

Abstract
The capacity of cells to correct their volume in response to hyposmotic stress via the efflux of inorganic and organic osmolytes is well documented. However, the ability of cell-surface receptors, in particular G-protein-coupled receptors (GPCRs), to regulate this homeostatic mechanism has received much less attention. Mechanisms that underlie the regulation of cell volume are of particular importance to cells in the central nervous system because of the physical restrictions of the skull and the adverse impact that even small increases in cell volume can have on their function. Increases in brain volume are seen in hyponatraemia, which can arise from a variety of aetiologies and is the most frequently diagnosed electrolyte disorder in clinical practice. In this review we summarize recent evidence that the activation of GPCRs facilitates the volume-dependent efflux of osmolytes from neural cells and permits them to more efficiently respond to small, physiologically relevant, reductions in osmolarity. The characteristics of receptor-regulated osmolyte efflux, the signalling pathways involved and the physiological significance of receptor activation are discussed. In addition, we propose that GPCRs may also regulate the re-uptake of osmolytes into neural cells, but that the influx of organic and inorganic osmolytes is differentially regulated. The ability of neural cells to closely regulate osmolyte homeostasis through receptor-mediated alterations in both efflux and influx mechanisms may explain, in part at least, why the brain selectively retains its complement of inorganic osmolytes during chronic hyponatraemia, whereas its organic osmolytes are depleted.
Stephen K. Fisher is a Professor in the Department of Pharmacology and a Research Professor in the Molecular and Behavioral Neuroscience Institute at the University of Michigan, Ann Arbor. He obtained his PhD degree from the University of Birmingham (UK) and postdoctoral training at the University of Michigan. He has a long-standing interest in signal transduction mechanisms in the nervous system and in particular, the role played by inositol lipids. His current research focus is that of the regulation of osmolyte fluxes in neural cells following activation of G-protein-coupled receptors and the signalling pathways involved.

Maintenance of cell volume is an ancient homeostatic mechanism necessary for the survival and proper function of the vast majority of cells. Alterations in cell volume can lead to a number of changes in cell function including excitability, cell-cycle progression, proliferation, apoptosis and metabolic regulation (Okada et al. 2009). However, regulation of cell volume is of particular significance to the central nervous system (CNS) because of the physical restrictions of the skull. Even small changes in brain cell volume can profoundly influence the spatial relationships between neurons, astrocytes and the extracellular space. A reduction in the latter, as occurs during brain cell swelling, will result in both an increased lateral diffusion and higher extracellular concentrations of neurotransmitters (Sykova, 2004; Thorne & Nicholson, 2006). Larger increases in brain cell volume can compress blood vessels leading to episodes of anoxia and ischaemia, and ultimately to a displacement of the brain parenchyma through the foramen magnum, leading to cardiac and respiratory arrest (Pasantes-Morales et al. 2002).
Brain cells can swell either as a result of changes in intracellular ion and water distribution (isotonic swelling, as occurs in stroke or traumatic head injury) or by a reduction in plasma osmolarity (hyposmotic swelling). The most common cause of hyposmotic swelling is hyponatraemia, which is defined as reduction in serum Na+ concentration from a normal value of 145 mequiv l−1 to 136 or below. Hyponatraemia is the most frequently encountered electrolyte disorder in clinical practice and can arise from a diverse array of aetiologies including congestive heart failure, syndrome of inappropriate secretion of vasopressin, liver cirrhosis, psychotic polydipsia or over-hydration, as may occur in athletes and military personnel (Bhardwaj, 2006; Lien & Shapiro, 2007). Although patients with mild hyponatraemia are often asymptomatic, reductions in plasma osmolarity of >10% can elicit symptoms of nausea and headache whereas seizures, coma, permanent brain damage and death can occur when plasma osmolarity is chronically reduced by >20%. The focus of this review is on the endogenous molecular mechanisms that restore brain volume when neural cells are subjected to hyposmotic stress. In particular, the emergence of a potentially important physiological role for cell surface receptors in this process, in both the efflux and re-uptake of osmolytes, is discussed.
Mechanisms for restoration of neural cell volume
During episodes of hyponatraemia, the reduction in plasma osmolarity results in an influx of water into the brain through the blood–brain barrier and into neural cells across their plasma membranes. However, the increases in brain water are less than expected from a perfect osmometer (Verbalis & Drutarosky, 1988). This reflects the presence of two homeostatic mechanisms that reduce the potential impact of water influx. The first is an increase in the flow of fluid from the interstitial space into the cerebrospinal fluid and from there to the systemic circulation. This phase accounts for the rapid loss of Na+ and Cl− ions from brain observed at the onset of hyponatraemia. The second, and more sustained mechanism, is an efflux of osmotically active solutes from the cells, primarily inorganic ions such as K+ and Cl− and small organic molecules, such as taurine, glutamate or myo-inositol. This in turn leads to the exit of osmotically obligated water and reduces cellular swelling. The mechanism whereby the increase in cell volume is detected by a hypothetical ‘volume sensor’, and the signal transduction events that elicit volume correction, remain largely unknown, with the exception that a tyrosine kinase has frequently been implicated.
Although organic osmolytes potentially contribute less than their inorganic counterparts to cell volume adaptation (35 and 65%, respectively), organic osmolytes are highly enriched in the CNS and their utilization minimizes changes in membrane potential associated with the efflux of inorganic osmolytes such as K+ or Cl−. However, it should be remembered that several of the quantitatively major organic osmolytes are ‘neuroactive’ and can activate their respective receptors on nearby neurons and glia. Thus, as discussed later, their release under conditions of volume correction is not without neurobiological consequences. The normalization of cell volume that occurs as a result of osmolyte loss is referred to as regulatory volume decrease (RVD). Volume-dependent osmolyte efflux is mediated via a variety of anion/cation channels, including K+ channels (both Ca2+-dependent and -independent), the K+ −Cl− co-transporter (KCC) and a putative volume-sensitive organic osmolyte and anion channel (VSOAC; Fig. 1). The latter is yet to be characterized at a molecular level, but is thought to be primarily a swelling-activated Cl− channel that also mediates the efflux of organic osmolytes (Okada, 2006; Kimelberg et al. 2006). Swelling-activated efflux of osmolytes has been monitored in a variety of neural preparations including primary cultures of neurons and astrocytes, neurotumour cell lines, nerve-ending preparations and brain slices (for review, see Fisher et al. 2008).
