

Abstract
Dysregulation of protein expression, function and/or aggregation is a hallmark of a number of neuropathological conditions. Among them, upregulation and/or de novo expression of the neuronal isoform of nitric oxide (NO) synthase (nNOS) commonly occurs in diverse neurodegenerative diseases and in axotomized motoneurons. We used adenoviral (AVV) and lentiviral (LVV) vectors to study the effects of de novo nNOS expression on the functional properties and synaptic array of motoneurons. AVV-nNOS injection into the genioglossus muscle retrogradely transduced neonatal hypoglossal motoneurons (HMNs). Ratiometric real-time NO imaging confirmed that transduced HMNs generated NO gradients in brain parenchyma (space constant: ∼12.3 μm) in response to a glutamatergic stimulus. Unilateral AVV-nNOS microinjection in the hypoglossal nucleus of adult rats induced axotomy-like changes in HMNs. Specifically, we found alterations in axonal conduction properties and the recruitment order of motor units and reductions in responsiveness to synaptic drive and in the linear density of synaptophysin-positive puncta opposed to HMN somata. Functional alterations were fully prevented by chronic treatment with nNOS or soluble guanylyl cyclase inhibitors. Synaptic and functional changes were also completely avoided by prior intranuclear injection of a neuron-specific LVV system for miRNA-mediated nNOS knock-down (LVV-miR-shRNA/nNOS). Furthermore, synaptic and several functional changes evoked by XIIth nerve injury were to a large extent prevented by intranuclear administration of LVV-miR-shRNA/nNOS. We suggest that nNOS up-regulation creates a repulsive NO gradient for synaptic boutons underlying most of the functional impairment undergone by injured motoneurons. This further strengthens the case for nNOS targeting as a plausible strategy for treatment of peripheral neuropaties and neurodegenerative disorders.
Introduction
Most neurodegenerative disorders and prion diseases have common cellular and molecular mechanisms, including dysregulation of protein expression, function and/or aggregation (Ross & Poirier, 2004). Alteration in the expression level of the neuronal nitric oxide (NO) synthase (nNOS) is a hallmark of Alzheimer's (AD) (Luth et al. 2000; Simic et al. 2000; Fernandez-Vizarra et al. 2004), Parkinson's (PD) (Eve et al. 1998), amyotrophic lateral sclerosis (ALS) (Moreno-Lopez & Gonzalez-Forero, 2006), and Huntington's diseases (HD) (Deckel et al. 2002; Perez-Severiano et al. 2002) in humans and/or in animal models, as well as after peripheral (Sunico et al. 2005; Moreno-Lopez & Gonzalez-Forero, 2006) and central traumatic lesions (Herdegen et al. 1993; Chen & Aston-Jones, 1994; Saxon & Beitz, 1994; Rao et al. 1999). Subsequently, dysregulation of this enzyme is a key event in a broad spectrum of neuropathological states. Unmasking the role of nNOS imbalance in functional alterations of neurons in neurodegenerative disorders deserves attention.
Synapse loss is the main factor underlying cognitive decline in patients and/or in animal models of AD (Small, 2008), PD (Emre, 2003), HD (Cepeda et al. 2007), multiple sclerosis (Centonze et al. 2009), and HIV dementia (Kim et al. 2008). In addition, synaptic stripping of motoneurons occurs in several motor pathologies, such as ALS, progressive muscular atrophy, and traumatically damaged adult motor axons (Ikemoto et al. 1994; Sasaki & Maruyama, 1994; Ince et al. 1995; Sunico et al. 2005, 2010). Interestingly, synaptic alterations, rather than neuronal cell death, lead to an age-dependent decline in neuronal function, which contributes to the progression and phenotypical characterization of these neuropathological conditions (Coleman et al. 2004; Morfini et al. 2009).
We have developed a model of acquired peripheral motor neuropathy induced by a traumatic insult that causes a well-characterized range of functional and synaptic impairments in the insulted motoneurons. The three major NOS isoforms are concomitantly up-regulated after this type of lesion. At the peripheral level of the injured nerve, nNOS accumulates in the growing motor axons, the endothelial isoform (eNOS) is overexpressed in vasa nervorum in the distal stump and around the injury site. The inducible isoform (iNOS) is also expressed by the recruited macrophages and phagocytic Schwann cells (Moreno-Lopez, 2010). At the central level, nNOS, but not iNOS, is expressed de novo in the soma of motoneurons after traumatic motor nerve injury (Sunico et al. 2005; Moreno-Lopez, 2010). However, eNOS induction has not been reported in injured motoneurons so far. On the contrary, nerve transection reduced eNOS mRNA levels at P7 in the lumbar spinal cord, and eNOS immunoreactivity was only detected in endothelial cells even after axotomy (Rogerio et al. 2006). Finally, motoneuron apoptosis induced by nerve avulsion was similar in eNOS knockouts to that in wild-type mice (Martin et al. 2005). All this data strongly imply that nNOS is the major NO source responsible for pathophysiological central events after motor nerve injury.
In addition, de novo expression of nNOS is sufficient to induce synaptic withdrawal leading to a drastic reduction in synaptic strength on motoneurons in vitro (Sunico et al. 2010). All these effects occur within days after the axotomy of peripheral axons of these neurons, reaching maximum approximately 1 week after injury (Gonzalez-Forero et al. 2004; Sunico et al. 2005). Changes in functional properties of injured cells were prevented using various pharmacological agents targeting nNOS. NO-mediated disturbances involve changes in intrinsic membrane properties and anatomical synaptic deterioration that suggest a major pathological role of nNOS (Sunico et al. 2005; Gonzalez-Forero et al. 2007). However, nNOS is only one of the numerous proteins dysregulated after nerve damage (Singh et al. 2009). Thus, the actual role for nNOS is less clear within the complex, synergistic and/or antagonistic actions of multiple dysregulated proteins.
We modelled this pathological scenario using virally induced de novo expression of nNOS in HMNs together with complementary attempts to down-regulate nNOS expression in damaged HMNs using virally mediated gene knock-down. Our results show that overexpression of nNOS in motoneurons is a key signal for most pathophysiological changes associated with axonal damage. Moreover, they strengthen the case for nNOS as a molecular target for therapy of neurodegenerative disorders.
Methods
Neonatal (P3–P10) and adult (250–400 g) male Wistar rats obtained from authorized suppliers (Animal Supply Services, University of Cádiz, Spain and Animal Supply Unit, School of Medical Sciences, Bristol University, UK) were cared for and handled in accordance with the guidelines of the European Union Council (86/609/UE), Spanish regulations on the use of laboratory animals (BOE 67/8509-12; BOE 1201/2005) and the Animals (Scientific Procedures) Act 1986 of the UK, and approved by the local Animal Care and Ethics Committees. The experiments were also in compliance with the policies and regulations of The Journal of Physiology (Drummond, 2009). Surgical procedures were carried out under aseptic conditions and after ensuring a sufficient depth of anaesthesia evaluated by testing for the absence of withdrawal reflexes. After viral injection or nerve crushing animals received one post-operative injection of penicillin (20,000 i.u. kg−1; i.m.) to prevent infection. Pirazolone (0.1 mg kg−1; i.m.) was given on awakening for post-operative analgesia (Gonzalez-Forero et al. 2004). Statistical tests applied to each data set are indicated in the figure legends. Data values are presented as means ±s.e.m.
Retrograde adenoviral transduction of HMNs
This study used three replication-deficient recombinant adenoviral vectors (1010–1011 pfu ml−1) directing the expression of the enhanced green fluorescent protein (AVV-eGFP), monomeric red fluorescent protein (AVV-mRFP) or nNOS (AVV-nNOS) under the constitutive control of the human cytomegalovirus (hCMV) promoter. Adult and neonatal rats were anaesthetized using diethyl ether and then 50–300 μl (for adult rats) or 5–20 μl (for pups at P3) of the adenoviral suspension (AVV-eGFP, AVV-eGFP/AVV-mRFP or AVV-eGFP/AVV-nNOS; 1:1) containing 4% dimethylsulphoxide (DMSO) was injected into three spots on the tip of the tongue (Fig. 1A). Adult rats were then allowed to survive for 5–7 days, and pups for 3–7 days.
