

Abstract
Although normal dopaminergic tone has been shown to be essential for the induction of cortico-striatal and mesolimbic theta oscillatory activity, the influence of norepinephrine on these brain networks remains relatively unknown. To address this question, we simultaneously recorded local field potentials and single-neuron activity across 10 interconnected brain areas (ventral striatum, frontal association cortex, hippocampus, primary motor cortex, orbital frontal cortex, prelimbic cortex, dorsal lateral striatum, medial dorsal nucleus of thalamus, substantia nigra pars reticularis, and ventral tegmental area) in a combined genetically and pharmacologically induced mouse model of hyponoradrenergia. Our results show that norepinephrine (NE) depletion induces a novel state in male mice characterized by a profound disruption of coherence across multiple cortico-striatal circuits and an increase in mesolimbic cross-structural coherence. Moreover, this brain state is accompanied by a complex behavioral phenotype consisting of transient hyperactivity, stereotypic behaviors, and an acute 12-fold increase in grooming. Notably, treatment with a norepinephrine precursors (l-3,4-dihydroxyphenylalanine at 100 mg/kg or l-threo-dihydroxyphenylserine at 5 mg/kg) or a selective serotonin reuptake inhibitor (fluoxetine at 20 mg/kg) attenuates the abnormal behaviors and selectively reverses the circuit changes observed in NE-depleted mice. Together, our results demonstrate that norepinephrine modulates the dynamic tuning of coherence across cortico-striato-thalamic circuits, and they suggest that changes in coherence across these circuits mediate the abnormal generation of hyperactivity and repetitive behaviors.
Introduction
Altered catecholamine signaling is thought to play a key role in mediating the cognitive deficits observed across several brain disorders. Whereas dopamine (DA) dysfunction contributes to the deficits observed in Parkinson's disease (Carlsson, 1972), schizophrenia (Laruelle and Abi-Dargham, 1999), attention-deficit hyperactivity disorder (Jucaite et al., 2005), and bipolar disorder (Horschitz et al., 2005), noradrenergic dysfunction has been implicated in mediating endophenotypes observed in depression (Charney, 1998), posttraumatic stress disorder (Debiec and LeDoux, 2006), and Tourette's syndrome (Hedderick et al., 2009). Recent studies in hyperdopaminergic mice [dopamine transporter knock-out mice (DAT-KO mice)] and mice acutely depleted of dopamine (dopamine-depleted DAT-KO mice) have demonstrated that dopamine signaling plays a critical role in regulating neural oscillatory activity within multiple brain structures (Costa et al., 2006; Dzirasa et al., 2006). For instance, hyperdopaminergia increases hippocampal, striatal, and cortical theta (4–11 Hz) and gamma (33–55 Hz) oscillatory activity, whereas profound hypodopaminergia suppresses theta and gamma oscillatory activity across these regions (Costa et al., 2006; Dzirasa et al., 2006). Importantly, these studies also demonstrate that dopamine signaling modulates the dynamic synchronization of neural activity within and between brain structures. For example, hyperdopaminergia potentiates high-frequency synchrony between hippocampus and prefrontal cortex (Dzirasa et al., 2009), whereas dopamine depletion induces a significant increase in low-frequency synchronous neuronal firing across cortico-striatal circuits (Costa et al., 2006; Fuentes et al., 2009). Together, these studies suggest that changes in neural synchrony may play a critical role in mediating the motor and cognitive deficits observed in patients with neurological disorders such as Parkinson's disease.
In the same context, here we tested the hypothesis that noradrenergic dysfunction may also play a key role in regulating synchronous brain activity leading to the manifestation of aberrant behaviors. To test this hypothesis, we simultaneously recorded local field potential (LFP) and single-neuron activity across 10 interconnected brain areas in a combined genetically and pharmacologically induced mouse model of hyponoradrenergia, developed on the basis of the norepinephrine (NE) transporter knock-out (NET-KO) mouse. NET-KO mice lack the gene encoding the norepinephrine transporter, a transmembrane protein that is responsible for regulating the reuptake of synaptic NE and replenishing NE stores in the presynaptic terminal (Gainetdinov and Caron, 2003). Like other members of the plasma membrane monoamine transporter family, the NE transporter plays a vital role in controlling noradrenergic tone. Studies from knock-out mice have revealed that the loss of transporter function generally results in significant elevation of extracellular levels and decreased intraneuronal storage of the cognate monoamine (Gainetdinov and Caron, 2003). Thus, as a result of the loss of the NE transporter, NET-KO mice exhibit a twofold increase in extracellular NE levels as well as profound depletion of intraneural NE stores (Xu et al., 2000). This highlights the dependence of intracellular NE stores on both de novo synthesis and NET-mediated recycling of released NE.
Materials and Methods
Animal care and use.
The wild-type (WT) and NET-KO littermates were generated from heterozygotes that had been backcrossed over 10 generations onto the C57BL/6J background. Mice were housed three to five per cage and maintained in a humidity- and temperature-controlled room with water available ad libitum. Twelve male NET-KO mice and six male WT littermate controls were separated into individual cages and surgically implanted with recording electrodes. Experiments were initiated after a 2 week recovery. All electrophysiological recordings were conducted in a 10.75 × 10.75 inch open-field test environment (MED-OFA-MS; Med Associates) after a 12–24 h habituation period.
All studies were conducted with approved protocols from the Duke University Institutional Animal Care and Use Committee and were in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Surgery.
NET-KO mice and WT littermate controls were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) and placed in a stereotaxic device. Ground screws were secured to the cranium. A total of 56 microwires were arranged in array bundles and implanted as shown in Table 1.
Table 1.
Stereotaxic coordinates for all implanted areas (all coordinates are measured from bregma)
| Area | AP (mm) | ML (mm) | DV (mm) |
|---|---|---|---|
| Ventral striatum | +1.25 | +1.15 | −4.0 |
| Frontal association cortex | +2.95 | −1.5 | −0.5 |
| Hippocampus | −2.3 | +1.75 | −1.2 |
| Motor cortex | +1.2 | −1.75 | −0.5 |
| Prelimbic cortex | +2.5 | +0.25 | −0.75 |
| Orbital frontal cortex | +2.6 | −1.1 | −1.7 |
| Thalamus | −1.6 | +0.3 | −2.9 |
| Dorsolateral striatum | +0.6 | −2.0 | −2.0 |
| Substantia nigra | −3.2 | −1.35 | −4.2 |
| VTA | −3.2 | +0.3 | −4.25 |
AP, Anteroposterior; ML, mediolateral; DV, dorsoventral.
Implanted electrodes were anchored to ground screws using dental acrylic. Six WT and nine NET-KO animals were implanted in all 10 brain areas. The remaining animals were implanted in only four to six areas to increase the number of neurons isolated per brain area.
Drugs used for experiments.
All drugs were purchased from Sigma: α-methyl-p-tyrosine (αMT) at 250 mg/kg intraperitoneally, a tyrosine hydroxylase (TH) inhibitor; l-3,4-dihydroxyphenylalanine (l-DOPA) at 100 mg/kg intraperitoneally, an aromatic l-amino acid decarboxylase precursor; l-threo-dihydroxyphenylserine (l-DOPS) at 5 mg/kg intraperitoneally, an NE-specific precursor; and fluoxetine at 20 mg/kg intraperitoneally, a selective serotonin reuptake inhibitor.
Measurement of brain tissue catecholamine content.
After removing the cerebellum and the olfactory bulb, the frontal cortex region (approximately +2.3 mm anterior to bregma) and striatum of adult mice were dissected homogenized in 0.1 m HClO4 containing 100 ng/ml 3,4-dihydroxybenzylamin (DHBA) as an internal standard. Homogenates were centrifuged for 10 min at 10,000 × g. Supernatants were filtered through 0.22 mm filter and analyzed for levels of DA and NE using HPLC with electrochemical detection. Monoamines were separated on a microbore reverse-phase column (C-18, 5 mm, 1 × 150 mm; Unijet; BAS) with a mobile phase consisting of 0.03 m citrate–phosphate buffer with 2.1 mm octyl sodium sulfate, 0.1 mm EDTA, 10 mm NaCl, and 17% methanol, pH 3.6, at a flow rate of 90 ml/min and detected by a 3 mm glass carbon electrode (Unijet; BAS) set at 0.8 V. The volume of injection was 5 ml.