Figure 1. General schematic representation of events leading to swelling-activated (basal)- and GPCR-mediated osmolyte efflux.
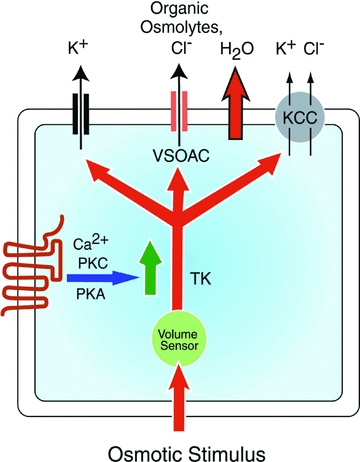
A reduction in osmolarity results in an influx of water and an increase in cell volume, which in turn triggers a hypothetical ‘volume sensor’. It has been proposed that the latter may represent changes in macromolecular crowding and/or changes in the cytoskeleton. Activation of the volume sensor elicits a tyrosine kinase (TK)-dependent activation of K+ channels (both Ca2+-dependent and -independent), VSOAC and, on occasions, the KCC transporter. This results in the efflux of K+, Cl−, organic osmolytes and osmotically obligated water. The latter results in a restoration of cell volume or ‘regulatory volume decrease’. Volume-dependent efflux of inorganic and organic osmolytes can also be significantly increased by activation of GPCRs, the magnitude of which is often dependent on Ca2+ availability and PKC activity. Note that GPCR activation of osmolyte release is strongly dependent on prior activation of the volume sensor and little or no increase is seen under isotonic or hypertonic conditions. Pharmacological evidence indicates that the same or similar channels mediate osmolyte efflux under basal- and GPCR-stimulated conditions.
A role for cell-surface receptors in the regulation of volume-sensitive osmolyte efflux
Although volume regulation has previously been considered to be an intrinsic property of cells, it is now evident that many extracellular agonists, acting via cell-surface receptors, can profoundly influence this process (for reviews see Fisher et al. 2008; Franco et al. 2008; Vasquez-Juarez et al. 2008). An indication that GPCRs could potentially be involved in osmoregulation in neural cells was obtained from experiments in which agents that mimic the activation of certain categories of cell-surface receptors, for example by increasing the concentration of cytosolic Ca2+ or activities of protein kinase C (PKC) or protein kinase A (PKA), were observed to potentiate volume-dependent osmolyte release from cells. However, Bender et al. (1993) provided the first direct demonstration of the involvement of G-protein-coupled receptors (GPCRs) in volume control when they reported that RVD in cultured astrocytes exposed to hypotonic media was accelerated by the addition of endothelin or noradrenaline (norepinephrine). However, only in the last few years has the potential contribution of cell-surface receptors to the regulation of osmolyte fluxes in the CNS been systematically examined. A significant number of pharmacologically distinct receptors have now been identified to regulate osmolyte fluxes and volume control in a variety of different neural preparations (for summary, see Table 1). Although a diverse array of receptors has been identified, a number of common characteristics are apparent. First, with some exceptions (e.g. AMPA, vascular endothelial growth factor and erythropoeitin receptors), most documented examples of receptor regulation of osmolyte fluxes and/or volume control involve the activation of GPCRs and, where examined, the effects are mediated via pharmacologically distinct receptor subtypes. Under hyposmotic conditions, receptor activation almost invariably leads to a facilitation of osmolyte efflux and, when monitored, an enhancement of RVD (see Ramos-Mandujano et al. 2007). However, it remains possible that a category of GPCRs may also serve to negatively regulate osmolyte efflux and volume correction, as has been observed for the adenosine A2B receptor in pituicytes (Pierson et al. 2007). The GPCRs coupled to an increase in osmolyte efflux typically respond to neurotransmitters or neuromodulators (e.g. neuropeptides, cholinergic or adrenergic agonists, ATP, thrombin or lysophospholipids) at nanmolar or micromolar concentrations, i.e. well within the concentration range found in vivo.
Table 1.
Receptor-mediated regulation of osmolyte fluxes or cell volume under hyposmotic conditions
| Receptor | Subtype | Signalling pathways | Cell type | Parameter | Reference |
|---|---|---|---|---|---|
| Adenosine | A1 | PKA | Retina | Cell volume | Uckermann et al. 2006 |
| A1 | PKA/PI3K/Akt | Retina | Cell volume | Wurm et al. 2008 | |
| A2B | PKA | Pituicytes | Taurine flux | Pierson et al. 2007 | |
| Purinergic | P2Y | ND | Retina | Cell volume | Uckermann et al. 2006; Wurm et al. 2008 |
| P2Y | Ca2+/PKC/CaM/CaMKII | Cortical astrocytes | d-Asp flux | Mongin & Kimelberg, 2002, 2005 | |
| P2Y | ND | Cortical astrocytes | d-Asp/Cl− flux | Abdullaev et al. 2006 | |
| P2Y | ND | Cortical astrocytes | Cl− flux | Darby et al. 2003 | |
| ND | Ca2+ | Pituicytes | Taurine flux | Rosso et al. 2004 | |
| ND | ND | Hippocampal neurons | Taurine/Cl− flux | Li & Olson, 2004 | |
| ND | ND | Substantia nigra | Taurine flux | Morales et al. 2007 | |
| Bradykinin | ND | Ca2+ | Pituicytes | Taurine flux | Rosso et al. 2004 |
| Endothelin | ND | PPI hydrolysis | Cortical astrocytes | Cell volume | Bender et al. 1993 |
| Erythropoietin | ND | Jak-2/ERK1/2 | Retina | Cell volume | Krugel et al. 2010 |
| Glutamate | AMPA | ND | Substantia nigra | Taurine flux | Morales et al. 2007 |
| mGluR | ND | Retina | Cell volume | Uckermann et al. 2006; Wurm et al. 2008 | |
| LPA | ND | Ca2+/PKC | SH-SY5Y | Taurine flux | Heacock et al. 2006 |
