base steps.20–21,29 As described in the Introduction, the bisbenzimidazole monocation, DB183, has been found to bind strongly to TTAA sites as a dimer while the related dication binds as a monomer.20 Both compounds interact with the reverse sequence, AATT, significantly more strongly than to TTAA as with other minor groove binders.21 These initial results and the quite different properties of AATT and TTAA21–22 suggest that it should be possible to design compounds with specificity for TTAA over other AT sites. Although significant selectivity among AT binding sites has been difficult to obtain, is an obvious advantage for development of a library of minor groove binders to target DNA.
We initially selected DB293 as a development compound with potential for binding to TTAA sites as a dimer based on its unusual ability to recognize the ATGA sequence as a stacked, antiparallel dimer.23, 30–32 This is true in spite of the fact that DB293 is a dication with two charged amidine groups and most dications will not form stacked complexes with DNA.33 In the work reported here a biosensor-SPR method was used for quantitative evaluation of the binding of DB293 to AATT and TTAA sites in a similar sequence context. We were encouraged by the finding that DB293, even though it is a dication, binds well to TTAA as a cooperative dimer, although it still binds 2–3 times more strongly to AATT (Fig. 2 and Table 1). This result suggested that modifications of the DB293 structure would be a promising route for design of improved agents for TTAA binding. To determine the contribution of each part of the DB293 structure to TTAA binding, a set of initial modified analogs were synthesized (Fig. 1 and Table 1) and their contribution to dimer formation and binding were evaluated by the biosensor-SPR method.
Compounds related to DB293 but without a benzimidazole group, such as DB75, DB832 (Table 1) and several linear triaryl compounds related to DB111128, were found to bind quite weakly to TTAA. Conversion of the benzimidazole to a benzoxazole (DB828) removes the benzimidazole -NH function that can H-bond to AT base pairs and as a result, the affinities for both AATT and TTAA are significantly reduced. In a particularly important result, the benzimidazole of DB293 was replaced by an indole with the -NH pointed opposite from the amidine group orientation (DB1478). This compound has a five-fold reduction in affinity for TTAA and binds preferentially to AATT. On the other hand, an indole with the -NH pointed in the same direction as the amidine (DB1878) binds more strongly to TTAA, but with somewhat less affinity than the benzimidazole. Its selectivity for TTAA is similar to DB293. For the compounds of Table 1, as well as with DB183, the benzimidazole group is clearly a key motif for binding with significant affinity to TTAA. Based on the indole results, it appears that both the benzimidazole -NH and the benzimidazole amidine can H-bond with DNA in the dimer complex.
The influence of the amidine groups of DB293 was evaluated by their conversion to guanidinium groups (DB704, Table 1) or conversion of one amidine to an amine (DB501 Table 1). Both changes drastically weaken binding to TTAA relative to DB293. The AATT affinity is also reduced but by a smaller extent. The monocations of DB293 bind weakly to both AATT and TTAA and these results are quite different from those obtained with DB183/DB185 and with polyamide minor groove binders. The dicationic polyamide, netropsin, for example, cannot form a stacked minor groove dimer but the monocation distamycin can form a cooperative dimer in AT sequences that can open to accommodate a stacked dimer.33–34 Clearly, whatever charge repulsion exists in the stacked DB293 complex with TTAA costs much less Gibbs energy (ΔG0) than is gained by the amidine interactions in the complex. The polyanionic backbone of DNA thus provides sufficient shielding of the four positive charges of the stacked dimer to allow it to form in the TTAA minor groove.
To determine the importance of the furan to binding in DB293, it was converted to a pyridine (DB809) with the pyridine N in a similar position to the furan O. Somewhat surprisingly, DB809 binds as a low cooperativity dimer with a ten-fold reduction in affinity for TTAA. Similar results were also obtained with a phenyl replacing the furan. There appear to be two primary possible reasons for this drop in affinity. First, the bond angle differences between the central furan versus the central pyridine or phenyl result in a larger radius of curvature for the furan derivatives. Second, the phenyl-furan-benzimidazole system is essentially planar while the phenyl-pyridine interaction results in a torsional angle of 20–25 degrees between the two six member rings and an even larger angle for the phenol derivative (DB837) in Table 1. Such a twist could significantly reduce the stacking energetics in the six-six system relative to furan derivatives. All of the six-six ring compounds in Table 1 show poor affinity for TTAA as well as reduced affinity for AATT relative to DB293.