Behavioral measurements and drug treatments.
Initial behavioral measurements were conducted in unimplanted male WT and NET-KO mice. After a 12–24 h habituation period to the behavioral recording chamber, animals were treated with saline, and behavioral measurements were recorded for 1 h. Mice were then treated with αMT (250 mg/kg, i.p.), and behavioral measurements were recorded for an additional 4 h. Forward locomotion and stereotypic counts were automatically scored using the Activity Monitor software (Med Associates). Total grooming time was scored in 30 min intervals using video recordings. A mouse was considered to be grooming when it engaged in the following behaviors: face wiping, full body grooming paws/tail/genitals/back, or scratching and rubbing head/ears. Given the profound increase in grooming behaviors displayed by αMT-treated NET-KO mice compared with WT animals, it was not feasible to keep the observer blind to the genotype of the animals.
Drug treatments (l-DOPA, l-DOPS, and fluoxetine) were given 2 h after treatment with α-methyl-p-tyrosine to correspond with the period of maximum grooming displayed by the NET-KO mice. Behaviors were monitored for 30 min after treatment with l-DOPA or l-DOPS and for 1 h after treatment with fluoxetine. NET-KO mice were subjected to norepinephrine depletion an average of two times each, and animals were allowed to recover at least 1 full week between successive treatments.
Neuronal and LFP data acquisition.
Neuronal activity was recorded using the Multi-Neuron Acquisition Processor system (Plexon). At the end of the recording, cells were sorted using an offline sorting algorithm, based on cluster analysis (Plexon), to confirm the quality of the recorded cells. Only clearly defined cell clusters with clear refractory periods on the interspike interval histogram (interspike intervals larger than 1.3 ms for this study) were used for analysis. LFPs were preamplified (500×), filtered (0.5–400 Hz), and digitized at 500 Hz using a Digital Acquisition card (National Instruments) and a Multi-Neuron Acquisition Processor (Plexon). It is important to note that LFP recordings were associated with a significant phase offset that varied across frequencies (Nelson et al., 2008). These phase offsets were corrected using the LFPAlign function (Plexon).
Behaviors were recorded with a video cassette recorder. Video images were recorded from four angles (two above and two horizontal angles), loaded onto a cassette recorder, and synchronized with the neural recordings using a millisecond-precision timer.
Determination of delta and theta power oscillatory power.
For electrophysiological comparisons, we selected 20 min recording periods immediately after treatment with saline and a 20 min period 1.5 h after treatment with αMT. The average power was determined for each brain area using a fast Fourier transform. These power values observed during the depletion period were then normalized to the values observed during the baseline period, giving a power ratio value for each frequency of interest (delta, 1–4 Hz; theta, 4–11 Hz).
Significant changes in neural oscillatory power were determined in two steps. First, we used a two-tailed paired t test to determine the effect of αMT treatment in NET-KO mice for each area and frequency range. This yielded a total of two sets of 20 p values (10 areas, two frequency ranges, for two genotypes). The significance threshold was then determined using a false discovery rate (FDR) correction at α = 0.05 (Benjamini et al., 2001), allowing us to investigate whether there was an overall effect of norepinephrine depletion within a given brain area in WT and NET-KO mice. We then compared power ratios for each brain area and frequency across genotype, yielding 20 additional p values. The significance threshold of these values was then determined using an FDR correction at α = 0.05.
Determination of norepinephrine depletion effects on cross-structure coherence.
For electrophysiological comparisons, we selected a 20 min recording period immediately after treatment with saline and a 20 min period 1.5 h after treatment with αMT. Cross-structure coherence was calculated using the MATLAB mscohere function at a 2 s sliding window with a 0.5 s step. This process yielded an average coherence values, ranging from 0 to 1, for each brain area pair of interest. The optimal coherence frequency for each LFP pair of interest was then determined empirically from cross-structural coherence plots (see Fig. 3) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). These optimal frequencies are shown in Table 2.
Figure 3.
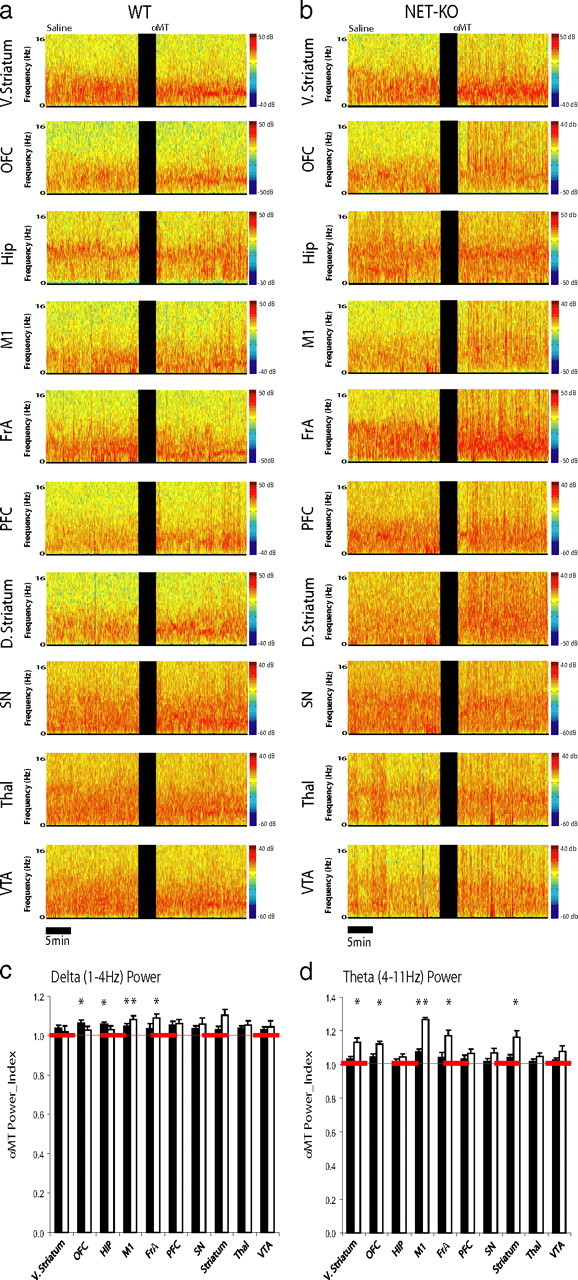
NE depletion selectively affects theta oscillatory power in NET-KO mice. a, b, LFPs were recorded from ventral striatum, frontal association cortex, hippocampus, motor cortex, orbital frontal cortex, prefrontal cortex, dorsal striatum, substantia nigra, medial dorsal thalamus, and VTA in WT (a; left column) and NET-KO (b; right column) mice. Each plot depicts LFP power spectra recorded during a baseline behavioral period juxtaposed with LFP power spectra recorded 1.5 h after treatment with αMT. c, d, The effect of norepinephrine depletion on LFP oscillatory power. Graphs depict delta (c) and theta (d) oscillatory power after NE depletion normalized to baseline oscillatory power. *p < 0.05 for comparisons within genotype (see Materials and Methods); n = 6 and 9 for αMT-treated WT and NET-KO mice, respectively.
Table 2.