| ND | ND | SH-SY5Y | K+ flux | Foster et al. 2008 | |
| NA | ND | PPI hydrolysis | Cortical astrocytes | Cell volume | Bender et al. 1993 |
| β-AR | PKA | Cortical astrocytes | Taurine flux | Moran et al. 2001 | |
| mAChR | M3 | PKC/Ca2+/Tyr kinases | SH-SY5Y | d-Asp/taurine flux | Heacock et al. 2004 |
| M3 | Ca2+/PKC | SH-SY5Y | Inositol flux | Loveday et al. 2003 | |
| M3 | Ca2+/PKC | SH-SY5Y | Cl− flux | Cheema et al. 2007 | |
| M3 | Ca2+/PKC | SH-SY5Y | K+ flux | Foster et al. 2008 | |
| S1P | ND | Ca2+/PKC | SH-SY5Y | Taurine flux | Heacock et al. 2006 |
| ND | ND | SH-SY5Y | K+ flux | Foster et al. 2008 | |
| PAR | PAR-1 | ND | Cortical astrocytes | Taurine flux | Cheema et al. 2005 |
| PAR-1 | Ca2+/PKC/CaM/PI3K | Cortical astrocytes | d-Asp flux | Ramos-Mandujano et al. 2007 | |
| ND | PPI hydrolysis | Cortical astrocytes | Cell volume | Bender et al. 1993 | |
| PAR-1 | Ca2+/PKC | SH-SY5Y | Taurine flux | Cheema et al. 2007 | |
| PAR-1 | ND | SH-SY5Y | Cl− flux | Cheema et al. 2007 | |
| PAR-1 | ND | SH-SY5Y | K+ flux | Foster et al. 2008 | |
| PAR-1 | Ca2+/PKC | 1321N1 astrocytoma | Taurine flux | Cheema et al. 2005 | |
| PAR-1 | Ca2+/Rho GTPase | 1321N1 astrocytoma | ATP flux | Blum et al. 2010 | |
| Vasopressin | V1A | Ca2+ | Pituicytes | Taurine flux | Rosso et al. 2004 |
| V1A | ND | Neocortex | Cell volume | Niermann et al. 2001 | |
| V1A | ND | Cortical astrocytes | Cell volume | Sarfaraz & Fraser, 1999 | |
| VEGF | VEGFR-2 | Ca2+/PLC/PKC/src | Retina | Cell volume | Wurm et al. 2008 |
Abbreviations: β-AR, β-adrenergic-receptor; d-Asp, d-aspartate; CaM, calmodulin; CaMKII, calmodulin-dependent protein kinase II; mGluR, metabotropic glutmate receptor; NA, noradrenaline; ND, not determined; PLC, phospholipase C; PPI, phosphoinositide; VEGF, vascular endothelial growth factor; VEGFR-2, vascular endothelial growth factor receptor 2. Jak-2, Janus kinase-2 and ERK1/2, Extracellular signal regulated kinases 1 or 2.
A second characteristic of receptor-stimulated osmolyte efflux is that the release of organic osmolytes and Cl− can be blocked by inclusion of known inhibitors of VSOAC, such as DCPIB (4-[(2-butyly-6,7- dichloro-2-cyclopentenyl-2,3-dihydo-1-oxo-1H-inden-5- yl)oxy] butanoic acid), DDF (1,9-dideoxyforskolin) or NPPB, (4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid). This is the same pharmacological profile as is observed under basal (swelling-activated) conditions and indicates that the same or similar channels mediate osmolyte release under both conditions. A third characteristic is that osmolyte efflux is only significantly increased following receptor activation under hyposmotic conditions, whereas little or no increase is observed under isotonic or hypertonic conditions (see Fig. 2A). From this observation it can be concluded that GPCRs serve to regulate, rather than initiate, the activity of channels that are involved in osmolyte release. Fourth, as initially described by Mongin & Kimelberg (2002) for ATP-stimulated release of d-aspartate from primary cultures of astrocytes, receptor activation appears to lower the threshold osmolarity at which osmolyte release occurs. An additional example of this phenomenon is shown in Fig. 2 for human SH-SY5Y neuroblastoma cells, a model neuronal cell line. For these cells, no measurable increase in the basal (swelling-activated) release of either taurine or Rb+ (used as a marker for K+) over that monitored under isotonic conditions is observed until the osmolarity of the medium is reduced by ∼30%. Similar large, non-physiologically relevant, reductions in osmolarity have frequently been required to reliably monitor osmolyte efflux from other neural preparations under basal conditions. In contrast, the activation of muscarinic cholinergic receptors (mAChRs) on SH-SY5Y cells permits the release of osmolytes in a volume-dependent manner at relatively modest reductions in osmolarity (>6% for taurine and >9% for Rb+). Activation of other GPCRs present on SH-SY5Y cells such as sphingosine 1-phosphate (S1P), lysophosphatidic acid (LPA) and protease-activated-1 (PAR-1) receptors also reduced the threshold osmolarity at which taurine was released (Heacock et al. 2006; Cheema et al. 2007). The ability of GPCRs to increase not only the magnitude of osmolyte release but also the threshold osmolarity at which the release occurs, may provide a physiological mechanism whereby neural cells are able to respond to reductions in osmolarity in the range that is encountered in hyponatraemia.
Figure 2. Basal- and oxotremorine-M (Oxo-M)-stimulated efflux of taurine and Rb+ as a function of osmolarity.
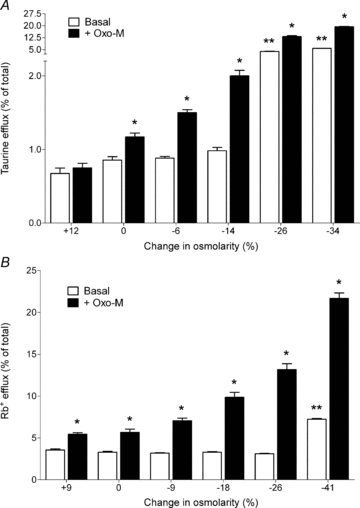
SH-SY5Y neuroblastoma cells, prelabelled overnight with either [14C]taurine or 86Rb+, were incubated in isotonic, hypertonic or hypotonic media for either 20 min (taurine) or 5 min (Rb+) in the absence (open bars) or presence (filled bars) of 100 μm Oxo-M, a muscarinic cholinergic agonist. Results are expressed as taurine or Rb+ efflux (percentage of total soluble radioactivity) and are the means ±s.e.m. for 4–13 experiments. A, taurine efflux; B, Rb+ efflux. *Different from basal efflux, P < 0.05 by paired Student's t test. **Different from basal efflux monitored under isotonic conditions, P < 0.01 by one-way ANOVA, followed by Dunnett's multiple comparison test. Data are taken from Heacock et al. (2004) and Foster et al. (2008).