Phenyl substituted DB293 analogs such as DB915, DB995, DB928 (Table 1) exhibit slightly weaker dimer formation with TTAA than DB293. SPR results show that these compounds form the specific 2:1 stacked dimer complexes in TTAA while they form 1:1 complexes in AATT. DB992, and similar analogues, with different substituents ortho to the amidine bind approximately ten fold more weakly than DB293 to TTAA, probably due to larger twist of their phenyl amidine group. If the phenyl is replaced by a benzimidazole (DB270) binding to TTAA drops by ten fold while binding to AATT is similar to DB293. Conversion of the phenyl to a furan in DB293 to give DB1003, however, gave significantly improved specificity for TTAA relative to DB293. The highly cooperative binding of DB1003 to TTAA is seen in the concave Scatchard plot (Fig. 5). Strong binding of DB1003 is also supported by the large positive induced CD for DB1003 binding to TTAA (Fig. 3). Of all the modified derivatives of DB293, DB1003 is the most improved in terms of selectivity between AATT and TTAA. It is 6 times more selective for TTAA as compared to 0.5 in the case of DB293 (Fig. 9). With the benzimidazole of DB1003 replaced with an indole (DB1871) that has the -NH pointed in the same direction as the amidine, the binding to both TTAA and AATT is very similar to that for DB1003. This result again demonstrates that it is critical to TTAA binding to have an -NH pointed in the same direction as the amidine.
Fig. 9.

Comparative plot of selectivity (KTTAA/KAATT) of studied compounds against AATT and TTAA sequences. KTTAA = (√ K1×K2); KAATT is equilibrium binding constant for AATT site.
The DNase I footprinting studies support the strong selectivity of DB1003 for -TTAA- binding sites. As shown in Figure 8, the selectivity is observed even in the presence of competing AT binding sites that are generally quite favorable for minor groove compounds. DB75 and other classical-type minor groove binders interact with the sequences in Figure 8 in almost the inverse manner to DB1003 and avoid the TTAA sites that are favored by DB1003. The same conclusions thus arise from both the high resolution studies on small DNA hairpin duplexes and the combinatorial-type analysis of binding specificity in high molecular weight duplex DNA by DNase I.
All of these results show that small compound structural changes, at positions throughout the molecular structure of the parent compound, DB293, can have profound effects on DNA recognition. The AATT minor groove which is deep and narrow, does not allow more than one molecule to bind without a significant free energy cost. On the other hand, TTAA with a wider groove and disordered array of water molecules provides weaker van der Waals interaction and lower entropy for single small molecule binding.21 One strategy for enhancing both the specificity and the affinity in TTAA recognition would be to recognize bases in both strands in the same complex. The TTAA minor groove cannot be strongly recognized by single small molecules but its recognition by molecules, such as DB1003, which can bind as stacked dimers, is a promising strategy. The excellent selectivity of DB1003 can easily be seen by the comparison of histogram results in Figure 9 where the SPR selectivity ratio from Table 1 is plotted for a panel of compounds with different structures. DB1003 and the related indole have selectivity for TTAA versus AATT sites above 5 fold while most compounds have selectivity below 1 and are selective for the AATT sequence. This selectivity is strongly supported by the footprinting results in Fig. 8 where the selectivity, based on concentrations required to observe a footprint, is even greater. DB293 and closely related derivatives have selectivity between 0.5–2 and bind to TTAA as dimer complexes, but they also bind well to AATT and that keeps their selectivity low.
As noted in the Introduction, clustered local 4–5 base pair sites offer an attractive method to enhance selective biological effects. Single sites of small molecule binding, in general, have much less significant effects. We have also recently found that with DB293 inhibition of transcription factor binding is much stronger for dimer than for monomer binding35 and this makes the DB1003 dimer reported here particularly attractive. The biological and inhibition results likely depend, at least in part, on effects of minor grove binding on DNA topology, which is a factor of compound structure and DNA sequence.36 Whether biological effects appear on binding of small molecules to DNA is thus a complex function of both the number of locally bound small molecules, the site range covered, and the local effects produced on binding. Linking molecular binding units to target larger numbers of base pairs is a method that we are also currently exploring to increase selectivity.37 Although it is not yet clear how many base pairs will be required in a local sequence cluster and what effects they must have on the DNA structure to generate a biological response, it is clear that agents that have improved selectivity will be required. DB1003, which has excellent selectivity for TTAA sites in long DNA sequences (Fig. 8) and binds as a dimer to enhance the potential to perturb protein binding, is an excellent model for development of such agents.