The optimal coherence frequency for each LFP pair of interest determined empirically from cross-structural coherence plots
| Structure 1 | Structure 2 | Coherence band |
|---|---|---|
| Ventral striatum | FrA | 1–8 Hz |
| Ventral striatum | Thalamus | 1–11 Hz |
| Hippocampus | Thalamus | 6–11 Hz |
| Thalamus | FrA | 1–6 Hz |
| M1 | FrA | 1–8 Hz |
| PFC | Ventral striatum | 1–8 Hz |
| Ventral striatum | OFC | 1–11 Hz |
| Dorsal striatum | M1 | 1–7 Hz |
| Ventral striatum | VTA | 1–11 Hz |
| Dorsal striatum | FrA | 1–6 Hz |
| Dorsal striatum | SN | 1–6 Hz |
| Thalamus | SN | 6–11 Hz |
| Thalamus | PFC | 1–7 Hz |
| Thalamus | OFC | 1–8 Hz |
| VTA | PFC | 1–7 Hz |
The effect of norepinephrine depletion on cross-structure coherence was then determined in two steps. First, the treatment effect of αMT on cross-structure coherence in WT and NET-KO mice was determined by two-tailed paired t tests. This yielded a total of 15 p values per genotype (one for each cross-structure comparison). The significance threshold was then determined within genotype using an FDR correction at α = 0.05 (Benjamini et al., 2001). Second, the relative effect of profound NE depletion was determined by comparing treatment effects across genotype using an unpaired t test. The significance threshold for these p values was also determined using an FDR correction at α = 0.05 (Benjamini et al., 2001). For all cross-structure comparisons that displayed significance across genotype, the effect of norepinephrine depletion on cross-structure coherence was given by the following:
 |
Although this process successfully identified the robust effects of norepinephrine depletion on cross-structure coherence, it is plausible that small effects were missed as a result of the large number of comparisons made.
Norepinephrine depletion effects on single-neuron firing rates.
To determine the effects of αMT on single-neuron firing rates, we generated a firing-rate distribution across the 20 min recording segments (60 s bins) and calculated the 99% confidence interval based on a Poisson distribution. We considered that a neuron changed its firing rate if the mean firing rate of that neuron was outside of the 99% confidence interval observed during the previous recording period (i.e., the depletion period was compared with baseline recording period). To determine whether NE depletion altered population firing rates, similar methods were applied to the mean firing rate observed within each 60 s bin across all brain areas and animals within genotype.
Drug treatment effects on cross-structural coherence in NE-depleted NET-KO mice.
After the baseline and norepinephrine depletion recording periods, animals were treated with l-DOPA (100 mg/kg, i.p.), l-DOPS (5 mg/kg, i.p.), or fluoxetine (20 mg/kg, i.p.), and additional LFP data were recorded for 20 min. For each drug treatment, recordings were initially conducted in two animals implanted across all 10 brain areas. Power analysis was then conducted using a one-tailed paired t test on the cross-structural coherence values measured before (NE-depleted state) and after drug treatment, to determine which cross-structural comparisons (NE depleted vs drug treatment) were powered to display a partial or complete reversal of the NE depletion effect using a total of eight or less mice. This was done to limit the number of eventual comparisons made for each drug treatment. Next, three to five additional animals were implanted across the specific brain areas of interest to increase statistical power for the cross-structural comparisons determined to be adequately powered. Finally, p values obtained for each previously identified cross-structural comparison of interest were subject to an FDR correction at α = 0.05.
The effect of drug treatment on cross-structure coherence was then given by the following:
 |
Results
Catecholamine depletion in WT and NET-KO mice
In mice with normal transporter-mediated recycling, inhibition of catecholamine synthesis with the TH inhibitor αMT (250 mg/kg) results in only a partial (50–60%) reduction in neuronal levels of the TH-dependent monoamines dopamine and NE (Sotnikova et al., 2005). However, in mice lacking the dopamine transporter, the same treatment completely eliminated neuronal dopamine and caused extreme akinesia and rigidity (Sotnikova et al., 2005). Similarly, because NET-KO mice are unable to recycle NE and intracellular NE concentrations are solely dependent on de novo synthesis, treatment with αMT (250 mg/kg, i.p.) reduced the cortical concentration of NE to <5% of that observed in control animals [NE-depleted NET-KO mice (NDN mice); ANOVA of cortical NE tissue content: genotype effect, F(1,33) = 523.13, p < 0.0001; drug effect, F(1,33) = 91.26, p < 0.0001]. Importantly, both WT and NET-KO mice displayed a normal pattern of partial reduction in striatal dopamine (40–60%; ANOVA of striatal DA tissue content: genotype effect, F(1,21) = 0.02, p = 0.89; drug effect, F(1,21) = 62.13, p < 0.0001) and cortical dopamine (65–75%; ANOVA of cortical DA tissue content: genotype effect, F(1,29) = 0.51, p = 0.48) and drug effect (F(1,29) = 33.56, p < 0.0001) levels after treatment with αMT (Fig. 1).
Figure 1.

Treatment with αMT induces cortical NE depletion in NET-KO mice. Treatment with αMT (250 mg/kg) reduced cortical NE content in NET-KO mice to <5% of that observed in saline-treated WT control mice (left); n = 12 and 11 for saline-treated WT and NET-KO, respectively, and n = 6 and 5 for αMT-treated WT and NET-KO, respectively. Treatment with αMT (250 mg/kg) produced an equivalent 40–50% reduction in striatal dopamine content in WT and NET-KO mice (right); n = 12 and 10 for saline-treated WT and NET-KO, respectively, and n = 4 and 4 for αMT-treated WT and NET-KO, respectively. Treatment with αMT (250 mg/kg) produced an equivalent 70–75% reduction in cortical dopamine content in WT and NET-KO mice (right); n = 6 and 5 for saline-treated WT and NET-KO, respectively, and n = 5 and 5 for αMT-treated WT and NET-KO, respectively. Importantly, there was no difference in cortical dopamine levels observed between saline-treated NET-KO mice and WT controls; p > 0.05. Catecholamine levels were measured 2 h after treatment with saline or αMT as described previously (Wang et al., 1997); *p < 0.05, two-tailed Mann–Whitney U test, WT versus NET-KO.
NE depletion has limited effects on cortical and subcortical LFP oscillatory power
To investigate the role that noradrenergic dysfunction plays in regulating synchronous neural activity, we conducted electrophysiological recordings across multiple structures of the cortico-striatal and mesolimbic circuits in six WT and 12 NET-KO mice treated with αMT. Microelectrode arrays were implanted in each animal across 10 brain areas [i.e., the ventral striatum/nucleus accumbens (v. striatum), frontal association cortex (FrA), hippocampus (Hip), primary motor cortex (M1), orbital frontal cortex (OFC), prelimbic cortex (PFC), dorsal lateral striatum (d. striatum), medial dorsal nucleus of thalamus (Thal), substantia nigra pars reticularis (SN), and ventral tegmental area (VTA); for coordinates, see Materials and Methods] (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Mice implanted with microelectrodes recovered well after this operation and behaved normally, allowing us to simultaneously record LFPs and single-neuron activity across multiple cortico-striato-thalamo-cortical circuits in awake, nonrestrained animals (Fig. 2a–c). To investigate the effects of NE depletion on neural oscillatory activity, we first quantified the effect of partial NE and DA depletion on neural oscillatory activity in WT mice (Fig. 3a,b). Our results showed that treatment with αMT yielded a 4–6% increase in the FrA, M1, and Hip delta power and a 7% increase in M1 theta power in WT mice (partial NE and DA depletion) (Fig. 3c,d). In contrast, NET-KO mice treated with αMT (profound NE depletion and partial DA depletion) displayed a 8–9% increase in M1 and FrA delta oscillatory power and a 12–26% increase in theta power within v. striatum, FrA, M1, OFC, and d. striatum (Fig. 3c,d). Together, these results demonstrated that NE depletion induces theta oscillatory activity across multiple cortical and subcortical brain areas. Importantly, this was in stark contrast to previous studies that demonstrated that treatment with αMT suppresses cortical and subcortical theta oscillatory activity in DAT-KO mice (partial NE depletion, profound DA depletion) (Costa et al., 2006; Dzirasa et al., 2006).
Figure 2.