Hyposmolarity has also been reported to enhance the release of several potential ligands for GPCRs, thereby increasing the likelihood of their activation (Tuz et al. 2004; Tuz & Pasantes-Morales, 2005; Blum et al. 2010). One such ligand is ATP, which is released at an increased rate from cells in response to hyposmotic stress (Corriden & Insel, 2010). ATP then contributes to RVD by triggering the efflux of both inorganic and organic osmolytes, an effect mediated via purinergic receptors. A role for ATP (and also its breakdown product, ADP) in osmolyte homeostasis has been frequently observed for primary cultures of astrocytes and neurons, neurotumour cells and the retina (Table 1). The importance of purinergic mediation of volume control in the retina is indicated by the fact that retinal glial cells do not swell in response to hyposmolarity unless the P2Y receptor is pharmacologically blocked. Thus in response to hyposmolarity, glia (and possibly nearby neurons) are thought to release ATP, which, in an autocrine or paracrine fashion, serves to minimize cell swelling (Wurm et al. 2010). In contrast, retinal glial cells obtained from P2Y knockout mice exhibit swelling in response to hyposmolarity, but this can be countered by administration of ATP. At least in retinal glial cells, it appears that the final mediator of volume correction is not ATP itself, but adenosine, derived from the breakdown of ATP. Adenosine is proposed to facilitate K+ and Cl− efflux via activation of adenosine A1 receptors. Consistent with this mechanism is the observation that volume regulation is impaired in glial cells deficient in adenosine A1 receptors (Wurm et al. 2010). In addition to its efflux under hyposmotic conditions, the release of ATP can be further increased following activation of the PAR-1 receptor, mediated by thrombin (Blum et al. 2010). Since activation of the PAR-1 receptor itself has previously been documented to facilitate osmolyte release and volume correction (Cheema et al. 2005; Ramos-Mandujano et al. 2007; Blum et al. 2010), it is conceivable that a release of ATP may contribute to some of the effects on osmolyte efflux elicited by activation of other GPCRs. However, not all cells possess P2Y receptors (e.g. SH-SY5Y neuroblastoma or 1321N1 astrocytoma cells) and thus, an obligatory role for ATP release in regulating osmolyte efflux following GPCR activation appears unlikely.
Signal transduction pathways that mediate GPCR-mediated stimulation of osmolyte efflux
Many of the GPCRs that have thus far been identified to facilitate osmolyte efflux are also those known to regulate Ca2+ homeostasis and the activation of PKC. Examples of receptors in this category are the P2Y purinergic, M3 mAChR, PAR-1, LPA, S1P and V1A vasopressin receptors. Accordingly, attenuation of the ability of GPCRs to regulate osmolyte release or modulate volume control is consistently observed following depletion of intra/extracellular Ca2+ or inhibition of PKC. If intracellular Ca2+ is depleted and PKC inhibited, receptor regulation of osmolyte efflux is essentially abolished. It should be stressed that the Ca2+- and PKC-dependence of receptor-stimulated efflux is in marked contrast to that observed for swelling-activated efflux, which appears to be relatively independent of both parameters. These observations are consistent with the proposal that distinct up-steam mechanisms underlie basal- and receptor-mediated osmolyte efflux, even though the same or similar channels are used for the exit of osmolytes from cells under both conditions (Mongin & Kimelberg, 2005; Heacock et al. 2006). Although the evidence for a requirement for Ca2+ in osmoregulation is compelling, the source of this Ca2+ (extra- or intracellular) appears to be both receptor- and cell-type-specific (Cheema et al. 2005; Mongin & Kimelberg, 2005, Heacock et al. 2006; Wurm et al. 2010). Moreover, no simple relationship exists between the magnitude of increase in Ca2+ concentration elicited by activation of individual GPCRs in a specific cell type and the extent of osmolyte release (Heacock et al. 2006). An additional complexity is that the release of inorganic osmolytes (K+, Cl−) appears less sensitive to the removal of Ca2+ than that of organic osmolytes, a result that suggests that distinct biochemical requirements may exist for the two classes of osmolytes within the same cell (Cheema et al. 2007; Foster et al. 2008). It is also evident that osmolyte release can be facilitated by Ca2+-mobilizing receptors that operate via either phospholipase C-dependent or -independent mechanisms (Heacock et al. 2006). The down-stream effectors for Ca2+ have yet to be identified but may include calmodulin (Mongin & Kimelberg, 2005; Ramos-Mandujano et al. 2007) and/or PKC. There is also evidence for the involvement of two conventional (Ca2+- and diacylglycerol-dependent) isoforms of PKC (α and β) in ATP-mediated d-aspartate release from astrocytes (Rudkouskaya et al. 2008).
Other signalling pathways have also been implicated in receptor-mediated osmolyte efflux. These include phosphatidylinositol 3-kinase (PI3K; Ramos-Mandujano et al. 2007), tyrosine kinases (Heacock et al. 2004; Mongin & Kimelberg, 2005) and protein kinase A. Evidence for involvement of the latter pathway has been obtained for β-adrenergic stimulation of taurine efflux from cortical astrocytes (Moran et al. 2001) and for adenosine A1 receptor-mediated volume changes in retinal glial cells (Uckermann et al. 2006; Wurm et al. 2010). In summary, it appears likely that cell volume regulation by GPCRs involves the activation of several distinct signalling pathways.
Is osmolyte influx an additional mechanism whereby GPCRs can regulate osmolyte homeostasis?