In summary, the key features for the DB1003 minor-groove dimer from the information provided by the compound structure-binding results in Table 1 and Figure 9 are: (i) a favorable stacking surface provided by the benzimidazole-furan-furan pi system that can interact well with the wider minor groove in TTAA sites, (ii) an inward pointing benzimidazole (or indole) -NH and amidine, which provide four H-bond donors in a stacked dimer to interact with the TA base pairs at the floor of the groove, (iii) appropriate curvature of the stacked dimer complex to match the curvature of the minor groove. Given the low toxicity and excellent cell uptake of diamidines in this series38 dimer discovery presents an opportunity to develop new therapeutics. This discovery also suggests that compounds can be developed to selectively recognize a variety of four base pair AT sequences while excluding binding to GC sequences.
MATERIALS AND METHODS
Compounds, DNAs, and Buffers
The syntheses of DB75, DB293, DB828, DB270, DB837, DB853, DB1242, DB704, and DB501 have been published.39–43 The syntheses of the remaining compounds will be published elsewhere. Purity of all compounds was verified by NMR and elemental analysis. SPR, CD and Tm experiments are performed with CGTTAACG, and a CGAATTCG hairpin oligomer DNAs from IDT (Integrated DNA Technologies) (Fig. 1). Solutions for CD and SPR were prepared in degassed cacodylic acid buffer of pH 6.25 containing 0.01M cacodylic acid, 0.001M EDTA and 0.1 M NaCl. Tm experiments were done under the same conditions with 0M NaCl. The concentration of the DNA solutions was determined spectrophotometrically at 260 nm using extinction coefficients per nucleotide of AATT duplex DNA. The extinction coefficients were calculated on a per strand basis by the nearest-neighbor method and divided by the number of nucleotides per strand.44
SPR-Biosensor Binding Determinations
SPR (Surface plasmon resonance) measurements were performed with a four-channel BIAcore 2000 optical biosensor system (BIAcore Inc.). 5′-biotin labeled DNA samples (Fig. 1) were immobilized onto streptavidin-coated sensor chips (BIAcore SA) as previously described.24 Three flow cells were used to immobilize the DNA oligomer samples, while a fourth cell was left blank as a control. The SPR experiments were performed at 25°C in filtered, degassed, 10 mM cacodylic acid buffer (pH 6.25) containing 100 mM NaCl, 1 mM EDTA. Steady state binding analysis was performed with multiple injections of different compound concentrations over the immobilized DNA surface at a flow rate of 25 μl/min and 25°C. Solutions of known ligand concentration were injected through the flow cells until a constant steady-state response was obtained. Compound solution flow was then replaced by buffer flow resulting in dissociation of the complex. The reference response from the blank cell was subtracted from the response in each cell containing DNA to give a signal (RU, resonance units) that is directly proportional to the amount of bound compound. The predicted maximum response per bound compound in the steady-state region (RUmax) was determined from the DNA molecular weight, the amount of DNA on the flow cell, the compound molecular weight, and the refractive index gradient ratio of the compound and DNA, as previously described.45–46 The number of binding sites and the equilibrium constant were obtained from fitting plots of RU versus Cfree.
The stoichiometry of the reaction can be calculated as follows:
RU is the observed (experimental) response in the plateau region and RUmax is the predicted maximum response for a single small molecule binding to a nucleic acid site. Dividing the observed steady-state response RU by the calculated response RUpred-max yields a stoichiometry normalized binding isotherm. The calculated value (r) was fitted to an appropriate binding model, either a single site model (K2 = 0) or with a two site model:
| (1) |
Where r represents the moles of bound compound per mole of DNA hairpin duplex, K1 and K2 are macroscopic binding constants, and Cfree is the free compound concentration in equilibrium with the complex (the concentration in the flow solution).24
Circular Dichroism
CD spectra were obtained on a Jasco J-810 spectrometer. The software supplied by Jasco provided instrument control, data acquisition and manipulation. A DNA solution in cacodylic acid buffer with 100 mM NaCl was scanned in 1 cm quartz cuvettes and solution of the compounds were added to DNA at all the desired ratios and the complexes were rescanned.