Figure 1. Experimental and analytical procedures.
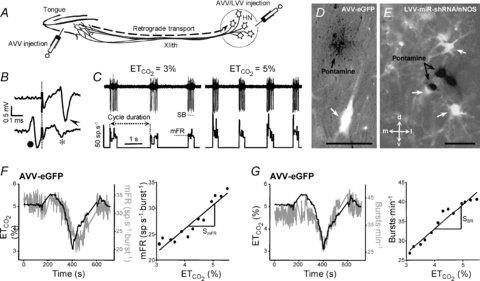
A, schematic diagram of viral administration. Adenoviral vectors (AVV) were injected either into the tip of the tongue or into the hypoglossal nucleus (HN). Lentiviral vectors (LVV) were only administered into the HN. B, HMNs were identified by their antidromic activation from the electrode implanted in the XIIth nerve and by the collision test between spontaneous orthodromic (filled circle) and antidromic (arrowhead) evoked action potentials. When the stimulus was triggered by a spontaneous spike at a short latency, the antidromic action potential was occluded (asterisk). Dotted line indicates stimulus artifact. C, characterization of firing properties of HMNs. Traces represent the extracellularly recorded spike discharge for a control inspiratory HMN and the instantaneous firing rate (FR, in spikes s−1; bin width = 25 μs) recorded at the indicated end-tidal CO2 (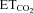 ). Mean firing rate (mFR) and number of spikes (SB) in each burst, as well as cycle duration, were measured. D and E, photomicrographs showing pontamine microiontophoretically ejected just after extracellular recordings of HMNs in transfected animals with the indicated viral vectors. Note that effectively infected hypoglossal neurons were identified close to the site of recording marked by pontamine. d, dorsal; l, lateral; m, medial; v, ventral. Calibration bars: 100 μm. F and G, left panels, time course of changes in the
). Mean firing rate (mFR) and number of spikes (SB) in each burst, as well as cycle duration, were measured. D and E, photomicrographs showing pontamine microiontophoretically ejected just after extracellular recordings of HMNs in transfected animals with the indicated viral vectors. Note that effectively infected hypoglossal neurons were identified close to the site of recording marked by pontamine. d, dorsal; l, lateral; m, medial; v, ventral. Calibration bars: 100 μm. F and G, left panels, time course of changes in the 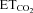 (left y-axis, black lines) and mFR (spikes s−1; F) or BR (bursts min−1; G) (right y-axis; grey lines) of a HMN recorded 6 days after AVV-eGFP injection into the HN. Note that mFR and BR paralleled changes in
(left y-axis, black lines) and mFR (spikes s−1; F) or BR (bursts min−1; G) (right y-axis; grey lines) of a HMN recorded 6 days after AVV-eGFP injection into the HN. Note that mFR and BR paralleled changes in 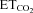 . Right panels, plots showing the relationships between mFR per burst (F) or BR (G) and
. Right panels, plots showing the relationships between mFR per burst (F) or BR (G) and 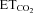 for the same motoneuron after grouping and averaging data at 0.2% intervals of
for the same motoneuron after grouping and averaging data at 0.2% intervals of 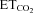 . The slopes of the regression lines represent the neuronal sensitivity or gain to
. The slopes of the regression lines represent the neuronal sensitivity or gain to 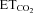 changes (SmFR, in spikes s−1%−1; SBR, in bursts min−1%−1).
changes (SmFR, in spikes s−1%−1; SBR, in bursts min−1%−1).
Viral microinjection into the hypoglossal nucleus
Adult rats were anaesthetized with 1.5–3% isoflurane in 100% O2 for microinjection into the hypoglossal nucleus (HN). Animals were placed in a stereotaxic frame and a midline dorsal incision exposed the caudal dorsal medulla. Glass micropipettes with tips broken to ∼25 μm were used to perform unilateral microinjections of the vectors: 1.2 μl of AVV-eGFP, AVV-eGFP/AVV-mRFP or AVV-eGFP/AVV-nNOS (1:1 ratio) was injected 200 μm lateral to the midline and 1.2 mm below the dorsal surface of the medulla at the obex level. Intranuclear microinjections were performed over 5 min intervals using an oil-filled system (Fig. 1A). The incision was sutured and cleaned with an aseptic solution (povidone-iodine). Animals were used for unitary extracellular recordings of HMNs (see below) 5–7 days after adenoviral administration. The effectiveness and location of the injection was analysed histologically post mortem. Some experimental groups were also used to perform immunohistochemical studies.
For nNOS knock-down, we used a recently developed binary LVV system. This system requires co-operative action of two viral vectors. The first vector expresses tetracycline-sensitive transactivator Tet-off under control of an enhanced synapsin-1 promoter (Liu et al. 2008). In this study, the second LVV harbours an expression cassette for a miRNA30 (miR30)-based short hairpin (shRNA) interference system (Stegmeier et al. 2005) under control of a Tet-sensitive promoter. The system expresses a miRNA30-like hairpin targeting the gene of choice as a fusion with the eGFP, which facilitates targeting of the RNA duplex into the RNA-induced silencing pathway. Tet-off is able to bind tetracycline or similar molecules such as doxycycline (Dox); this renders it unable to bind to the Tet-sensitive promoter and blocks the expression of the hairpin. For simplicity, this system is hereafter referred to as LVV-miR-shRNA/nNOS. LVV injections into the HN were performed 3 days before XIIth nerve crushing (see below) or AVV administration. Injection coordinates and volumes were as described for AVV. Titres of LVV used in this study were 2–6 × 109 TU ml−1. Details for the lentiviral construct and efficacy are shown in the online Supplemental Material, Fig. S1.
XIIth nerve crushing
Three days after the intranuclear microinjection of LVV in the HN, rats were anaesthetized with isoflurane (as above). The right XIIth nerve was isolated from surrounding tissue and the common nerve trunk was then thoroughly crushed with microdissecting forceps, applied for 30 s. The lesion was made just proximal to the bifurcation into lateral and medial branches. The incision was sutured and cleaned (as above).
In vitro assessment of NO synthesis and diffusion
Under deep halothane anaesthesia, P3 rat pups were injected with AVV-nNOS and/or AVV-eGFP viral vectors (∼5 × 1011 pfu ml−1, 6 μl pup−1, into the tongue). Three to seven days later the pups were deeply anaesthetized under halothane atmosphere and killed by decapitation. The brainstem was rapidly removed from the skull and immersed in ice-cold (4°C), oxygenated (95% O2 and 5% CO2) recording artificial cerebrospinal fluid (aCSF) solution containing (in mm): 24 glucose, 24 NaHCO3, 125 NaCl, 5 KCl, 1.25 MgSO4·7H2O, 1.25 KH2PO4 and 2.5 CaCl2·2H2O. Transverse slices (250 μm thick) through the brainstem at the level of the HN were made using a vibrating-blade microtome (MA752; Campden Instruments), and collected and stored in oxygenated aCSF. After 1 h incubation at 36°C, slices were transferred into the recording chamber for in vitro imaging experiments.