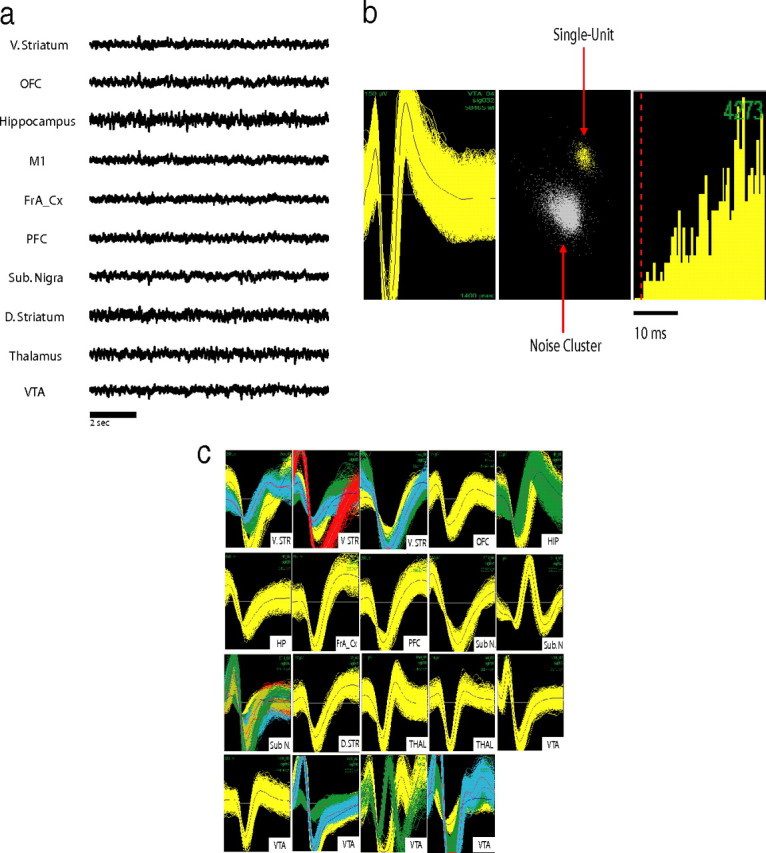
Multisite electrophysiological recording in mice. a, LFP recordings from WT baseline waking period. LFPs were simultaneously recorded from ventral striatum (V. STR), frontal association cortex (FrA_Cx), hippocampus (HP), M1, OFC, PFC, substantial nigra (Sub. N), dorsal lateral striatum (D. STR), medial dorsal thalamus (THAL), and VTA. b, Example of an isolated single unit in VTA. From left to right, Depiction of the extracellularly recorded wave form of the unit (x-axis, 1400 ms; y-axis, 150 mV), interspike interval histogram, and projection of the clusters correspondent to the unit and the noise based on analysis of the first two principal components of the waveforms recorded (x-axis, PC1; y-axis, PC2). c, Simultaneous recording of 37 single units across nigrostriatal and mesolimbic brain circuits in an awake behaving mouse.
NE depletion disrupts cross-structural coherence
Because synchronous theta oscillations have been implicated in mediating long-range communication across brain areas (Seidenbecher et al., 2003; Jones and Wilson, 2005), we measured whether low-frequency LFP coherence (for exact frequencies, see Materials and Methods) between anatomically connected brain areas, instead of within-area power, was modified by treatment with αMT in mice WT (Fig. 4). Our results indicated that treatment with αMT reduced LFP oscillatory synchrony across all of the cortico-striatal and FrA-thalamo-cortical circuits examined in WT mice (Fig. 5a), suggesting that partial NE and DA depletion dampens cross-structural coherence across these circuits. Next, we examined the effect of αMT on cortico-striatal circuits in NET-KO mice. Although treatment with αMT also reduced M1-striatal and FrA-striatal coherence in the NET-KOs (Fig. 5b), profound NE depletion paradoxically elevated OFC-striato-thalamo-cortical, VTA-striatal, and PFC-thalamic cross-structural coherence. To further quantify the effect of profound NE depletion on these circuits, we measured the treatment effect of αMT in NET-KO mice (partial DA depletion, profound NE depletion) relative to that observed in WT mice (partial DA and NE depletion). We observed that profound NE depletion further reduced coherence across FrA-M1 and M1-striatal circuits. In contrast, NE depletion increased coherence across OFC-striato-thalamo-cortical, striato-thalamo-PFC, VTA-striato-thalamic, and PFC-VTA neural circuits (Fig. 5c). These results suggested that functional interactions between some cortical areas and striatum were sustained by NE, whereas others were suppressed. Most importantly, these findings demonstrated that NE depletion selectively alters the synchronous timing of oscillatory activity between anatomically connected brain structures that define particular neural circuits, without disrupting the power of oscillatory activity within each of the distinct brain structures that form these circuits. Incidentally, we also found that NE depletion did not induce changes in theta coherence between Hip and any of the cortical areas examined in WT or NET-KO mice (p > 0.05).
Figure 4.
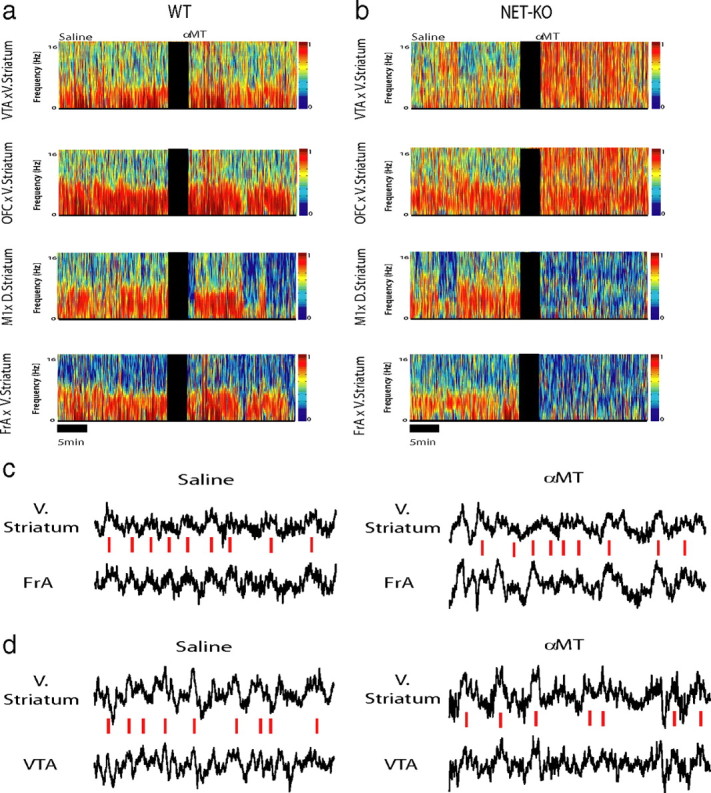
Norepinephrine depletion alters cross-structural coherence. Cross-structural coherence was measured in WT and NET-KO animals after treatment with αMT (250 mg/kg, i.p.). a, b, Each plot depicts select cross-coherence values observed during a baseline behavioral period juxtaposed with cross-coherence values observed 1.5 h after treatment with αMT in WT (left) and NET-KO (right) mice. Treatment with αMT dramatically altered cross-structural coherence across several brain areas in NET-KO mice but not WT mice. c, d, Sample LFP traces recorded from NET-KO mouse before (left) and after (right) treatment with αMT. Red dashed lines highlight theta synchrony during baseline periods. Note that v. striatum theta oscillation peaks fail to synchronize with OFC theta oscillations peaks after treatment with αMT (c) and that v. striatum and VTA theta oscillations become synchronized after treatment with αMT (d). Each trace depicts 2.5 s of continuous LFP activity; n = 6 and 9 for αMT-treated WT and NET-KO mice, respectively.
Figure 5.