When monitored under acute conditions in vitro, the rate of efflux of inorganic osmolytes from neural cells under both basal- and receptor-stimulated conditions either exceeds, or is comparable to, that observed for organic osmolytes. In contrast, chronic hyponatraemia in vivo results in a disproportionately greater loss of organic osmolytes than of inorganic osmolytes from the brain (Melton et al. 1987; Pasantes-Morales et al. 2002; Massieu et al. 2004). Similarly, if primary astrocytes are chronically cultured in a hyposmotic medium, organic osmolytes such as taurine or aspartate are lost from the cells whereas K+ is retained (Olson, 1999). One potential explanation for this discrepancy is that the influx of inorganic and organic osmolytes, like that of their efflux, is also volume dependent, but that the two classes of osmolyte are differentially regulated by hyposmolarity. In keeping with this possibility, previous studies have indicated that hyposmotic swelling of astrocytes results in a stimulation of K+ influx (Mongin et al. 1994, 1996) whereas, in contrast, a down-regulation of the taurine transporter (TauT) in hippocampal neurons has been reported in response to a reduced osmolarity (Olson & Martinho, 2006). However, whether the influx of osmolytes, like that of their efflux, is also subject to receptor regulation in an osmosensitive manner, remains to be determined. In a recent series of studies, we have systematically examined this possibility by monitoring the influx of three distinct osmolytes, namely K+, taurine and glutamate in SH-SY5Y neuroblastoma. Agonist occupancy of several GPCRs (mAChR, LPA receptor, PAR-1 and S1P receptor) in this cell line under isotonic conditions resulted in an increase in the uptake of K+ (monitored as 86Rb+ flux) that was further enhanced when the osmolarity was reduced (Foster et al. 2008). A similar increase in K+ influx was also obtained following activation of PAR-1, LPA and S1P receptors in primary cultures of astrocytes, cells that are known to actively regulate K+ concentrations in the extracellular space. Thus activation of GPCRs appears to facilitate both the efflux and influx of K+ in an osmosensitive manner. However, whereas K+ channels and the KCC transporter mediate the efflux of K+, influx occurs via the Na+ −K+ −2Cl− co-transporter (NKCC), ouabain-sensitive Na+–K+-ATPase and to a lesser extent KCC (Fig. 3A). A physiological role for K+ uptake under hyposmotic conditions was indicated by the observation that activation of mAChRs under either isotonic or mildly hypotonic conditions did not result in a change in the intracellular concentration of K+, unless the influx of K+ was concurrently prevented by inclusion of ouabain and furosemide (frusemide) (an inhibitor of NKCC). This result suggests that, under normal conditions, the agonist stimulation of K+ efflux from cells is countered by an equivalent increase in K+ influx. Only under more pronounced hypotonic conditions (>30% reduction in osmolarity) were reductions in intracellular K+ concentration observed, due to the predominance of the efflux pathway (Foster et al. 2008).
Figure 3. Diagrammatic representation of the ability of GPCRs present in SH-SY5Y cells to regulate the efflux and uptake of K+ (A), taurine (B) or glutamate (C) under isotonic (Iso) or hypotonic (Hypo) conditions.
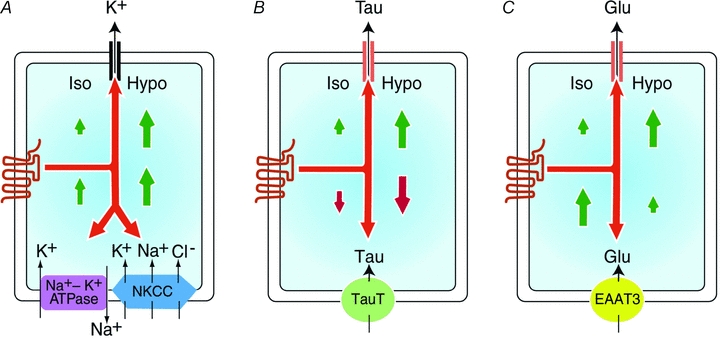
Although only a limited efflux of osmolytes is observed following the activation of GPCRs under isotonic conditions, the efflux of all three is markedly increased by hypotonicity (as indicated by the relative size of the arrows). The efflux of K+ is mediated primarily by K+ channel(s) whereas taurine (Tau) and glutamate (Glu) are released via the VSOAC. K+ uptake, which is principally mediated by Na+ −K+-ATPase and the NKCC transporters in this cell line, is facilitated by GPCR activation under conditions of isotonicity, and further enhanced by hyposmolarity. In contrast, under isotonic conditions, GPCR activation results in an inhibition of taurine uptake, which is mediated via the taurine transporter (TauT). This inhibition of TauT is further enhanced by hyposmolarity. Glutamate uptake, which is mediated by EAAT3, is markedly increased by GPCR activation under isotonic conditions. However, receptor-mediated stimulation of Glu uptake is progressively inhibited as osmolarity is reduced.
In contrast to the results obtained for K+ influx, activation of GPCRs in SH-SY5Y cells (mAChR, LPA, S1P or PAR-1 receptors) results in a reduction in the rate of taurine transport under isotonic conditions, an effect that is further enhanced by hyposmolarity (Fig. 3B). Similar results were obtained for primary cultures of astrocytes. The receptor-mediated reduction in taurine transport in SH-SY5Y cells was Ca2+ dependent and resulted from a reduction in the Vmax for transport while the Km value for taurine uptake remained unchanged (Foster et al. 2009). The loss of taurine from cells under hypotonic conditions thus reflects not only an increased rate of efflux through VSOAC, but also its reduced re-uptake from the extracellular space. These observations could explain, in part at least, the dramatic loss of taurine observed from the brain during chronic hyponatraemia (Massieu et al. 2004). As TauT-mediated uptake involves the co-transport of two Na+ ions and one Cl− ion, the inhibition of taurine re-uptake will also have the net effect of preventing the uptake of four osmolyte equivalents per cycle. Taurine is considered to be a relatively inert organic osmolyte and a significant increase in its extracellular concentration might be expected to have only minimal cellular consequences. However, a similar increase in the concentration of extracellular glutamate, another quantitatively important organic osmolyte within the CNS, would be potentially deleterious, due to the known ability of glutamate to contribute to the rate of propagation of spreading depression, a slow wave of astrocytic and neuronal depolarization that leads to synaptic depression (Basarsky et al. 1999). Higher concentrations of extracellular glutamate can also elicit excitotoxic death in neurons. Accordingly, blockade of glutamate release by pharmacological inhibition of VSOAC activity can significantly reduce the infarct size following focal cerebral ischaemia (Zhang et al. 2008). To determine whether similar or distinct mechanisms exist for glutamate and taurine homeostasis, glutamate uptake into SH-SY5Y cells (monitored as d-[3H]aspartate influx) was measured as a function of osmolarity and receptor activation. Glutamate uptake was substantially increased under isotonic conditions by activation of mAChRs and to a lesser extent by endothelin or LPA receptors (Foster et al. 2010). The mAChR-mediated enhancement of glutamate uptake, which required the presence of Ca2+ and activation of PKC and PI3K, was mediated by a cellular redistribution to the plasma membrane of the excitatory amino acid transporter 3 (EAAT3), a neuron-specific transporter. However, the ability of the mAChR to facilitate glutamate uptake was significantly attenuated by hyposmolarity (>45% following a 21% reduction in osmolarity). As observed for the taurine transporter, the osmosensitive attenuation of glutamate uptake reflects a reduction in the Vmax for transport without a change in Km and was accompanied by an altered trafficking pattern of the transporter (Fig. 3C; Foster et al. 2010). Endothelin-1-stimulated increases in glutamate uptake into C6 glioma cells (mediated by EAAT3) were similarly attenuated by hyposmolarity indicating that receptor-mediated regulation of glutamate transport may be a general, rather than cell-specific, phenomenon. The regulation of EAAT3-mediated glutamate uptake could potentially have physiological consequences in the intact brain. Thus under conditions of isotonicity or mild hypotonicity, activation of GPCRs may permit EAAT3-containing neurons to maintain relatively low concentrations of extracellular glutamate. However, when osmolarity is further reduced, a combination of glutamate efflux, along with a progressive inhibition of glutamate re-uptake, could result in an increased accumulation of extracellular glutamate.