Real-time visualization of NO production was performed using ratiometric confocal imaging of the NO-sensitive fluorescent probe 1,2-diaminoanthraquinone sulphate (DAA; 50 mg ml−1 in DMSO 4%; Invitrogen/Molecular Probes), with Alexa Fluor 633 hydrazide (10 mm in H2O; Invitrogen/Molecular Probes) used as the reference dye. eGFP-fluorescent neurons were identified and a mixture of DAA and Alexa 633 (1:1) was introduced into the adjacent parenchyma using a patch pipette with broken tip (∼50 μm; Fig. 2A) pressurized by air from a glass capillary. Dyes were imaged using a Leica SP (Heidelberg, Germany) confocal microscope with a 40× water immersion objective, in separate channels for DAA and Alexa 633. A 488 nm argon laser was used for excitation. DAA fluorescence was detected within a 500–550 nm band set by the SP system, and Alexa 633 fluorescence within 650–700 nm. The sensitivity of the photomultipliers, the pinhole value and the temperature (32°C) of the perfusate were maintained constant throughout the recordings.
Figure 2. AVV transfection of HMNs with functional nNOS.
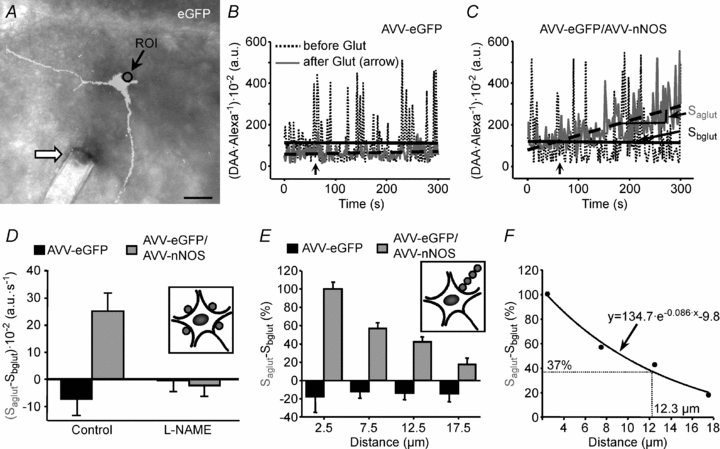
A, merged DIC and eGFP channels showing the tip of a pipette (arrow) ejecting dyes close to an eGFP-transfected HMN. The circle indicates the region of interest (ROI) used to construct the plot illustrated in B. Calibration bar: 50 μm. B and C, time courses of the DAA/Alexa 633 ratio within a 5 μm diameter ROI placed just at the border of transfected HMNs by means of the indicated adenoviruses before and after addition to the perfusate of l-glutamate (500 μm; arrows). Slopes of the regression lines adjusted before (Sbglut) and after (Saglut) glutamate addition are indicated in C. The slope increase after Glut application in C is the resultant of a rise in DAA relative to Alexa fluorescence. This indicates that NO synthesized by transduced HMN interacts with the NO-sensitive dye DAA. D, averaged Saglut–Sbglut obtained from HMNs transfected with the specified adenoviruses and incubated with the indicated drugs. Drugs were added to the bath 5 min before glutamate. Inset, illustration signalling the location of ROIs analysed per motoneuron. The average of Saglut–Sbglut was taken as the representative value for a HMN. The number of analysed HMNs per condition was as follows: AVV-eGFP, n = 4 from 3 pups; AVV-eGFP/AVV-nNOS, n = 3 from 3 pups; AVV-eGFP+l-NAME (2 mm), n = 3 from 3 pups; AVV-eGFP/AVV-nNOS+l-NAME (2 mm), n = 4 from 2 pups. Prevention of the slope increase after Glut by pre-incubation with the NOS inhibitor l-NAME strongly suggests that the change in the slope is mediated by generation of NO. E, NO gradient in brain tissue surrounding nNOS expressing HMNs. Average Saglut–Sbglut obtained from HMNs transfected with the indicated adenoviruses relative to the distance of the centre of the ROI to the motoneuron border. Measures have been normalized relative to the value obtained in the ROI nearest to the motoneuron. Inset illustrates how ROIs were located for this type of measure. The number of analysed HMNs per condition was as follows: AVV-eGFP, n = 6 from 6 pups; AVV-eGFP/AVV-nNOS, n = 13 from 10 pups. F, data presented in E were well fitted to the exponential decay equation used to calculate the theoretical space constant of the NO gradient created around AVV-nNOS transfected motoneurons.
Analysis of the recordings was made off-line, using the Leica confocal software. Measurements were performed within four circular regions of interest (ROIs) of 5 μm in diameter, adjacent to the soma of the motoneuron and at four different orientations from the pipette ejecting fluorophores, with the centre of each consecutive circle at 2.5, 7.5, 12.5 and 17.5 μm from the soma. Results were expressed as the ratio of the fluorescence intensity of DAA and Alexa 633 (DAA/Alexa 633) × 100 in arbitrary units (a.u.), before and after adding l-glutamate (500 μm) to the perfusate. The rate of NO synthesis induced by glutamate in transfected HMNs was defined as the increase in the slope of the ratio (DAA/Alexa 633) × 100 before and after glutamate application.
For more information about dyes characterization for ratiometric imaging of NO in vitro, see Material and Methods and Fig. S2 in the Supplemental Material.
Unitary extracellular recordings of HMNs
Five to seven days after XIIth nerve crushing or microinjection of AVV in the HN, adult animals were prepared for extracellular recordings as described previously (Gonzalez-Forero et al. 2004; Sunico et al. 2005; Montero et al. 2008). Briefly, rats were anaesthetized (1.5–3% isoflurane mixed with 100% O2) and additionally injected i.m. with atropine (0.2 mg kg−1) and dexamethasone sodium phosphate (0.8 mg kg−1). Teflon-isolated silver bipolar electrodes were fixed around the right XIIth nerves. To minimize differences in the inter-animal antidromic conduction distance, electrodes were placed ∼4 mm proximal to their bifurcation. Electrodes were electrically isolated from neighbouring tissue with Vaseline jelly and parafilm. The trachea, bladder, and femoral artery and vein were cannulated. Subsequently, animals were vagotomized, decerebrated, subject to neuromuscular block with gallamine triethiodide (20 mg kg−1, i.v., initially; 4 mg kg−1, i.v., as needed) and mechanically ventilated. After decerebration, isoflurane anaesthesia was discontinued. Expired CO2 and O2 were monitored continuously (Eliza duo; Gambro Engström, Bromma, Sweden). The end-tidal CO2 (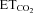 ) was changed (∼3 to ∼6%) as required by adjusting ventilation parameters (tidal volume and/or respiratory rate). Expired O2 (14–19%) was always higher than values below which hypoxia-induced alterations have been reported. Rectal temperature (37 ± 1°C) was continuously monitored and maintained. After decerebration and before neuronal recording, animals were allowed to stabilize for 30 min and
) was changed (∼3 to ∼6%) as required by adjusting ventilation parameters (tidal volume and/or respiratory rate). Expired O2 (14–19%) was always higher than values below which hypoxia-induced alterations have been reported. Rectal temperature (37 ± 1°C) was continuously monitored and maintained. After decerebration and before neuronal recording, animals were allowed to stabilize for 30 min and 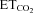 was maintained at 4.8–5.2%. A glass micropipette (1–3 MΩ) filled with a conductive solution of 3 m NaCl was then visually guided and advanced through the brainstem to the HN at least 200–300 μm in the rostro-caudal axis from the obex. The correct position of the micropipette was confirmed by recording the characteristic inspiratory pattern of the HN and the presence of the antidromic field potential elicited by electrical stimulation of the ipsilateral XIIth nerve (Fig. 1B,C). HMNs were positively identified by their antidromic activation from the XIIth nerve and by the collision test (Fig. 1B). The electrical signals were amplified and filtered at a bandwidth of 10 Hz to 10 kHz for display and digitalization purposes. HMNs’ responses to a change in
was maintained at 4.8–5.2%. A glass micropipette (1–3 MΩ) filled with a conductive solution of 3 m NaCl was then visually guided and advanced through the brainstem to the HN at least 200–300 μm in the rostro-caudal axis from the obex. The correct position of the micropipette was confirmed by recording the characteristic inspiratory pattern of the HN and the presence of the antidromic field potential elicited by electrical stimulation of the ipsilateral XIIth nerve (Fig. 1B,C). HMNs were positively identified by their antidromic activation from the XIIth nerve and by the collision test (Fig. 1B). The electrical signals were amplified and filtered at a bandwidth of 10 Hz to 10 kHz for display and digitalization purposes. HMNs’ responses to a change in 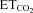 from hypocapnic (3–3.5%) to hypercapnic (5.5–6%) conditions were recorded using glass micropipettes filled with a 2% solution of pontamine sky blue (Sigma) in 0.5% sodium acetate. Only inspiratory HMNs discharging at basal conditions (
from hypocapnic (3–3.5%) to hypercapnic (5.5–6%) conditions were recorded using glass micropipettes filled with a 2% solution of pontamine sky blue (Sigma) in 0.5% sodium acetate. Only inspiratory HMNs discharging at basal conditions (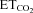 = 4.8–5.2%) were considered in this study (Fig. 1C). After recording, the micropipette was slightly (∼50 μm) withdrawn and pontamine was microiontophoretically (5 μA, 3–5 min) ejected. Animals were immediately perfused (see below). Post-mortem analysis confirmed placement of recording points close to transfected motoneurons (Fig. 1D and E). At least three animals were used per experimental condition.