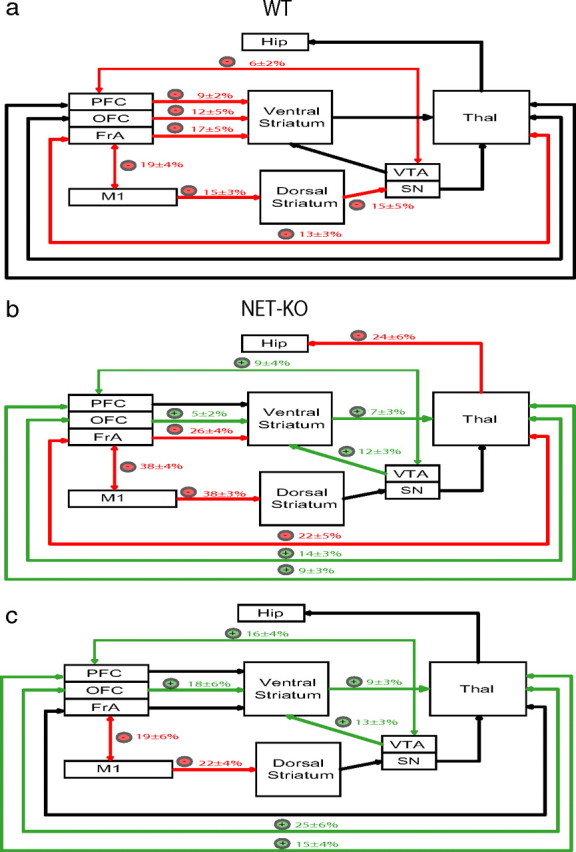
Dynamic changes in coherence across cortio-striatal and mesolimbic circuits after NE depletion. a, Partial norepinephrine and dopamine depletion in WT mice attenuated coherence (red) across multiple cortico-striatal, cortico-thalamic, and mesolimbic circuits. b, Profound norepinephrine and partial dopamine depletion in NET-KO mice attenuated coherence (red) across FrA-striato-thalamo-cortical and M1-striatal neural circuits (red) and potentiated coherence (green) across OFC-striato-thalamo-cortical, striato-thalamo-PFC, VTA-striato-thalamic, and PFC-VTA neural circuits. c, Treatment effect of αMT on coherence in NET-KO animals relative to WT mice. Only coherence differences that reached significance are highlighted (*p < 0.05 for comparisons within and across genotype; n = 6 and 9 for αMT-treated WT and NET-KO mice, respectively.
NE depletion increases neuronal firing rates across the mesolimbic circuit
Next, we investigated the effect of NE depletion on single-neural activity. Using our recording technique, we isolated 438 single neurons (ventral striatum/nucleus accumbens, 63 neurons; orbital frontal cortex, 35 neurons; prelimbic cortex, 44 neurons; d. striatum, 44 neurons; substantia nigra, 45 neurons; thalamus, 39 neurons; and VTA, 168 neurons) from 12 NET-KO mice and six WT mice treated with αMT. As expected, we found neurons that either increased (Type 1 neuron) or decreased (Type 2 neuron) their firing rates after NE depletion in each of the brain areas examined (Fig. 6a). When we examined the population firing rate, our results demonstrated that treatment with αMT decreased the mean firing rate observed in WT animals (Fig. 6b). Moreover, our findings demonstrated that the majority of single neurons observed across each brain region were primarily suppressed by partial NE and DA depletion (Type 2 neurons). In contrast, the population firing rate observed in NET-KO animals was paradoxically increased after NE depletion (Fig. 6c). This change was primarily attributable to a decrease in the proportion of Type 2 neurons observed in v. striatum, FrA, PFC, and VTA in NET-KO mice (Fig. 6d,e). Together, these results demonstrated that the increased mesolimbic coherence observed in NDN mice was correlated with increased neuronal activity across several cortical and subcortical mesolimbic structures.
Figure 6.
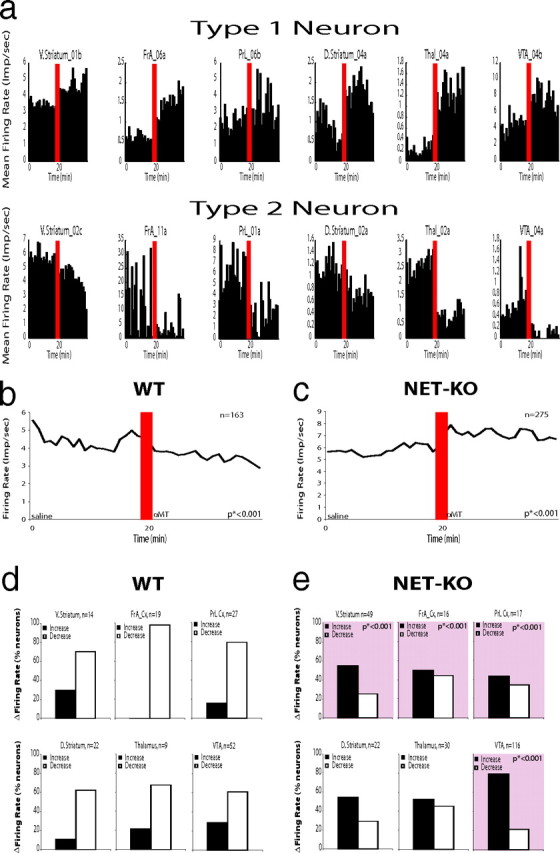
Norepinephrine depletion alters single-unit figure rates across cortico-striatal circuits. a, We found neurons in v. striatum, d. striatum, OFC, thalamus, and VTA that increased their firing rates after norepinephrine depletion (Type 1 neurons) and neurons in each of these areas that decreased their firing rates after norepinephrine depletion (Type 2 neurons). b, Treatment with αMT reduced the population firing rate observed in WT mice (n = 163; *p < 0.001). c, Treatment with αMT significantly increased the population firing rate observed in NET-KO mice (n = 275; *p < 0.001). d, WT mice displayed more Type 2 neurons than Type 1 neurons within each brain area investigated. e, NET-KO mice displayed a significantly decreased proportion of Type 2 neurons within v. striatum, orbital frontal cortex, and VTA compared with WT mice (*p < 0.001 for comparisons between genotype; see squares with pink highlights).
Profound NE depletion induces repetitive behaviors
Next, we examined the behavioral profiles in WT and NET-KO mice after treatment with αMT. Whereas partial NE and DA depletion led to decreased forward locomotion in WT mice [mixed ANOVA of distance traveled found the main effects of genotype (F(1,54) = 18.36, p = 0.002) and drug (F(4,54) = 12.42, p < 0.0001), as well as the genotype × drug interaction (F(4,54) = 13.12, p < 0.0001) to be significant], treatment of NET-KO mice with αMT paradoxically induced a complex behavioral phenotype that involved a transient (for up to 3 h) increase in locomotor activity and stereotypic behaviors [i.e., sniffing and rearing; quantified via repetitive beam breaks in an activity monitor; mixed ANOVA of stereotypy counts found the main effects of genotype (F(1,54) = 13.24, p = 0.005) and drug (F(4,54) = 6.01, p = 0.0008), as well as the genotype × drug interaction (F(4,54) = 6.94, p = 0.0003) to be significant] (Fig. 7a,b) and occasional hindlimb extensions (data not shown). NET-KO mice also exhibited a potent increase in grooming behaviors beginning 1 h after treatment with αMT [mixed ANOVA of grooming time found the main effects of genotype (F(1,54) = 79.44, p < 0.0001) and drug (F(4,54) = 27.12, p < 0.0001), as well as the genotype × drug interaction (F(4,54) = 27.62, p < 0.0001) to be significant] (Fig. 7c). To test whether these behaviors were attributable to depletion of the animals' catecholamine levels, we treated NDN mice with the dopamine and NE precursor l-DOPA (100 mg/kg, i.p.). This dose was chosen because it has been shown to partially restore catecholamine levels depleted by treatment with αMT (Dzirasa et al., 2006). Treatment with l-DOPA (100 mg/kg, i.p.) attenuated the hyperactivity and increased grooming behaviors observed in the NDN mice, suggesting that acute changes in DA and or NE signaling likely contributed to these behaviors observed in NDN mice. Next, to ensure that this effect was not simply attributable to the restoration of partially affected dopaminergic tone, we treated NDN mice with the synthetic NE-specific precursor l-DOPS (5 mg/kg, i.p.) (Thomas et al., 1998). Interestingly, our results demonstrated that treatment with l-DOPS attenuated the increased grooming behaviors but not the hyperactivity or increased stereotypic behaviors seen in NDN mice (Fig. 7d). Because WT mice treated with αMT never displayed hyperactivity or the grooming behaviors exhibited by NDN mice although they demonstrated a similar degree of DA depletion, our results suggested that depletion of NE played a critical role in generating the grooming behaviors observed in NDN mice. Moreover, our results suggested that the stereotypic behaviors and hyperactivity displayed by these animals likely resulted from complex interactions between dopaminergic and noradrenergic neuromodulatory systems. Overall, these findings suggest that significant reductions in noradrenergic tone may be sufficient to generate profound alterations of synchronous oscillations between interconnected structures that define multiple cortico-striatal and mesolimbic circuits and ultimately lead to the induction of repetitive grooming behaviors in NET-KO mice.