In addition to glutamate, other quantitatively major organic osmolytes such as aspartate, glycine or GABA are neuroactive and thus their release from osmotically swollen cells, which is enhanced following the activation of GPCRs, can be expected to impact nearby neurons and glia. Even taurine, which is generally considered to be relatively inert, can act as a ligand at glycine, GABAA, GABAB and N-methyl-d-aspartate receptors. Thus the ability of GPCRs to facilitate the release of organic osmolytes constitutes a ‘double-edged sword’ in that both the beneficial effects of volume reduction and the potentially harmful pathophysiological effects of higher extracellular organic osmolyte concentrations need to be considered. An ability of neural cells to selectively retain or release individual organic osmolytes via differential regulation of their re-uptake might provide a mechanism whereby selectivity of organic osmolyte release is attained. However, the full physiological significance of tonicity-mediated changes in receptor-stimulated osmolyte uptake remains to be directly evaluated in vivo.
Summary and conclusions
Regulation of osmolyte fluxes and cell volume following GPCR activation has been documented in both neural and non-neural cells. However, this regulatory mechanism is likely to be of particular physiological significance to the CNS given the prevalence of GPCRs (∼300 identified) and the vulnerability of the brain to osmotic disturbances. The identification of GPCRs that not only regulate the efflux of osmolytes, but also their re-uptake, may provide neural cells with the ability to respond to small, physiologically relevant, reductions in osmolarity and to selectively retain or relinquish individual osmolytes.
Acknowledgments
This work was supported by NIH Grants NS23831 (S.K.F.), NS 034709 (R.F.K.) and GM007767 (D.J.F.)
Glossary
Abbreviations
- CNS
central nervous system
- EAAT3
excitatory amino acid transporter 3
- GABA
γ-amino butyric acid
- GPCR
G-protein-coupled receptor
- KCC
K+ −Cl− cotransporter
- LPA
lysophosphatidic acid
- mAChR
muscarinic cholinergic receptor
- NKCC
Na+ −K+ −2Cl− cotransporter
- PAR-1
protease-activated receptor-1
- PKA
cyclic AMP-dependent protein kinase A
- PKC
protein kinase C
- PI3K
phosphatidylinositol 3-phosphate
- RVD
regulatory volume decrease
- S1P
sphingosine 1-phosphate
- TauT
taurine transporter
- VSOAC
volume-sensitive organic osmolyte and anion channel
References
- Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK, Mongin AA. Pharmacological comparison of swelling-activated excitatory amino acid release and Cl− currents in cultured rat astrocytes. J Physiol. 2006;572:677–689. doi: 10.1113/jphysiol.2005.103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basarsky TA, Feighan D, MacVicar BA. Glutamate release through volume-activated channels during spreading depression. J Neurosci. 1999;19:6439–6445. doi: 10.1523/JNEUROSCI.19-15-06439.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AS, Neary JT, Norenberg MD. Role of phosphoinositide hydrolysis in astrocyte volume regulation. J Neurochem. 1993;61:1506–1514. doi: 10.1111/j.1471-4159.1993.tb13646.x. [DOI] [PubMed] [Google Scholar]
- Bhardwaj A. Neurological impact of vasopressin dysregulation and hyponatremia. Ann Neurol. 2006;59:229–236. doi: 10.1002/ana.20788. [DOI] [PubMed] [Google Scholar]
- Blum AE, Walsh BC, Dubyak GR. Extracellular osmolarity modulates G protein-coupled receptordependent ATP release from 1321N1 astrocytoma cells. Am J Physiol Cell Physiol. 2010;298:C386–C396. doi: 10.1152/ajpcell.00430.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema TA, Pettigrew VA, Fisher SK. Receptor regulation of the volume-sensitive efflux of taurine and iodide from human SH-SY5Y neuroblastoma cells: differential requirements for Ca2+ and protein kinase C. J Pharmacol Exp Ther. 2007;320:1068–1077. doi: 10.1124/jpet.106.115741. [DOI] [PubMed] [Google Scholar]
- Cheema TA, Ward CE, Fisher SK. Subnanomolar concentrations of thrombin enhance the volume-sensitive efflux of taurine from human 1321N1 astrocytoma cells. J Pharmacol Exp Ther. 2005;315:755–763. doi: 10.1124/jpet.105.090787. [DOI] [PubMed] [Google Scholar]
- Corriden R, Insel PA. Basal release of ATP: an autocrine-paracrine mechanism for cell regulation. Sci Signal. 2010;3:re1. doi: 10.1126/scisignal.3104re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby M, Kuzmiski JB, Panenka W, Feighan D, MacVicar BA. ATP released from astrocytes during swelling activates chloride channels. J Neurophysiol. 2003;89:1870–1877. doi: 10.1152/jn.00510.2002. [DOI] [PubMed] [Google Scholar]