= 4.8–5.2%) were considered in this study (Fig. 1C). After recording, the micropipette was slightly (∼50 μm) withdrawn and pontamine was microiontophoretically (5 μA, 3–5 min) ejected. Animals were immediately perfused (see below). Post-mortem analysis confirmed placement of recording points close to transfected motoneurons (Fig. 1D and E). At least three animals were used per experimental condition.
The histograms of instantaneous firing rate (FR, i.e. the reciprocal of the interval between two adjacent spikes) and the 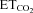 were generated from the original recordings. Bursts of action potentials were automatically selected using a macro, and several parameters were calculated. The mean FR (mFR, in spikes per second), the number of spikes per burst (SB) and the burst rate (BR, in bursts per minute) were measured in each motoneuron while varying the tidal volume and/or the respiratory rate to expose the animal to a range of
were generated from the original recordings. Bursts of action potentials were automatically selected using a macro, and several parameters were calculated. The mean FR (mFR, in spikes per second), the number of spikes per burst (SB) and the burst rate (BR, in bursts per minute) were measured in each motoneuron while varying the tidal volume and/or the respiratory rate to expose the animal to a range of 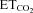 (see Fig. 1C). In this way, chemoreceptor input to the respiratory network can be modulated. Healthy HMNs faithfully follow this parameter (Fig. 1C, F and G) because they are synaptically excited by pre-motor respiratory neurons located within the medulla and pons (Ono et al. 1994; Peever et al. 2002).
(see Fig. 1C). In this way, chemoreceptor input to the respiratory network can be modulated. Healthy HMNs faithfully follow this parameter (Fig. 1C, F and G) because they are synaptically excited by pre-motor respiratory neurons located within the medulla and pons (Ono et al. 1994; Peever et al. 2002).
Data were processed statistically and the neuronal activity was correlated to the variable 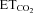 using linear regression (Fig. 1F and G). Data plots were fitted by linear functions with the equation: y = mx+n; where y is mFR, SB or BR; x is
using linear regression (Fig. 1F and G). Data plots were fitted by linear functions with the equation: y = mx+n; where y is mFR, SB or BR; x is 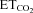 ; n is the intercept (I), i.e. the theoretical value when
; n is the intercept (I), i.e. the theoretical value when 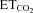 = 0%; and m is the slope, i.e. the neuronal or system gain or sensitivity (S) for each parameter relative to
= 0%; and m is the slope, i.e. the neuronal or system gain or sensitivity (S) for each parameter relative to 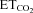 variation. Three equations characterizing the firing rate of a HMN in response to changes of
variation. Three equations characterizing the firing rate of a HMN in response to changes of 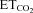 were obtained: (a) mFR = SmFR×
were obtained: (a) mFR = SmFR×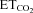 +ImFR; (b) SB = SSB×
+ImFR; (b) SB = SSB×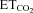 +ISB and (c) BR = SBR×
+ISB and (c) BR = SBR×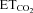 +IBR. The units for slopes in these equations were: spikes s−1%−1 for SmFR, spikes %−1 for SSB, and bursts min−1%−1 for SBR. Data were grouped and averaged in intervals of 0.2% of
+IBR. The units for slopes in these equations were: spikes s−1%−1 for SmFR, spikes %−1 for SSB, and bursts min−1%−1 for SBR. Data were grouped and averaged in intervals of 0.2% of 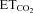 . Only regressions with r≥ 0.8 and P < 0.001 were considered for statistical analysis.
. Only regressions with r≥ 0.8 and P < 0.001 were considered for statistical analysis.
Histological procedures
After completion of the experiments, animals were anaesthetized with chloral hydrate (0.5 g kg−1; i.p.), injected intraventricularly with heparin, and perfused transcardially first with phosphate buffered saline (PBS), followed by 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, at 4°C. The brains were removed, postfixed for 2 h in the same solution, and cryoprotected overnight in 30% sucrose in phosphate buffer at 4°C. Serial coronal sections (30—50 μm thick) from brainstem were obtained using a cryostat and stored at −20°C in a cryoprotectant solution (glycerol and PBS, pH 7.4, 1:1 in volume) for later analysis or immunohistochemistry.
Immunolabelling against synaptophysin (Syn), an integral protein of synaptic vesicles, was performed to label pre-synaptic structures. Sections were rinsed in PBS and immersed in 2.5% (w/v) bovine serum albumin, 0.25% (w/v) sodium azide, and 0.1% (v/v) Triton X-100 in PBS for 30 min, followed by overnight incubation at 4°C with the antiserum. Polyclonal primary antibody used in this study was anti-Syn (1:200; Zymed Laboratories) developed in rabbit. Subsequently, the tissue was rinsed three times with PBS for 5 min each and incubated for 2 h at room temperature with anti-rabbit IgG labelled with cyanine 5 (Cy5; 1:400; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) developed in donkey. Finally, sections were washed with PBS and mounted on slides with a solution containing propyl gallate (0.1 mm in PBS/glycerol, 1:9 v/v). Omission of the primary antibody resulted in no detectable staining. For other immunohistochemistry studies performed, see supplementary Material and Methods.
Slides were analysed using a Leica confocal microscope. All neurons were acquired in a z-plane where optimal antibody diffusion was reached. Only neurons with a cell body perimeter larger than 65 μm were analysed. The pinhole opening was the same for all experimental conditions.
Drugs and treatments
In vitro procedures
The bath solution was supplemented with the NO donor 2-(N,N-diethylamino)-diazenolate-2-oxide (DEA/NO; 1 or 10 μm; Sigma) or the broad-spectrum NOS inhibitor Nω-nitro-l-arginine methyl ester (l-NAME, 2 mm, Sigma), alone or for 5 min before adding the glutamate.
In vivo treatments
Administration of chemicals began on the day of intranuclear administration of AVV. Rats were intraperitoneally injected with l-NAME (90 mg kg−1 day−1), the relatively specific nNOS inhibitor 7-nitroindazole (7-NI) (30 mg kg−1 day−1), or the specific soluble guanylyl cyclase (sGC) inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) (2 mg kg−1 day−1). Last injection was performed at least 18 h before recordings. Dox was added to the drinking water (2 mg ml−1 supplemented with 5% sucrose) for a group of animals receiving intranuclear injections of LVV. Dox treatment began 1 day before viral injection.