Figure 7.

Norepinephrine depletion induces repetitive grooming behaviors. a–c, Treatment with αMT induced locomotor hyperactivity, stereotypic behaviors, and excessive grooming in NET-KO mice (n = 5–6 per group; **p < 0.01, mixed ANOVA followed by Bonferroni's corrected t test). d, Treatment with l-DOPA (100 mg/kg) or l-DOPS (5 mg/kg) attenuated the increased grooming behaviors observed in NDN mice. Animals were treated with l-DOPA or l-DOPS 2 h after initial treatment with αMT (n = 5 per group; **p < 0.01, Bonferroni's corrected t test).
l-DOPA and l-DOPS selectively reverse circuit alterations induced by NE depletion
After demonstrating that treatment with l-DOPA or l-DOPS attenuated grooming behaviors in NDN mice, we examined the effect of these pharmacological agents on cross-structural coherence. We treated NDN mice with l-DOPA (100 mg/kg, i.p.) or l-DOPS (5 mg/kg, i.p.) 2 h after αMT treatment, and cross-structural coherence values were determined for brain area pairs affected by NE depletion. Using this analysis, we found that treatment with l-DOPA reversed the M1-striatal, PFC-thalamo-VTA, and OFC-striatal-thalamo-cortical coherence alterations induced by profound NE depletion (Fig. 8a). In contrast, treatment with l-DOPS only reversed the thalamo-OFC-cortico-striatal coherence alterations induced by profound NE depletion (Fig. 8b).
Figure 8.

DA and NE precursors selectively reverse circuit deficits in NDN mice. a, NDN mice were treated with l-DOPA (a; 100 mg/kg, i.p.) or l-DOPS (b; 5 mg/kg, i.p.), and cross-structural coherence values were determined for brain area pairs affected by NE depletion. Treatment with l-DOPA attenuated OFC-striato-thalamo-cortical, PFC-thalamo-VTA, and enhanced M1-striatal coherence. Treatment with l-DOPS attenuated thalamo-FrA-cortico-striatal coherence. n = 5–6 NET-KO mice per drug treatment.
Treatment with fluoxetine selectivity reverses circuit alterations induced by NE depletion
Treatment with fluoxetine has been shown to suppress pathological grooming behaviors in mice (Welch et al., 2007). Thus, we set out to determine the effect of fluoxetine on the behavioral alterations observed in NDN mice. Mice were treated with fluoxetine (20 mg/kg, i.p.) 2 h after treatment with αMT, and behavioral assessments were taken for 1 h. Interestingly, we found that fluoxetine treatment attenuated the hyperactivity (distance traveled per hour: 1086 ± 90 cm in NDN mice, 248 ± 53 cm in fluoxetine-treated NDN mice; t(1,10) = 7.94; p < 0.0001), stereotypic behaviors (stereotypy count per hour: 9624 ± 535 in NDN mice, 4848 ± 847 in fluoxetine-treated NDN mice; t(1,10) = 4.77; p < 0.001), and grooming behaviors (grooming time percentage per hour: 63.1 ± 2.8% in NDN mice, 4.4 ± 1.8% in fluoxetine-treated NDN mice; t(1,10) = 17.80; p < 0.0001) observed in NDN mice. Next, we performed coherence analysis in NDN mice treated with fluoxetine across brain areas shown to be affected by NE depletion. These results demonstrated that treatment with fluoxetine selectively reversed the VTA-striatal and OFC-striatal coherence alterations induced by profound NE depletion (Fig. 9). Together, these findings demonstrated that fluoxetine treatment attenuates the behavioral responses observed in NDN mice and reverses several of the circuit changes induced by NE depletion.
Figure 9.

Fluoxetine selectively reverses circuit deficits in NDN mice. NET-KO mice were treated with fluoxetine (20 mg/kg, i.p.) 2 h after treatment with αMT. Treatment with fluoxetine decreased the elevated OFC-striatal and VTA-striatal circuit coherence observed in NDN mice; n = 5.
Discussion
Our findings suggest that, in contrast to previous observations made in dopamine-depleted mice (Costa et al., 2006), profound NE depletion does not suppress theta frequency LFP oscillatory power within brain areas comprising various cortico-striatal circuits. However, it is important to note that profound NE depletion induces behavioral hyperactivity in NET-KO mice. Because exploratory behaviors are strongly correlated with increases in theta oscillatory activity (Vanderwolf, 1969; Winson, 1974), our findings do not eliminate the possibility that NE depletion itself may lead to small decreases in theta activity (McNaughton et al., 1980) that are masked by the increased behavioral profiles seen in NDN mice. Nevertheless, our findings do demonstrate that NE depletion peculiarly alters the synchronization of oscillations across a particular combination of interconnected brain areas.
Dynamic changes in cross-structure coherence are not attributable to volume conduction
Although previous studies have demonstrated that hippocampal theta oscillatory activity can be recorded from several cortical areas via volume conduction (Sirota et al., 2008), our data recorded simultaneously from 10 brain areas provide ample evidence that the changes in coherence observed between various brain regions after NE depletion are not simply the result of volume conduction. For instance, our results show that neither partial nor profound NE depletion alter hippocampal theta oscillatory power. Second, we found no changes in theta coherence between Hip and any of the cortical areas examined after NE depletion in WT or NET-KO mice. Third, although NE depletion did induce OFC and FrA theta oscillatory activity in NET-KO mice, cross-structural coherence within PFC-striatal, OFC-striatal, and FrA-striatal circuits was differentially affected by NE depletion. For example, NE depletion in NET-KO mice profoundly suppressed FrA-striatal coherence but enhanced OFC-striatal coherence within the theta frequency range. Moreover, coherence between thalamus and these three cortical areas was also differentially affected by NE depletion. Importantly, these phenomena were observed despite the anatomical proximity of the three simultaneously recorded cortical areas (∼1 mm separation). Finally, our results show that NE depletion enhances VTA-striatal coherence despite the fact that it does not induce VTA theta oscillatory power. Together, these results suggest that our findings observed in NE-depleted mice are the result of dynamic changes in cross-structural LFP synchrony attributable to changes in NE tone and not simply artifacts of volume conduction.
Circuit changes in NE-depleted mice correspond with repetitive behavioral disorders in humans
Partial NE and DA depletion (as observed in WT animals treated with αMT) led to a decrease in the neuronal firing rates observed across all of the brain areas examined. Moreover, our results showed that partial NE and DA depletion also suppressed coherence across all of the cortico-striatal circuits examined and decreased forward locomotion. In contrast, profound NE depletion (as observed in NDN mice) led to an paradoxical increase in the firing rate observed across many of the mesolimbic areas examined, dynamic changes in coherence across cortico-striatal and mesolimbic circuits, and a complex behavioral phenotype characterized by hyperactivity, stereotypic behaviors, and grooming. Importantly, inhibition of NE neurons in locus ceruleus via the local injection of pharmacological agents has also been shown recently to induce hyperactivity in mice (Stone et al., 2009).