- Fisher SK, Cheema TA, Foster DJ, Heacock AM. Volume-dependent osmolyte efflux from neural tissues: regulation by G-protein-coupled receptors. J Neurochem. 2008;106:1998–2014. doi: 10.1111/j.1471-4159.2008.05510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DJ, Heacock AM, Fisher SK. Muscarinic receptor stimulation of d-aspartate uptake into human SH-SY5Y neuroblastoma cells is attenuated by hypoosmolarity. J Pharmacol Exp Ther. 2010;333:297–309. doi: 10.1124/jpet.109.164277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DJ, Heacock AM, Keep RF, Fisher SK. Activation of muscarinic cholinergic receptors on human SH-SY5Y neuroblastoma cells enhances both the influx and efflux of K+ under conditions of hypo-osmolarity. J Pharmacol Exp Ther. 2008;325:457–465. doi: 10.1124/jpet.107.135475. [DOI] [PubMed] [Google Scholar]
- Foster DJ, Vitvitsky VM, Banerjee R, Heacock AM, Fisher SK. Muscarinic receptor regulation of osmosensitive taurine transport in human SH-SY5Y neuroblastoma cells. J Neurochem. 2009;108:437–449. doi: 10.1111/j.1471-4159.2008.05773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Panayiotidis MI, de la Paz LD. Autocrine signaling involved in cell volume regulation: the role of released neurotransmitters and plasma membrane receptors. J Cell Physiol. 2008;216:14–28. doi: 10.1002/jcp.21406. [DOI] [PubMed] [Google Scholar]
- Heacock AM, Dodd MS, Fisher SK. Regulation of volume-sensitive osmolyte efflux from human SH-SY5Y neuroblastoma cells following activation of lysophospholipid receptors. J Pharmacol Exp Ther. 2006;317:685–693. doi: 10.1124/jpet.105.098467. [DOI] [PubMed] [Google Scholar]
- Heacock AM, Kerley D, Gurda GT, VanTroostenberghe AT, Fisher SK. Potentiation of the osmosensitive release of taurine and d-aspartate from SH-SY5Y neuroblastoma cells after activation of M3 muscarinic cholinergic receptors. J Pharmacol Exp Ther. 2004;311:1097–1104. doi: 10.1124/jpet.104.072553. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Macvicar BA, Sontheimer H. Anion channels in astrocytes: biophysics, pharmacology and function. Glia. 2006;54:747–757. doi: 10.1002/glia.20423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krugel K, Wurm A, Linnertz R, Pannicke T, Wiedemann P, Reichenbach A, Bringmann A. Erythropoietin inhibits osmotic swelling of retinal glial cells by Janus kinase- and extracellular signal-regulated kinases1/2-mediated release of vascular endothelial growth factor. Neuroscience. 2010;165:1147–1158. doi: 10.1016/j.neuroscience.2009.11.035. [DOI] [PubMed] [Google Scholar]
- Li G, Olson JE. Extracellular ATP activates chloride and taurine conductances in cultured hippocampal neurons. Neurochem Res. 2004;29:239–246. doi: 10.1023/b:nere.0000010452.26022.a7. [DOI] [PubMed] [Google Scholar]
- Lien YH, Shapiro JI. Hyponatremia: clinical diagnosis and management. Am J Med. 2007;120:653–658. doi: 10.1016/j.amjmed.2006.09.031. [DOI] [PubMed] [Google Scholar]
- Loveday D, Heacock AM, Fisher SK. Activation of muscarinic cholinergic receptors enhances the volume-sensitive efflux of myo-inositol from SH-SY5Y neuroblastoma cells. J Neurochem. 2003;87:476–486. doi: 10.1046/j.1471-4159.2003.02021.x. [DOI] [PubMed] [Google Scholar]
- Massieu L, Montiel T, Robles G, Quesada O. Brain amino acids during hyponatremia in vivo: clinical observations and experimental studies. Neurochem Res. 2004;29:73–81. doi: 10.1023/b:nere.0000010435.06586.e2. [DOI] [PubMed] [Google Scholar]
- Melton JE, Patlak CS, Pettigrew KD, Cserr HF. Volume regulatory loss of Na, Cl, and K from rat brain during acute hyponatremia. Am J Physiol Renal Physiol. 1987;252:F661–F669. doi: 10.1152/ajprenal.1987.252.4.F661. [DOI] [PubMed] [Google Scholar]
- Mongin AA, Aksentsev SL, Orlov SN, Kvacheva ZB, Mezen NI, Fedulov AS, Konev SV. Swelling-induced activation of Na+,K+,2Cl− cotransport in C6 glioma cells: kinetic properties and intracellular signalling mechanisms. Biochim Biophys Acta. 1996;1285:229–236. doi: 10.1016/s0005-2736(96)00165-4. [DOI] [PubMed] [Google Scholar]
- Mongin AA, Aksentsev SL, Orlov SN, Slepko NG, Kozlova MV, Maximov GV, Konev SV. Swelling-induced K+ influx in cultured primary astrocytes. Brain Res. 1994;655:110–114. doi: 10.1016/0006-8993(94)91603-9. [DOI] [PubMed] [Google Scholar]
- Mongin AA, Kimelberg HK. ATP potently modulates anion channel-mediated excitatory amino acid release from cultured astrocytes. Am J Physiol Cell Physiol. 2002;283:C569–C578. doi: 10.1152/ajpcell.00438.2001. [DOI] [PubMed] [Google Scholar]
- Mongin AA, Kimelberg HK. ATP regulates anion channel-mediated organic osmolyte release from cultured rat astrocytes via multiple Ca2+-sensitive mechanisms. Am J Physiol Cell Physiol. 2005;288:C204–C213. doi: 10.1152/ajpcell.00330.2004. [DOI] [PubMed] [Google Scholar]
- Morales I, Dopico JG, Sabate M, Gonzalez-Hernandez T, Rodriguez M. Substantia nigra osmoregulation: taurine and ATP involvement. Am J Physiol Cell Physiol. 2007;292:C1934–C1941. doi: 10.1152/ajpcell.00593.2006. [DOI] [PubMed] [Google Scholar]
- Moran J, Morales-Mulia M, Pasantes-Morales H. Reduction of phospholemman expression decreases osmosensitive taurine efflux in astrocytes. Biochim Biophys Acta. 2001;1538:313–320. doi: 10.1016/s0167-4889(01)00082-9. [DOI] [PubMed] [Google Scholar]