Results
AVV-nNOS retrogradely transfects HMNs with functional nNOS
In an initial group of experiments, the functionality of the adenoviral-directed nNOS transgene product in HMNs was tested. Ratiometric real-time imaging of NO was performed in slices from P7–P10 pups that were injected with AVV-eGFP or AVV-eGFP/AVV-nNOS (1:1) into the tip of the tongue at P3. Under these conditions, the maximum number of eGFP-identified HMNs was two per rat; in most selected slices, only one transfected HMN was present (Fig. 2A). This minimized contaminant effects of feasible eGFP/nNOS-expression in neighbouring motoneurons. The increment of the slope of the DAA/Alexa 633 ratio after perfusion with glutamate was essentially nil in HMNs transfected with eGFP alone (Saglut–Sbglut = −7.2 ± 6.1 × 10−2 a.u. s−1; Fig. 2B and D) in areas adjacent to the cell membrane. This demonstrates that under these conditions endogenous NO, if synthesized, was undetectable. However, a drastic increase in the slope of DAA/Alexa 633 after glutamate addition was recorded in regions next to the eGFP/nNOS-transfected motoneurons (Saglut–Sbglut = 25.1 ± 6.7 × 10−2 a.u. s−1; Fig. 2C and D). This increase was prevented by adding the NOS inhibitor l-NAME to the bath from 5 min before adding glutamate (Saglut–Sbglut = −0.5 ± 4.0 × 10−2 a.u. s−1; Fig. 2D). Therefore, the AVV-expressed nNOS in HMNs was functional and able to synthesize NO in response to a glutamatergic stimulus.
Much of the pathological effect of NO produced in HMNs seems to depend on its retrograde/paracrine action on the synaptic inputs from pre-motor respiratory neurons (Sunico et al. 2005, 2010). Therefore, to estimate the generation of a gradient for NO synthesized by transfected motoneurons in the surrounding parenchyma, we analysed series of ROIs whose centre points were separated by 5 μm (Fig. 2E). Maximal increment in the slope was taken as 100% in the ROI exactly on the border of the eGFP/nNOS-transfected HMN. The remaining ROIs were normalized relative to the value recorded at the motoneuron edge. Figure 2F shows the relationship between the distance from the membrane of the HMN and the change in DAA/Alexa 633 ratio. Data values were well fitted by an exponential decay function (r = 0.99; P < 0.0001). These experimental conditions yielded a space constant of 12.3 μm (i.e. where 37% of the value measured in the ROI at the edge of the soma was reached), indicating that HMN activation, such as that induced by glutamate, leads to NO synthesis in nNOS-transfected motoneurons. This creates a NO concentration gradient around these motoneurons that could easily affect not only the synaptic boutons directly contacting the membrane of HMNs but also targets within at least a few micrometres. In eGFP-transfected motoneurons these NO gradients were absent (Fig. 2E).
Transgenic nNOS expression in HMNs induces axotomy-like alterations in antidromic latency
Directly at the site of injection, both HMNs and local astrocytes expressed AVV-delivered transgene. However, at 200 μm and further away from the injection site the only cells expressing transgenes were neurons (Fig. S3G–I). This is explained by the differences in sizes of HMNs and astrocytes. Indeed, AVV can enter large neurons via any of their dendrites and possibly even proximal axons; astrocytes, being small cells with relatively short processes, only internalize vectors directly at the site of injection. Therefore, recording micropipettes were first visually placed directly above the obex, at the level where the injections were made, and then moved rostrally or caudally for at least 200 μm to avoid recordings at the site of injection of viral vectors (see supplementary Results and Fig. S3F–I). The HN was located using the antidromic field potential produced by the electrical stimulation of the ipsilateral XIIth nerve (Figs 1B and 3A). The antidromic spike of HMNs was summated on the antidromic field. To ascertain that the recorded neuron was a motoneuron, the collision test was performed (Gonzalez-Forero et al. 2004).
Figure 3. AVV-nNOS injection into the HN mimics antidromic latency alterations induced by XIIth nerve crushing.
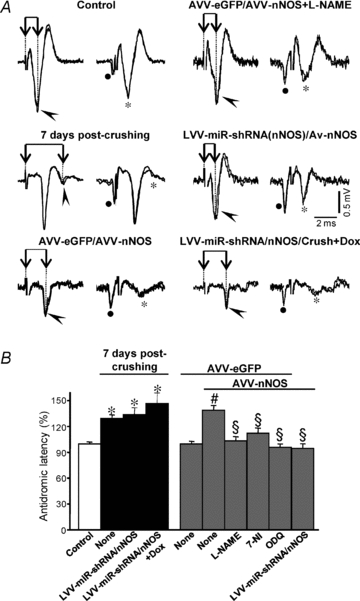
A, representative antidromic activations (arrowheads) and collisions (asterisks) in motoneurons recorded at the indicated conditions. Activation latency was measured as the time difference between XIIth nerve stimulation artifact (dotted line) and the negative peak of the antidromic spike (connected arrows). B, average antidromic activation latency (in percent) measured at the indicated conditions. Control condition was taken as 100%. The number of analysed HMNs per condition was as follows: control, n = 112; 7 days post-crushing+None, n = 55; 7 days post-crushing+LVV-miR-shRNA/nNOS, n = 15; 7 days post-crushing+LVV-miR-shRNA/nNOS+Dox, n = 15; AVV-eGFP, n = 30; AVV-eGFP/AVV-nNOS, n = 41; AVV-eGFP/AVV-nNOS+l-NAME, n = 33; AVV-eGFP/AVV-nNOS+7-NI, n = 38; AVV-eGFP/AVV-nNOS+ODQ, n = 35; AVV-eGFP/AVV-nNOS+LVV-miR-shRNA/nNOS, n = 25. Significant differences (P < 0.05; one-way ANOVA; post hoc Tukey's test) relative to control (*), AVV-eGFP- (#) or AVV-eGFP/AVV-nNOS-injected (§) groups. AVV-nNOS-induced latency alterations were fully prevented by systemic administration of 7-NI or ODQ and intranuclear injection of LVV-miR-shRNA/nNOS. However, lentivirus was not protective against crushing-provoked changes in latency.
The mean antidromic latency of HMNs recorded 5–7 days after AVV-eGFP injection in the HN was similar (1.15 ± 0.05 ms; Fig. 3B) to that in our previous studies under the control condition (1.1 ± 0.02 ms; Gonzalez-Forero et al. 2004). A significant increase in mean antidromic latency occurred after AVV-eGFP/AVV-nNOS injection (38.9 ± 5.6% relative to eGFP-transfected animals; Fig. 3), similar to that reported for HMNs 7 days after axonal injury (29.5 ± 4.1% relative to control/intact animals; Gonzalez-Forero et al. 2004). This was prevented by a chronic administration of the NOS inhibitor l-NAME beginning the day of AVV-eGFP/AVV-nNOS injection (3.4 ± 4.8%; Fig. 3). Administration of 7-NI, a relatively specific nNOS inhibitor, also protected against the changes in the latency induced by AVV-eGFP/AVV-nNOS transduction (12.2 ± 5.9%; Fig. 3B). Systemic administration of ODQ, a specific inhibitor of sGC (−3.9 ± 3.6%; Fig. 3B) prevented reduction in this parameter. Finally, neuron-specific nNOS knock-down using LVV-miR-shRNA/nNOS prevented alterations in latency of AVV-nNOS administration (−4.2 ± 5.0%; Fig. 3).
These results demonstrate that de novo expression of nNOS in HMNs results in changes very similar to those previously demonstrated after traumatic nerve injury. These changes could involve slowing of the axonal conduction velocity as differences in conduction distance between animals were minimized by placing stimulation electrodes at a fixed distance proximal to the nerve bifurcation. Since motoneurons express NO-responsive sGC (Montero et al. 2008), it is very likely that nNOS-synthesized NO triggers changes in the axonal conduction properties autocrinely, by a cGMP-mediated mechanism. Surprisingly, lentivirus injection in the HN 3 days before XIIth nerve crushing did not prevent the latency increase induced by axonal injury (Fig. 3).