Thermal melting (Tm) experiments
Tm experiments were conducted with a Cary 300 UV–visible spectrophotometer in 1-cm quartz cuvettes. The absorbance of the DNA–compound complex was monitored at 260 nm as a function of temperature and DNA without compound was used as a control. Cuvettes were mounted in a thermal block, and the solution temperatures were monitored by a thermistor in a reference cuvette with a computer-controlled heating rate of 0.5 °C/min. Experiments were conducted in 0.01 M cacodylic acid (Dimethylarsinic acid) at pH 6.25. The concentration of the DNA was about 3×10−6 M in hairpin. For experiments with complexes a ratio of 3:1 compound per oligomer duplex was generally used.
DNase I Footprinting
DNase I footprinting experiments were essentially performed as described in35, with the following modifications. The MS1-198 bp DNA fragment was obtained from double digestion of the pUC18-MS1 plasmid 47 at SacI and HindIII restriction sites for 1 hour with the corresponding enzymes in their respective buffers. The TTAA-69 bp DNA fragment was obtained from EcoRI and PstI double digestion of the pUC19-TTAA vector. This vector derived from pUC19 (Stratagene) by insertion between SalI and BamHI restriction sites of a double-stranded DNA obtained from hybridization of the two phosphorylated oligonucleotides 5′-GATCCCACTGCTTAACTGGCTTCGACTCACTATAGGGAGACCCG and 5′-TCGACGGGTCTCCCTATAGTGAGTCGAAGCCAGTTAAGCAGTGG (Eurogentec, Belgium) containing both TTAA and TATA sequences (underlined). The generated DNA fragments were 3′-end labeled using α-[32P]-dATP (3000 Ci/mmol each, GE Healthcare, Buckinghamshire, England) and 10 units of Klenow enzyme (BioLabs) for 30 min at 37°C. The resulting radio-labeled DNA fragments were separated on a 6 % native polyacrylamide gel using electrophoresis in TBE buffer (89 mM Tris base, 89 mM boric acid, 2.5 mM Na2 EDTA, pH 8.3). The DNA fragments were removed from gel by crushing the portion of gel and dialyzing it over-night against 400 μl of elution buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 100 mM NaCl) prior to filtration through a Millipore 0.22 μm membrane and subsequent ethanol precipitation. Increasing concentrations of the DB1003 as indicated in the legend of figures (μM) were incubated 15 min at 37°C with the radio-labeled DNA fragments to ensure equilibrium prior to subsequent digestion upon addition of 0.001 unit/mL of DNase I (Sigma, France) for 3 min in digestion buffer (20 mM NaCl, 2 mM MgCl2, 2 mM MnCl2, pH 7.3). The reaction was stopped by freeze-drying and lyophilization. The dried pellets containing the cleaved DNA fragments were then dissolved in 4 μL of denaturing loading buffer (80% formamide solution containing tracking dyes), heated at 90 °C for 4 min and rapidly chilled on ice for 5 min prior to electrophoresis at 65 W in TBE buffer on a denaturing polyacrylamide gel (0.35 mm thick, 8% polyacrylamide containing 8 M urea). After migration during the appropriate delay, gels were soaked 10 min in 10% acetic acid, transferred to What man 3 MM paper and dried under vacuum at 80 °C. Dried gels were exposed overnight on storage screens and scans using a Molecular Dynamics STORM 860. Comparison of the relative position of the bands to the guanines sequencing standard (G-track, obtained from Maxam and Gilbert sequencing standard method) was used to identify the positions of protected bases. Quantifications of the footprints were performed using Image Quant 3.3 software.
Supplementary Material
Fig. 7.

Equilibrium binding studies. SPR binding plots for DB1878 in the presence of TTAA and AATT.
Acknowledgments
This research was supported by NIH grant AI064200 and GM61587 (W.D.W. and D.W.B.). Biacore instruments were purchased with partial support from the Georgia Research Alliance. M.-H.D.-C. thanks the Ligue Nationale contre le Cancer (Comité du Nord) and the Institut pour la Recherche sur le Cancer de Lille (IRCL) for grants, the Fonds européen de développement régional (FEDER, European Community) and the Région Nord-Pas de Calais for a grant and a fellowship to S.D., the CHRU de Lille and the Conseil Régional Nord-Pas de Calais for a Ph.D fellowship to R.N and the Institut de Médecine Prédictive et de Recherche Thérapeutique (IMPRT) – IFR114 for access to the Molecular Dynamics STORM 860 equipment.