Deficits in cortico-striatal signaling have been implicated in mediating repetitive behaviors in human disorders such as obsessive compulsive disorder (OCD) (Graybiel and Rauch, 2000; Aouizerate et al., 2004; Chamberlain et al., 2008; Greenberg et al., 2010). Although studies have yet to demonstrate that alterations in central noradrenergic signaling underlie the central pathophysiological mechanisms of OCD, our findings suggest that the primary circuits implicated in the disorder can be disrupted in mice via depletion of NE. Conversely, pharmacological agents that modulate noradrenergic tone have been shown to be effective in treating other repetitive behavior disorders such as Tourette's syndrome (Du et al., 2008). Some evidence also suggests that alterations in mesolimbic dopamine signaling may contribute to the pathophysiology of this disorder (Gilbert et al., 2006). Thus, these studies point to the involvement of at least two aberrant circuits in the generation of excessive repetitive behaviors: one circuit involving cortical-striatal projections and one putative dopaminergic circuit (Swerdlow et al., 2006). This is consistent with our findings in NE-depleted NET-KO mice that demonstrate that the excessive grooming behaviors displayed by these mice are associated with altered OFC-striatal coherence, enhanced VTA-striatal coherence, and increased VTA firing rates. Given that increased VTA activity has been correlated with higher expected reward value (Adcock et al., 2006), our data provide support for the hypothesis that excessive grooming and aberrant repetitive behaviors may result when OFC-striatal dysfunction is partnered with other circuit changes that generate increased expected outcome values (Damasio et al., 1990; Graybiel and Rauch, 2000; Gilbert et al., 2006). Moreover, our results significantly expand this hypothesis by suggesting that repetitive behaviors may result when cortical association loops are reinforced by (1) a loss of FrA-mediated inhibitory control attributable to decreased thalamic excitatory input, (2) diminished voluntary motor control attributable to altered cortical-mediated excitatory input via retrosplenial cortex, and (3) increased VTA-mediated motivational saliency possibly attributable to increased thalamically driven prefrontal cortical excitatory input (Fig. 10).
Figure 10.
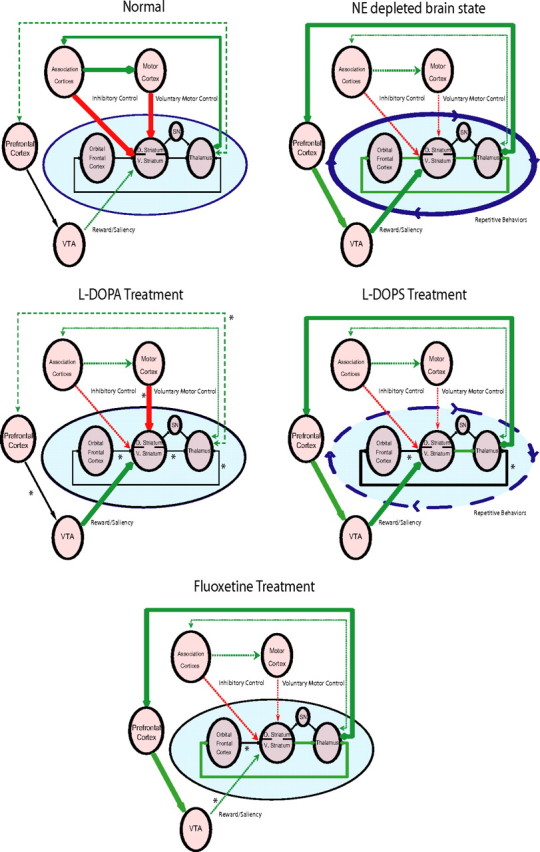
Model of cortico-striato-thalamic and mesolimbic circuit dysfunction underlying the aberrant generation of repetitive behaviors. Aberrant repetitive behaviors manifest when association cortico-striato-thalamic circuits are reinforced by a loss of FrA inhibitory control, decreased voluntary motor control, and increased VTA-mediated saliency/reward expectancy value.
Pharmacological agents work as selective circuit modulators
Our data recorded in NDN mice treated with various pharmacological agents also suggest that the enhanced grooming behaviors observed in these mice likely results from coordinated changes across multiple cortico-striatal and mesolimbic brain circuits. For example, treatment with the DA and NE precursor l-DOPA selectively attenuated OFC-striato-thalamo-cortical circuit coherence but enhanced M1-striatal circuit coherence. Additionally, treatment with the NE-specific precursor l-DOPS attenuated OFC-striatal and thalamo-OFC-cortical circuit coherence. Finally, treatment with the selective serotonin reuptake inhibitor fluoxetine attenuated VTA-striatal circuit coherence, demonstrating that circuit changes observed in NDN mice could be reversed via non-DA or NE-dependent mechanisms. Together, these findings provide strong support for a distributed network model of brain disease (a model in which pathological behaviors occur via alterations in the dynamic function of neuronal networks distributed across many brain areas), because each of the pharmacological agents that attenuated the excessive grooming behaviors observed in NDN mice reversed different components of the gross circuit changes induced by NE depletions. Notably, our approach can also be combined with novel optogenetic stimulation techniques to dissect the specific cell types and brain areas that drive the network and behavioral changes that result from NE depletion.
CNS diseases as disorders of brain timing
Overall, our findings suggest that norepinephrine plays a critical role in coordinating theta oscillation synchrony across extensive forebrain circuits formed by multiple cortical and subcortical structures. Moreover, our results demonstrate that acute norepinephrine depletion induces circuit changes in mice that are consistent with those implicated in mediating excessive repetitive behaviors in human disorders. It has been suggested recently that the behavioral deficits observed across neurological disease states such as Parkinson's disease may result from changes in the synchronous neural oscillations responsible for coordinating activity across vast distributed brain circuits (Fuentes et al., 2009). Here we show that repetitive behaviors observed in norepinephrine-depleted NET-KO mice are correlated with profoundly altered neural oscillatory network timing across OFC-striato-thalamic and mesolimbic circuits. These findings raise the hypothesis that errors in neural oscillatory timing may contribute in a major way to the behavioral manifestations observed across many psychiatric disorders as well. Thus, the present findings point to the potential that novel approaches aimed at readjusting altered levels of neural synchrony across specific forebrain circuits may lead to a new generation of treatments for a variety of psychiatric disorders.
Most importantly, in conjunction with the emerging body of literature supporting the use of electrophysiological techniques to probe neural circuits in mouse models of CNS disease, our findings highlight the critical need to develop tools that allow neural and neuronal activity to be quantified in freely behaving mice across many brain circuits simultaneously.
Footnotes
This work was supported by funding from the following: the Wakeman Foundation, the Ruth K. Broad Foundation, United Negro College Fund/Merck, and National Institute of Mental Health Grant P50MH060451 (K.D.); National Institutes of Health (NIH) Grants MH-60451 and MH-79201 (M.G.C.); and NIH Grants NS049534, MH060451, and MH084194 and the Safra Foundation (M.A.L.N.). Part of this work was also supported by unrestricted gifts from the Lennon Family Foundation and Lundbeck USA (M.G.C.). A special thanks to Freeman Hrabowski, Robert and Jane Meyerhoff, and the Meyerhoff Scholarship Program. We thank G. Lehew for technical assistance, L. Oliveira, T. Jones, and G. Wood for miscellaneous support, and S. Halkiotis for proofreading of this manuscript.