Transgenic nNOS expression in HMNs mimics axonal injury-induced discharge pattern changes
Baseline 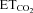
XIIth nerve lesion decreases basal activity of the motoneuron pool in spontaneously breathing rats (Gonzalez-Forero et al. 2004) in parallel with nNOS up-regulation in motoneurons (Sunico et al. 2005). However, a link between these two changes remains elusive. To evaluate this issue, unitary basal firing activity of HMNs under basal conditions (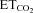 = 4.8–5.2%) was recorded in decerebrated and vagotomized animals under neuromuscular blockade 5–7 days after AVV microinjection into the HN. As in the previous section, recordings were made at least 200 μm from the injection site, where nNOS expression in astrocytes was absent (see Supplementary Results and Fig. S3). Burst parameters were measured and averaged using a minimum of 30 bursts per motoneuron to characterize the basal activity of the motoneuron pool. In AVV-eGFP-transfected animals, as in controls, most HMNs showed a characteristic respiratory pattern of bursts of action potentials synchronized with the inspiratory phase of breathing (Fig. 4A and C). This pattern was characterized in this condition by a mFR per burst of 35.8 ± 1.9 sp s−1, and a BR of 48.1 ± 1.4 bursts min−1 (Table 1). In contrast, mFR in basal conditions was significantly decreased after intranuclear injection of AVV-eGFP/AVV-nNOS (26.7 ± 1.4 sp s−1), similar to typical response 7 days after nerve injury (Fig. 4B and D; Table 1). In contrast, BR, an index of the respiratory network integrity, was not altered after virus application (50.9 ± 1.4 bursts min−1; Table 1). Therefore, nNOS de novo expression induced severe changes in HN basal activity, but structures involved in the rhythmogenesis of breathing operated as in control or eGFP-transfected conditions (Table 1). Chronic treatment with l-NAME, 7-NI or ODQ, beginning on the day of AVV-eGFP/AVV-nNOS microinjection, prevented reduction in mFR and, except for l-NAME, did not modify BR at basal
= 4.8–5.2%) was recorded in decerebrated and vagotomized animals under neuromuscular blockade 5–7 days after AVV microinjection into the HN. As in the previous section, recordings were made at least 200 μm from the injection site, where nNOS expression in astrocytes was absent (see Supplementary Results and Fig. S3). Burst parameters were measured and averaged using a minimum of 30 bursts per motoneuron to characterize the basal activity of the motoneuron pool. In AVV-eGFP-transfected animals, as in controls, most HMNs showed a characteristic respiratory pattern of bursts of action potentials synchronized with the inspiratory phase of breathing (Fig. 4A and C). This pattern was characterized in this condition by a mFR per burst of 35.8 ± 1.9 sp s−1, and a BR of 48.1 ± 1.4 bursts min−1 (Table 1). In contrast, mFR in basal conditions was significantly decreased after intranuclear injection of AVV-eGFP/AVV-nNOS (26.7 ± 1.4 sp s−1), similar to typical response 7 days after nerve injury (Fig. 4B and D; Table 1). In contrast, BR, an index of the respiratory network integrity, was not altered after virus application (50.9 ± 1.4 bursts min−1; Table 1). Therefore, nNOS de novo expression induced severe changes in HN basal activity, but structures involved in the rhythmogenesis of breathing operated as in control or eGFP-transfected conditions (Table 1). Chronic treatment with l-NAME, 7-NI or ODQ, beginning on the day of AVV-eGFP/AVV-nNOS microinjection, prevented reduction in mFR and, except for l-NAME, did not modify BR at basal 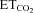 (Fig. 4E; Table 1). This last effect was not associated with changes in the other burst parameters measured in the l-NAME-treated group, indicating that both effects were independent. Additionally, l-NAME treatment of intact/control animals for 7 days did not alter mFR or BR of HMNs relative to untreated rats (not shown).
(Fig. 4E; Table 1). This last effect was not associated with changes in the other burst parameters measured in the l-NAME-treated group, indicating that both effects were independent. Additionally, l-NAME treatment of intact/control animals for 7 days did not alter mFR or BR of HMNs relative to untreated rats (not shown).
Figure 4. AVV-nNOS injection into the HN mimics effects of XIIth nerve crushing on the basal firing activity of the HMNs.
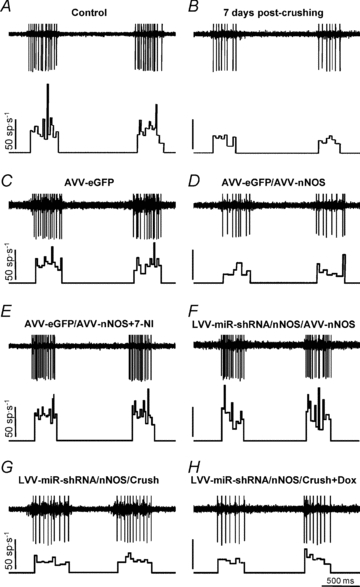
Representative examples showing the discharge activity of HMNs recorded at basal conditions (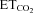 = 4.8–5.2%) at the indicated conditions. For each panel, traces are the raw signals (top) of extracellularly recorded spike activity and the histogram of instantaneous firing rate (FR, in spikes s−1; bottom). Whereas AVV-nNOS induced alterations in basal activity of HMNs were prevented by 7-NI or LVV-miR-shRNA/nNOS, crushing-evoked impairment was not avoided by LVV-miR-shRNA/nNOS.
= 4.8–5.2%) at the indicated conditions. For each panel, traces are the raw signals (top) of extracellularly recorded spike activity and the histogram of instantaneous firing rate (FR, in spikes s−1; bottom). Whereas AVV-nNOS induced alterations in basal activity of HMNs were prevented by 7-NI or LVV-miR-shRNA/nNOS, crushing-evoked impairment was not avoided by LVV-miR-shRNA/nNOS.
Table 1.
Firing properties of inspiratory HMNs in basal conditionsa
| Experimental conditions | n | mFR (sp s−1) | SB (sp burst−1) | BR (bursts min−1) |
|---|---|---|---|---|
| Intact (control) | 47 | 35.7 ± 2.1 | 16.7 ± 1.0 | 45.8 ± 0.8 |
| 7 days post-crushing | 47 | 24.9 ± 2.3* | 11.1 ± 0.9* | 44.7 ± 1.1 |
| AVV-eGFP | 51 | 35.8 ± 1.9 | 16.7 ± 0.9 | 48.1 ± 1.4 |
| AVV-eGFP/AVV-nNOS | 55 | 26.7 ± 1.4*# | 12.1 ± 0.8*# | 50.9 ± 1.4 |
| AVV-eGFP/AVV-nNOS+L-NAME | 32 | 41.6 ± 2.6§ | 20.0 ± 1.6§ | 40.1 ± 1.5#§ |
| AVV-eGFP/AVV-nNOS+7-NI | 34 | 46.0 ± 2.8*#§ | 18.8 ± 1.5§ | 45.4 ± 2.3 |
| AVV-eGFP/AVV-nNOS+ODQ | 28 | 38.2 ± 2.9§ | 17.1 ± 1.4§ | 45.4 ± 1.8 |
| LVV-miR-shRNA/nNOS/AVV-nNOS | 20 | 45.0 ± 2.7§ | 20.0 ± 1.6§ | 44.9 ± 1.7 |
| LVV-miR-shRNA/nNOS/Crush | 31 | 23.7 ± 2.0* | 10.3 ± 0.8* | 50.4 ± 0.9 |
| LVV-miR-shRNA/nNOS/Crush +Dox | 26 | 26.6 ± 2.1* | 8.9 ± 1.0* | 68.6 ± 3.1* |
Basal conditions were considered to be when 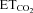 = 4.8–5.2%.