Footnotes
Supplementary data associated with this article can be found, in the online version, at to be added.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dervan PB. Design of sequence-specific DNA-binding molecules. Science. 1986;232:464–71. doi: 10.1126/science.2421408. [DOI] [PubMed] [Google Scholar]
- 2.Wilson WD, Tanious FA, Mathis A, Tevis D, Hall JE, Boykin DW. Antiparasitic compounds that target DNA. Biochimie. 2008;90:999–1014. doi: 10.1016/j.biochi.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soeiro MN, de Castro SL. Trypanosoma cruzi targets for new chemotherapeutic approaches. Expert Opin Ther Targets. 2009;13:105–21. doi: 10.1517/14728220802623881. [DOI] [PubMed] [Google Scholar]
- 4.Motta MC. Kinetoplast as a potential chemotherapeutic target of trypanosomatids. Curr Pharm Des. 2008;14:847–54. doi: 10.2174/138161208784041051. [DOI] [PubMed] [Google Scholar]
- 5.Hebert MD. Targeting the gene in Friedreich ataxia. Biochimie. 2008;90:1131–9. doi: 10.1016/j.biochi.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Gottesfeld JM. Small molecules affecting transcription in Friedreich ataxia. Pharmacol Ther. 2007;116:236–48. doi: 10.1016/j.pharmthera.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burnett R, Melander C, Puckett JW, Son LS, Wells RD, Dervan PB, Gottesfeld JM. DNA sequence-specific polyamides alleviate transcription inhibition associated with long GAA.TTC repeats in Friedreich’s ataxia. Proc Natl Acad Sci U S A. 2006;103:11497–502. doi: 10.1073/pnas.0604939103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neidle S. The structures of quadruplex nucleic acids and their drug complexes. Curr Opin Struct Biol. 2009;19:239–50. doi: 10.1016/j.sbi.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Sun D, Hurley LH. The importance of negative superhelicity in inducing the formation of G-quadruplex and i-motif structures in the c-Myc promoter: implications for drug targeting and control of gene expression. J Med Chem. 2009;52:2863–74. doi: 10.1021/jm900055s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gatto B, Palumbo M, Sissi C. Nucleic acid aptamers based on the G-quadruplex structure: therapeutic and diagnostic potential. Curr Med Chem. 2009;16:1248–65. doi: 10.2174/092986709787846640. [DOI] [PubMed] [Google Scholar]
- 11.De Cian A, Lacroix L, Douarre C, Temime-Smaali N, Trentesaux C, Riou JF, Mergny JL. Targeting telomeres and telomerase. Biochimie. 2008;90:131–55. doi: 10.1016/j.biochi.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Tidwell RR, Boykin DW. Dicationic DNA minor-groove binders as antimicrobial agents. In: Demeunynck M, Bailly C, Wilson WD, editors. DNA and RNA Binders: From Small Molecules to Drugs. Vol. 2. WILEY-VCH; Weinheim: 2003. pp. 414–460. 2 vols. [Google Scholar]
- 13.Wenzler T, Boykin DW, Ismail MA, Hall JE, Tidwell RR, Brun R. New treatment option for second stage African sleeping sickness: In vitro and in vivo efficacy of aza analogs of DB289. Antimicrob Agents Chemother. 2009;10:4185–92. doi: 10.1128/AAC.00225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purfield AE, Tidwell RR, Meshnick SR. The diamidine DB75 targets the nucleus of Plasmodium falciparum. Malar J. 2009;8:104. doi: 10.1186/1475-2875-8-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lansiaux A, Tanious F, Mishal Z, Dassonneville L, Kumar A, Stephens CE, Hu Q, Wilson WD, Boykin DW, Bailly C. Distribution of furamidine analogues in tumor cells: targeting of the nucleus or mitochondria depending on the amidine substitution. Cancer Res. 2002;62:7219–29. [PubMed] [Google Scholar]
- 16.Nguyen B, Neidle S, Wilson WD. A role for water molecules in DNA-ligand minor groove recognition. Acc Chem Res. 2009;42:11–21. doi: 10.1021/ar800016q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neidle S. DNA minor-groove recognition by small molecules. Nat Prod Rep. 2001;18:291–309. doi: 10.1039/a705982e. [DOI] [PubMed] [Google Scholar]