References
- Adcock RA, Thangavel A, Whitfield-Gabrieli S, Knutson B, Gabrieli JD. Reward-motivated learning: mesolimbic activation precedes memory formation. Neuron. 2006;50:507–517. doi: 10.1016/j.neuron.2006.03.036. [DOI] [PubMed] [Google Scholar]
- Aouizerate B, Guehl D, Cuny E, Rougier A, Bioulac B, Tignol J, Burbaud P. Pathophysiology of obsessive-compulsive disorder: a necessary link between phenomenology, neuropsychology, imagery and physiology. Prog Neurobiol. 2004;72:195–221. doi: 10.1016/j.pneurobio.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- Carlsson A. Biochemical and pharmacological aspects of Parkinsonism. Acta Neurol Scand Suppl. 1972;51:11–42. [PubMed] [Google Scholar]
- Chamberlain SR, Menzies L, Hampshire A, Suckling J, Fineberg NA, del Campo N, Aitken M, Craig K, Owen AM, Bullmore ET, Robbins TW, Sahakian BJ. Orbitofrontal dysfunction in patients with obsessive-compulsive disorder and their unaffected relatives. Science. 2008;321:421–422. doi: 10.1126/science.1154433. [DOI] [PubMed] [Google Scholar]
- Charney DS. Monoamine dysfunction and the pathophysiology and treatment of depression. J Clin Psychiatry. 1998;59(Suppl 14):s11–s14. [PubMed] [Google Scholar]
- Costa RM, Lin SC, Sotnikova TD, Cyr M, Gainetdinov RR, Caron MG, Nicolelis MA. Rapid alterations in corticostriatal ensemble coordination during acute dopamine-dependent motor dysfunction. Neuron. 2006;52:359–369. doi: 10.1016/j.neuron.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Damasio AR, Tranel D, Damasio H. Individuals with sociopathic behavior caused by frontal damage fail to respond autonomically to social stimuli. Behav Brain Res. 1990;41:81–94. doi: 10.1016/0166-4328(90)90144-4. [DOI] [PubMed] [Google Scholar]
- Debiec J, LeDoux JE. Noradrenergic signaling in the amygdala contributes to the reconsolidation of fear memory: treatment implications for PTSD. Ann N Y Acad Sci. 2006;1071:521–524. doi: 10.1196/annals.1364.056. [DOI] [PubMed] [Google Scholar]
- Du YS, Li HF, Vance A, Zhong YQ, Jiao FY, Wang HM, Wang MJ, Su LY, Yu DL, Ma SW, Wu JB. Randomized double-blind multicentre placebo-controlled clinical trial of the clonidine adhesive patch for the treatment of tic disorders. Aust N Z J Psychiatry. 2008;42:807–813. doi: 10.1080/00048670802277222. [DOI] [PubMed] [Google Scholar]
- Dzirasa K, Ribeiro S, Costa R, Santos LM, Lin SC, Grosmark A, Sotnikova TD, Gainetdinov RR, Caron MG, Nicolelis MA. Dopaminergic control of sleep–wake states. J Neurosci. 2006;26:10577–10589. doi: 10.1523/JNEUROSCI.1767-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzirasa K, Ramsey AJ, Takahashi DY, Stapleton J, Potes JM, Williams JK, Gainetdinov RR, Sameshima K, Caron MG, Nicolelis MA. Hyperdopaminergia and NMDA receptor hypofunction disrupt neural phase signaling. J Neurosci. 2009;29:8215–8224. doi: 10.1523/JNEUROSCI.1773-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes R, Petersson P, Siesser WB, Caron MG, Nicolelis MA. Spinal cord stimulation restores locomotion in animal models of Parkinson's disease. Science. 2009;323:1578–1582. doi: 10.1126/science.1164901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Caron MG. Monoamine transporters: from genes to behavior. Annu Rev Pharmacol Toxicol. 2003;43:261–284. doi: 10.1146/annurev.pharmtox.43.050802.112309. [DOI] [PubMed] [Google Scholar]
- Gilbert DL, Christian BT, Gelfand MJ, Shi B, Mantil J, Sallee FR. Altered mesolimbocortical and thalamic dopamine in Tourette syndrome. Neurology. 2006;67:1695–1697. doi: 10.1212/01.wnl.0000242733.18534.2c. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Rauch SL. Toward a neurobiology of obsessive-compulsive disorder. Neuron. 2000;28:343–347. doi: 10.1016/s0896-6273(00)00113-6. [DOI] [PubMed] [Google Scholar]
- Greenberg BD, Gabriels LA, Malone DA, Jr, Rezai AR, Friehs GM, Okun MS, Shapira NA, Foote KD, Cosyns PR, Kubu CS, Malloy PF, Salloway SP, Giftakis JE, Rise MT, Machado AG, Baker KB, Stypulkowski PH, Goodman WK, Rasmussen SA, Nuttin BJ. Deep brain stimulation of the ventral internal capsule/ventral striatum for obsessive-compulsive disorder: worldwide experience. Mol Psychiatry. 2010;15:64–79. doi: 10.1038/mp.2008.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedderick EF, Morris CM, Singer HS. Double-blind, crossover study of clonidine and levetiracetam in Tourette syndrome. Pediatr Neurol. 2009;40:420–425. doi: 10.1016/j.pediatrneurol.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Horschitz S, Hummerich R, Lau T, Rietschel M, Schloss P. A dopamine transporter mutation associated with bipolar affective disorder causes inhibition of transporter cell surface expression. Mol Psychiatry. 2005;10:1104–1109. doi: 10.1038/sj.mp.4001730. [DOI] [PubMed] [Google Scholar]
- Jones MW, Wilson MA. Theta rhythms coordinate hippocampal-prefrontal interactions in a spatial memory task. PLoS Biol. 2005;3:e402. doi: 10.1371/journal.pbio.0030402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucaite A, Fernell E, Halldin C, Forssberg H, Farde L. Reduced midbrain dopamine transporter binding in male adolescents with attention-deficit/hyperactivity disorder: association between striatal dopamine markers and motor hyperactivity. Biol Psychiatry. 2005;57:229–238. doi: 10.1016/j.biopsych.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A. Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies. J Psychopharmacol. 1999;13:358–371. doi: 10.1177/026988119901300405. [DOI] [PubMed] [Google Scholar]
- McNaughton N, Kelly PH, Gray JA. Unilateral blockade of the dorsal ascending noradrenergic bundle and septal elicitation of hippocampal theta rhythm. Neurosci Lett. 1980;18:67–72. doi: 10.1016/0304-3940(80)90214-1. [DOI] [PubMed] [Google Scholar]
- Nelson MJ, Pouget P, Nilsen EA, Patten CD, Schall JD. Review of signal distortion through metal microelectrode recording circuits and filters. J Neurosci Methods. 2008;169:141–157. doi: 10.1016/j.jneumeth.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidenbecher T, Laxmi TR, Stork O, Pape HC. Amygdalar and hippocampal theta rhythm synchronization during fear memory retrieval. Science. 2003;301:846–850. doi: 10.1126/science.1085818. [DOI] [PubMed] [Google Scholar]
- Sirota A, Montgomery S, Fujisawa S, Isomura Y, Zugaro M, Buzsáki G. Entrainment of neocortical neurons and gamma oscillations by the hippocampal theta rhythm. Neuron. 2008;60:683–697. doi: 10.1016/j.neuron.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotnikova TD, Beaulieu JM, Barak LS, Wetsel WC, Caron MG, Gainetdinov RR. Dopamine-independent locomotor actions of amphetamines in a novel acute mouse model of Parkinson disease. PLoS Biol. 2005;3 doi: 10.1371/journal.pbio.0030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EA, Lin Y, Sarfraz Y, Quartermain D. Marked behavioral activation from inhibitory stimulation of locus coeruleus alpha1-adrenoceptors by a full agonist. Brain Res. 2009;1291:21–31. doi: 10.1016/j.brainres.2009.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Bongiovanni MJ, Tochen L, Shoemaker JM. Separable noradrenergic and dopaminergic regulation of prepulse inhibition in rats: implications for predictive validity and Tourette Syndrome. Psychopharmacology (Berl) 2006;186:246–254. doi: 10.1007/s00213-006-0374-7. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Marck BT, Palmiter RD, Matsumoto AM. Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine beta-hydroxylase. J Neurochem. 1998;70:2468–2476. doi: 10.1046/j.1471-4159.1998.70062468.x. [DOI] [PubMed] [Google Scholar]
- Vanderwolf CH. Hippocampal electrical activity and voluntary movement in the rat. Electroencephalogr Clin Neurophysiol. 1969;26:407–418. doi: 10.1016/0013-4694(69)90092-3. [DOI] [PubMed] [Google Scholar]
- Wang YM, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, Miller GW, Wightman RM, Caron MG. Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron. 1997;19:1285–1296. doi: 10.1016/s0896-6273(00)80419-5. [DOI] [PubMed] [Google Scholar]
- Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding JD, Feliciano C, Chen M, Adams JP, Luo J, Dudek SM, Weinberg RJ, Calakos N, Wetsel WC, Feng G. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448:894–900. doi: 10.1038/nature06104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winson J. Patterns of hippocampal theta rhythm in the freely moving rat. Electroencephalogr Clin Neurophysiol. 1974;36:291–301. doi: 10.1016/0013-4694(74)90171-0. [DOI] [PubMed] [Google Scholar]
- Xu F, Gainetdinov RR, Wetsel WC, Jones SR, Bohn LM, Miller GW, Wang YM, Caron MG. Mice lacking the norepinephrine transporter are supersensitive to psychostimulants. Nat Neurosci. 2000;3:465–471. doi: 10.1038/74839. [DOI] [PubMed] [Google Scholar]