= 4.8–5.2%.
n represents the number of motoneurons included in the study. Data values are expressed as means ±s.e.m. At least 3 animals were used for each experimental condition. Significant differences relative to intact/control
AVV-eGFP-
or AVV-eGFP/AVV-nNOS-injected
groups (P < 0.05; one-way ANOVA; post hoc Tukey's test).
mFR, mean firing rate; SB, number of spikes per burst; BR, burst rate.
nNOS-induced effects on mFR were fully prevented by administration of LVV-miR-shRNA/nNOS at 3 days before AVV-nNOS injection into the HN, indicating that nNOS expression in neurons, not in glial or endothelial cells, affects mFR (Fig. 4F; Table 1; also see supplementary Results and Fig. S3). However, LVV-miR-shRNA/nNOS did not prevent reduction in basal activity of HMNs induced by XIIth nerve injury (Fig. 4G and H; Table 1).
Response to 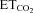 changes
changes
XIIth nerve lesion decreased the chemosensory-mediated responsiveness of HMNs to 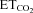 changes (Gonzalez-Forero et al. 2004). This was related to nNOS up-regulation in the injured motoneuron and NO synthesis by nNOS through cGMP (Sunico et al. 2005). However, it remained to be established whether nNOS up-regulation is the sole trigger to evoke all the downstream molecular events leading to changes in the motoneuron discharge pattern after injury. The inspiratory activity of motoneurons was modulated by chemoreceptor-driven changes in response to alterations in
changes (Gonzalez-Forero et al. 2004). This was related to nNOS up-regulation in the injured motoneuron and NO synthesis by nNOS through cGMP (Sunico et al. 2005). However, it remained to be established whether nNOS up-regulation is the sole trigger to evoke all the downstream molecular events leading to changes in the motoneuron discharge pattern after injury. The inspiratory activity of motoneurons was modulated by chemoreceptor-driven changes in response to alterations in 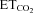 . The activity of HMNs increased when
. The activity of HMNs increased when 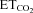 rose and decreased when
rose and decreased when 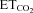 was lowered (Figs 1C–E and 5A). Relationships obtained between mFR per burst or BR and
was lowered (Figs 1C–E and 5A). Relationships obtained between mFR per burst or BR and 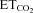 were linear (Figs 1C and D and 6A and B). The slope of the regression line represented the sensitivity (S) or gain of mFR or BR to
were linear (Figs 1C and D and 6A and B). The slope of the regression line represented the sensitivity (S) or gain of mFR or BR to 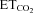 (SmFR and SBR, respectively) (Fig. 1C and D). Under control conditions, these rates averaged 9.2 ± 0.9 sp s−1%−1 and 5.6 ± 0.8 bursts min−1%−1, respectively (Fig. 6; Table 2). Thus, the SmFR can be taken as an index of synaptic efficacy on HMNs. Parameters characterizing chemosensory-mediated responsiveness of HMNs are presented in Table 2.
(SmFR and SBR, respectively) (Fig. 1C and D). Under control conditions, these rates averaged 9.2 ± 0.9 sp s−1%−1 and 5.6 ± 0.8 bursts min−1%−1, respectively (Fig. 6; Table 2). Thus, the SmFR can be taken as an index of synaptic efficacy on HMNs. Parameters characterizing chemosensory-mediated responsiveness of HMNs are presented in Table 2.
Figure 5. AVV-nNOS injection into the HN mimics effects of XIIth nerve crushing on the chemoreceptor-modulated inspiratory activity of HMNs.
Illustrative examples of the time courses of the mean firing rate modulation (mFR, in spikes (sp) s−1 burst−1; right y-axis; left panels) and burst rate (BR, in bursts min−1; right y-axis; right panels) relative to  levels (left y-axis) for a control HMN (A), a motoneuron recorded 7 days after XIIth nerve crushing (B), and 7 days after the injection of the indicated viruses into the HN (C–G). Chemical treatments began on viral injection day. A motoneuron recorded 7 days after XIIth nerve crushing in an animal pre-injected with the LVV-miR-shRNS/nNOS is also shown (H). Note that nNOS knock-down prevents mFR dissociation from
levels (left y-axis) for a control HMN (A), a motoneuron recorded 7 days after XIIth nerve crushing (B), and 7 days after the injection of the indicated viruses into the HN (C–G). Chemical treatments began on viral injection day. A motoneuron recorded 7 days after XIIth nerve crushing in an animal pre-injected with the LVV-miR-shRNS/nNOS is also shown (H). Note that nNOS knock-down prevents mFR dissociation from  .
.
Figure 6. Quantification of the effects of transgenic nNOS expression and viral nNOS knock-down in the HN on the chemoreceptor-modulated inspiratory activity of HMNs.
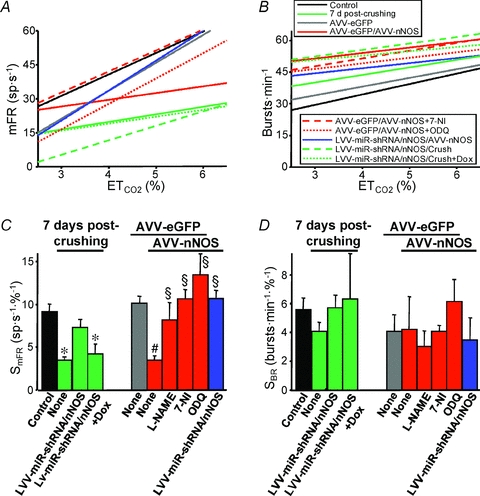
A and B, plots showing representative linear regression lines of mean firing rate (mFR, A) per burst or burst rate (BR, B) versus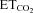 for motoneurons recorded at the indicated conditions. The slopes of the regression lines represent the neuronal sensitivity to or gain in response to
for motoneurons recorded at the indicated conditions. The slopes of the regression lines represent the neuronal sensitivity to or gain in response to 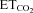 changes (SmFR, in spikes s−1%−1; SBR, in bursts min−1%−1). C and D, average values of SmFR (C) and SBR (D) obtained from the motoneuron pools recorded under the indicated conditions. The number of analysed HMNs per condition is indicated in Table 2. Significant differences (P < 0.05; one-way ANOVA; post hoc Tukey's test) relative to control (*), AVV-eGFP- (#) or AVV-eGFP/AVV-nNOS-injected (§) groups. Note that the sensitivity of HMNs mFR to
changes (SmFR, in spikes s−1%−1; SBR, in bursts min−1%−1). C and D, average values of SmFR (C) and SBR (D) obtained from the motoneuron pools recorded under the indicated conditions. The number of analysed HMNs per condition is indicated in Table 2. Significant differences (P < 0.05; one-way ANOVA; post hoc Tukey's test) relative to control (*), AVV-eGFP- (#) or AVV-eGFP/AVV-nNOS-injected (§) groups. Note that the sensitivity of HMNs mFR to 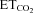 was similarly reduced after nerve crushing or AVV-nNOS intranuclear injection. In both cases alterations were fully prevented by LVV-miR-shRNA/nNOS.
was similarly reduced after nerve crushing or AVV-nNOS intranuclear injection. In both cases alterations were fully prevented by LVV-miR-shRNA/nNOS.
Table 2.