- 18.Bailly C, Waring MJ. The use of diaminopurine to investigate structural properties of nucleic acids and molecular recognition between ligands and DNA. Nucleic Acids Res. 1998;26:4309–14. doi: 10.1093/nar/26.19.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abu-Daya A, Brown PM, Fox KR. DNA sequence preferences of several AT-selective minor groove binding ligands. Nucleic Acids Res. 1995;23:3385–3392. doi: 10.1093/nar/23.17.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanious FA, Hamelberg D, Bailly C, Czarny A, Boykin DW, Wilson WD. DNA sequence dependent monomer-dimer binding modulation of asymmetric benzimidazole derivatives. J Am Chem Soc. 2004;126:143–53. doi: 10.1021/ja030403+. [DOI] [PubMed] [Google Scholar]
- 21.Hampshire AJ, Fox KR. The effects of local DNA sequence on the interaction of ligands with their preferred binding sites. Biochimie. 2008;90:988–98. doi: 10.1016/j.biochi.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Price MA, Tullius TD. How the structure of an adenine tract depends on sequence context: a new model for the structure of TnAn DNA sequences. Biochemistry. 1993;32:127–36. doi: 10.1021/bi00052a018. [DOI] [PubMed] [Google Scholar]
- 23.Bailly C, Tardy C, Wang L, Armitage B, Hopkins K, Kumar A, Schuster GB, Boykin DW, Wilson WD. Recognition of ATGA sequences by the unfused aromatic dication DB293 forming stacked dimers in the DNA minor groove. Biochemistry. 2001;40:9770–9779. doi: 10.1021/bi0108453. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen B, Tanious FA, Wilson WD. Biosensor-surface plasmon resonance: quantitative analysis of small molecule-nucleic acid interactions. Methods. 2007;42:150–61. doi: 10.1016/j.ymeth.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Collar CJ, Kumar A, Stephens CE, Boykin DW, Wilson WD. Heterocyclic diamidine interactions at AT base pairs in the DNA minor groove: effects of heterocycle differences, DNA AT sequence and length. J Phys Chem B. 2008;112:11809–18. doi: 10.1021/jp804048c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mazur S, Tanious FA, Ding D, Kumar A, Boykin DW, Simpson IJ, Neidle S, Wilson WD. A thermodynamic and structural analysis of DNA minor-groove complex formation. Journal of Molecular Biology. 2000;300:321–337. doi: 10.1006/jmbi.2000.3869. [DOI] [PubMed] [Google Scholar]
- 27.Rodger A, Norden B. Circular Dichroism and Linear Dichroism. Oxford University Press; New York: 1997. [Google Scholar]
- 28.Munde M, Ismail MA, Arafa R, Peixoto P, Collar CJ, Liu Y, Hu L, David-Cordonnier MH, Lansiaux A, Bailly C, Boykin DW, Wilson WD. Design of DNA minor groove binding diamidines that recognize GC base pair sequences: a dimeric-hinge interaction motif. J Am Chem Soc. 2007;129:13732–43. doi: 10.1021/ja074560a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laughton C, Luisi B. The mechanics of minor groove width variation in DNA, and its implications for the accommodation of ligands. J Mol Biol. 1999;288:953–63. doi: 10.1006/jmbi.1999.2733. [DOI] [PubMed] [Google Scholar]
- 30.Wang L, Bailly C, Kumar A, Ding D, Bajic M, Boykin DW, Wilson WD. Specific molecular recognition of mixed nucleic acid sequences: an aromatic dication that binds in the DNA minor groove as a dimer. Proc Natl Acad Sci U S A. 2000;97:12–6. doi: 10.1073/pnas.97.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Carrasco C, Kumar A, Stephens CE, Bailly C, Boykin DW, Wilson WD. Evaluation of the influence of compound structure on stacked-dimer formation in the DNA minor groove. Biochemistry. 2001;40:2511–21. doi: 10.1021/bi002301r. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Kumar A, Boykin DW, Bailly C, Wilson WD. Comparative thermodynamics for monomer and dimer sequence-dependent binding of a heterocyclic dication in the DNA minor groove. J Mol Biol. 2002;317:361–74. doi: 10.1006/jmbi.2002.5433. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Mitra SN, Rao ST, Sekar K, Sundaralingam M. A novel end-to-end binding of two netropsins to the DNA decamers d(CCCCCIIIII)2, d(CCCBr5CCIIIII)2 and d(CBr5CCCCIIIII)2. Nucleic Acids Res. 1998;26:5464–5547. doi: 10.1093/nar/26.23.5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pelton JG, Wemmer DE. Structural characterization of a 2:1 distamycin A.d(CGCAAATTGGC) complex by two-dimensional NMR. Proc Natl Acad Sci U S A. 1989;86:5723–7. doi: 10.1073/pnas.86.15.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peixoto P, Liu Y, Depauw S, Hildebrand MP, Boykin DW, Bailly C, Wilson WD, David-Cordonnier MH. Direct inhibition of the DNA-binding activity of POU transcription factors Pit-1 and Brn-3 by selective binding of a phenyl-furan-benzimidazole dication. Nucleic Acids Res. 2008;36:3341–53. doi: 10.1093/nar/gkn208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tevis DS, Kumar A, Stephens CE, Boykin DW, Wilson WD. Large, sequence-dependent effects on DNA conformation by minor groove binding compounds. Nucleic Acids Res. 2009;37:5550–8. doi: 10.1093/nar/gkp558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rahimian M, Kumar A, Say M, Bakunov SA, Boykin DW, Tidwell RR, Wilson WD. Minor groove binding compounds that jump a gc base pair and bind to adjacent AT base pair sites. Biochemistry. 2009;48:1573–83. doi: 10.1021/bi801944g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lansiaux A, Tanious F, Mishal Z, Dassonneville L, Kumar A, Stephens CE, Hu Q, Wilson WD, Boykin DW, Bailly C. Distribution of furamidine analogues in tumor cells: targeting of the nucleus or mitochondria depending on the amidine substitution. Cancer Res. 2002;62:7219–29. [PubMed] [Google Scholar]
- 39.Ismail MA, Arafa RK, Brun R, Wenzler T, Miao Y, Wilson WD, Generaux C, Bridges A, Hall JE, Boykin DW. Synthesis, DNA affinity, and antiprotozoal activity of linear dications: Terphenyl diamidines and analogues. J Med Chem. 2006;49:5324–32. doi: 10.1021/jm060470p. [DOI] [PubMed] [Google Scholar]
- 40.Hopkins KT, Wilson WD, Bender BC, McCurdy DR, Hall JE, Tidwell RR, Kumar A, Bajic M, Boykin DW. Extended aromatic furan amidino derivatives as anti-Pneumocystis carinii agents. J Med Chem. 1998;41:3872–8. doi: 10.1021/jm980230c. [DOI] [PubMed] [Google Scholar]
- 41.Ismail MA, Brun R, Wenzler T, Tanious FA, Wilson WD, Boykin DW. Dicationic biphenyl benzimidazole derivatives as antiprotozoal agents. Bioorg Med Chem. 2004;12:5405–13. doi: 10.1016/j.bmc.2004.07.056. [DOI] [PubMed] [Google Scholar]
- 42.Das BP, Boykin DW. Synthesis and Antiprotozoal Activity of 2,5-Bis(4-guanylphenyl)furans. J Med Chem. 1977;20:531–536. doi: 10.1021/jm00214a014. [DOI] [PubMed] [Google Scholar]
- 43.Hopkins KT, Wilson WD, Bender BC, McCurdy DR, Hall JE, Tidwell RR, Kumar A, Bajic M, Boykin DW. Extended aromatic furan amidino derivatives as anti-Pneumocystis carinii agents. J Med Chem. 1998;41:3872–8. doi: 10.1021/jm980230c. [DOI] [PubMed] [Google Scholar]
- 44.Fasman GD. Handbook of Biochemistry and Molecular Biology. In: Fasman G, editor. Nucleic Acids. 3. CRC Press; Cleveland, OH: 1975. p. 1. [Google Scholar]
- 45.Davis TM, Wilson WD. Determination of the refractive index increments of small molecules for correction of surface plasmon resonance data. Anal Biochem. 2000;284:348–353. doi: 10.1006/abio.2000.4726. [DOI] [PubMed] [Google Scholar]
- 46.Myszka DG. Kinetic, equilibrium, and thermodynamic analysis of macromolecular interactions with BIACORE. Methods Enzymol. 2000;323:325–340. doi: 10.1016/s0076-6879(00)23372-7. [DOI] [PubMed] [Google Scholar]
- 47.Lavesa M, Fox KR. Preferred binding sites for [N-MeCYs(3), N-MeCys(7)]TANDEM determined using a universal footprinting substrate. Anal Biochem. 2001;293:246–50. doi: 10.1006/abio.2001.5124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.