- Niermann H, Amiry-Moghaddam M, Holthoff K, Witte OW, Ottersen OP. A novel role of vasopressin in the brain: modulation of activity-dependent water flux in the neocortex. J Neurosci. 2001;21:3045–3051. doi: 10.1523/JNEUROSCI.21-09-03045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y. Cell volume-sensitive chloride channels: phenotypic properties and molecular identity. Contrib Nephrol. 2006;152:9–24. doi: 10.1159/000096285. [DOI] [PubMed] [Google Scholar]
- Okada Y, Sato K, Numata T. Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J Physiol. 2009;15:2141–2149. doi: 10.1113/jphysiol.2008.165076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JE. Osmolyte contents of cultured astrocytes grown in hypoosmotic medium. Biochim Biophys Acta. 1999;1453:175–179. doi: 10.1016/s0925-4439(98)00090-8. [DOI] [PubMed] [Google Scholar]
- Olson JE, Martinho E. Regulation of taurine transport in rat hippocampal neurons by hypo-osmotic swelling. J Neurochem. 2006;96:1375–1389. doi: 10.1111/j.1471-4159.2006.03652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasantes-Morales H, Franco R, Ordaz B, Ochoa LD. Mechanisms counteracting swelling in brain cells during hyponatremia. Arch Med Res. 2002;33:237–244. doi: 10.1016/s0188-4409(02)00353-3. [DOI] [PubMed] [Google Scholar]
- Pierson PM, Peteri-Brunback B, Pisani DF, Abbracchio MP, Mienville JM, Rosso L. A2b receptor mediates adenosine inhibition of taurine efflux from pituicytes. Biol Cell. 2007;99:445–454. doi: 10.1042/BC20070028. [DOI] [PubMed] [Google Scholar]
- Ramos-Mandujano G, Vazquez-Juarez E, Hernandez-Benitez R, Pasantes-Morales H. Thrombin potently enhances swelling-sensitive glutamate efflux from cultured astrocytes. Glia. 2007;55:917–925. doi: 10.1002/glia.20513. [DOI] [PubMed] [Google Scholar]
- Rosso L, Peteri-Brunback B, Poujeol P, Hussy N, Mienville JM. Vasopressin-induced taurine efflux from rat pituicytes: a potential negative feedback for hormone secretion. J Physiol. 2004;554:731–742. doi: 10.1113/jphysiol.2003.056267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudkouskaya A, Chernoguz A, Haskew-Layton RE, Mongin AA. Two conventional PKC isoforms, α and βI, are involved in the ATP-induced regulation of VRAC and glutamate release in cultured astrocytes. J Neurochem. 2008;105:2260–2270. doi: 10.1111/j.1471-4159.2008.05312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfaraz D, Fraser CL. Effects of arginine vasopressin on cell volume regulation in brain astrocyte in culture. Am J Physiol Endocrinol Metab. 1999;276:E596–E601. doi: 10.1152/ajpendo.1999.276.3.E596. [DOI] [PubMed] [Google Scholar]
- Sykova E. Extrasynaptic volume transmission and diffusion parameters of the extracellular space. Neuroscience. 2004;129:861–876. doi: 10.1016/j.neuroscience.2004.06.077. [DOI] [PubMed] [Google Scholar]
- Thorne RG, Nicholson C. In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci U S A. 2006;103:5567–5572. doi: 10.1073/pnas.0509425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuz K, Pasantes-Morales H. Hyposmolarity evokes norepinephrine efflux from synaptosomes by a depolarization- and Ca2+-dependent exocytotic mechanism. Eur J Neurosci. 2005;22:1636–1642. doi: 10.1111/j.1460-9568.2005.04344.x. [DOI] [PubMed] [Google Scholar]
- Tuz K, Pena-Segura C, Franco R, Pasantes-Morales H. Depolarization, exocytosis and amino acid release evoked by hyposmolarity from cortical synaptosomes. Eur J Neurosci. 2004;19:916–924. doi: 10.1111/j.0953-816x.2004.03209.x. [DOI] [PubMed] [Google Scholar]
- Uckermann O, Wolf A, Kutzera F, Kalisch F, Beck-Sickinger AG, Wiedemann P, Reichenbach A, Bringmann A. Glutamate release by neurons evokes a purinergic inhibitory mechanism of osmotic glial cell swelling in the rat retina: activation by neuropeptide Y. J Neurosci Res. 2006;83:538–550. doi: 10.1002/jnr.20760. [DOI] [PubMed] [Google Scholar]
- Vasquez-Juarez E, Ramos-Mandujano G, Hernandez-Benitez R, Pasantes-Morales H. On the role of G-protein coupled receptors in cell volume regulation. Cell Physiol Biochem. 2008;21:1–14. doi: 10.1159/000113742. [DOI] [PubMed] [Google Scholar]
- Verbalis JG, Drutarosky MD. Adaptation to chronic hypoosmolality in rats. Kidney Int. 1988;34:351–360. doi: 10.1038/ki.1988.188. [DOI] [PubMed] [Google Scholar]
- Wurm A, Lipp S, Pannicke T, Linnertz R, Krugel U, Schulz A, Farber K, Zahn D, Grosse J, Wiedemann P, Chen J, Schoneberg T, Illes P, Reichenbach A, Bringmann A. Endogenous purinergic signaling is required for osmotic volume regulation of retinal glial cells. J Neurochem. 2010;112:1261–1272. doi: 10.1111/j.1471-4159.2009.06541.x. [DOI] [PubMed] [Google Scholar]
- Wurm A, Pannicke T, Wiedemann P, Reichenbach A, Bringmann A. Glial cell-derived glutamate mediates autocrine cell volume regulation in the retina: activation by VEGF. J Neurochem. 2008;104:386–399. doi: 10.1111/j.1471-4159.2007.04992.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang H, Feustel PJ, Kimelberg HK. DCPIB, a specific inhibitor of volume regulated anion channels (VRACs) reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp Neurol. 2008;210:514–520. doi: 10.1016/j.expneurol.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]