Effects of viral gene manipulation on the coupling between 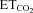 and discharge properties of inspiratory HMNs
and discharge properties of inspiratory HMNs
| Experimental conditions | n | SmFR (sp s−1%−1) | SSB (sp burst−1%−1) | SBR (bursts min−1%−1) |
|---|---|---|---|---|
| Intact (control) | 44 | 9.2 ± 0.9 | 4.6 ± 0.4 | 5.6 ± 0.8 |
| 7 days post-crushing | 44 | 3.5 ± 0.4* | 1.5 ± 0.2* | 4.1 ± 0.6 |
| AVV-eGFP | 25 | 10.1 ± 0.8 | 6.2 ± 0.6 | 4.1 ± 1.2 |
| AVV-eGFP/AVV-nNOS | 16 | 3.5 ± 0.5*# | 1.7 ± 0.4*# | 4.2 ± 2.3 |
| AVV-eGFP/AVV-nNOS+L-NAME | 16 | 8.2 ± 2.0§ | 5.0 ± 1.1§ | 3.1 ± 1.1 |
| AVV-eGFP/AVV-nNOS+7-NI | 10 | 10.7 ± 1.1§ | 6.4 ± 1.3§ | 4.1 ± 0.4 |
| AVV-eGFP/AVV-nNOS+ODQ | 12 | 13.4 ± 2.5§ | 6.4 ± 1.1§ | 6.2 ± 1.5 |
| LVV-miR-shRNA/nNOS/AVV-nNOS | 16 | 10.7 ± 1.0§ | 6.3 ± 0.5§ | 3.5 ± 1.6 |
| LVV-miR-shRNA/nNOS/Crush | 11 | 7.3 ± 0.9 | 3.7 ± 0.4 | 5.7 ± 0.9 |
| LVV-miR-shRNA/nNOS/Crush +Dox | 9 | 4.2 ± 1.2* | 1.8 ± 0.5* | 6.3 ± 3.2 |
Sensitivity (S) or gain parameters characterizing motoneuron activity were measured in response to 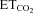 changes from 3–3.5% to 5.5–6.0%. n represents the number of motoneurons included in the study. Data values are expressed as means ±s.e.m. At least 3 animals were used per experimental condition. Significant differences relative to intact/control
changes from 3–3.5% to 5.5–6.0%. n represents the number of motoneurons included in the study. Data values are expressed as means ±s.e.m. At least 3 animals were used per experimental condition. Significant differences relative to intact/control
AVV-eGFP-
or AVV-eGFP/AVV-nNOS-injected
groups (P < 0.05; one-way ANOVA; post hoc Tukey's test).
After intranuclear injection of AVV-eGFP, SmFR and SBR of HMNs were similar to the control condition (Figs 5C and 6; Table 2). However, HMN transduction with nNOS induced a dramatic decrease in motoneuron sensitivity to the chemoreceptor-modulated inspiratory drive, affecting SmFR but not SBR (Figs 5D and 6; Table 2). This nNOS-induced alteration was equivalent to the effects on motoneuron sensitivity 7 days after XIIth nerve crushing (Figs 5B and 6; Table 2). The lack of effects on SBR is crucial for the validation of these results because this parameter is indicative of the integrity and functional state of chemosensors and pre-motor structures. Thus, virally mediated nNOS expression in the HN effectively mimics the outcome of nerve injury on chemosensory-mediated responsiveness of HMNs to 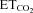 , with no effects on premotor respiratory network.
, with no effects on premotor respiratory network.
Chronic administration of the NOS inhibitor l-NAME to AVV-eGFP/AVV-nNOS-transduced rats, beginning on day of microinjection, maintained SmFR as in the animals receiving AVV-eGFP without affecting SBR (Figs 5E and 6; Table 2). Administration of 7-NI, a relatively specific inhibitor of nNOS, protected against the changes in activity induced by de novo nNOS expression (Fig. 6; Table 2). Impairment of synaptic functionality was also prevented by systemic administration of ODQ (Figs 5F and 6; Table 2). SBR was not altered by drugs, which indicates that responsiveness of the respiratory system was similarly preserved at all stages (Figs 5 and 6; Table 2). These results indicate that NO synthesized by nNOS transgene is sufficient to trigger molecular processes that lead to a reduction in motoneuron sensitivity to the afferent drive. This effect of NO is mediated by sGC activation.
Prior lentiviral injection in the HN fully prevented changes in SmFR induced by either AVV-nNOS administration or XIIth nerve crushing, without affecting SBR (Fig. 5G and H and 6; Table 2). This strongly supports the notion that AVV-nNOS-induced alterations were indeed due to HMN transduction and not due to transduction of some unspecified non-neuronal cells or affectation of pre-motor neurons. Furthermore, the ability of LVV-miR-shRNA/nNOS to block the effects of nerve damage on the discharge pattern of HMNs clearly demonstrates that these effects result from nNOS up-regulation in injured HMNs.
Transgenic nNOS expression changes the recruitment scheme of the HMN pool
Motor nerve injury is characteristically followed by alterations in threshold (Th) distribution and recruitment properties of injured HMNs in a NO/cGMP-dependent way (Gonzalez-Forero et al. 2004; Sunico et al. 2005). Recruitment Th for each recorded hypoglossal motor unit can be defined as the theoretical 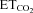 concentration at which the motoneuron begins to discharge (that is, the abscissa intercept in the mFR–
concentration at which the motoneuron begins to discharge (that is, the abscissa intercept in the mFR–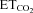 regression line) (Fig. 7A and B; Th). In the control group, the mean Th was –1.5 ± 0.9%, and its distribution was well fitted with a Gaussian function (r = 0.87; Fig. 7C). This recruitment scheme was distorted 7 days after nerve crushing (r = 0.57), and the mean Th was significantly reduced (−6.1 ± 1.7%; P < 0.05; one-way ANOVA; post hoc Tukey's test; Fig. 7C). Intranuclear administration of AVV-eGFP did not modify either Th distribution (r = 0.96) or mean Th (1.0 ± 0.5%) relative to the intact/control state. On the contrary, AVV-nNOS induced nerve injury-like changes in the recruitment pattern of the motoneuron pool (r = 0.30; mean Th: −5.2 ± 1.4%; P < 0.05; Fig. 7C). These effects were fully prevented by chronic treatment with 7-NI (r = 0.88; Th: −1.6 ± 0.7%) or ODQ (r = 0.86; Th: 1.2 ± 0.7%), or by preceding injection into the HN of LVV-miR-shRNA/nNOS (r = 0.85; Th: −1.1 ± 1.0%; Fig. 7C). Lentiviral administration 3 days before XIIth nerve crushing prevented changes in the recruitment scheme induced by axonal injury (r = 0.84; Th: 0.1 ± 0.9%), and this protective effect was blocked by Dox (r = 0.52; Th: −7.1 ± 4.9%). These results were further confirmed by the analysis of the cumulative sum histograms of active motor units relative to the
regression line) (Fig. 7A and B; Th). In the control group, the mean Th was –1.5 ± 0.9%, and its distribution was well fitted with a Gaussian function (r = 0.87; Fig. 7C). This recruitment scheme was distorted 7 days after nerve crushing (r = 0.57), and the mean Th was significantly reduced (−6.1 ± 1.7%; P < 0.05; one-way ANOVA; post hoc Tukey's test; Fig. 7C). Intranuclear administration of AVV-eGFP did not modify either Th distribution (r = 0.96) or mean Th (1.0 ± 0.5%) relative to the intact/control state. On the contrary, AVV-nNOS induced nerve injury-like changes in the recruitment pattern of the motoneuron pool (r = 0.30; mean Th: −5.2 ± 1.4%; P < 0.05; Fig. 7C). These effects were fully prevented by chronic treatment with 7-NI (r = 0.88; Th: −1.6 ± 0.7%) or ODQ (r = 0.86; Th: 1.2 ± 0.7%), or by preceding injection into the HN of LVV-miR-shRNA/nNOS (r = 0.85; Th: −1.1 ± 1.0%; Fig. 7C). Lentiviral administration 3 days before XIIth nerve crushing prevented changes in the recruitment scheme induced by axonal injury (r = 0.84; Th: 0.1 ± 0.9%), and this protective effect was blocked by Dox (r = 0.52; Th: −7.1 ± 4.9%). These results were further confirmed by the analysis of the cumulative sum histograms of active motor units relative to the 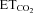 threshold (see supplementary Results and Fig. S4). Together, these results suggest that the decrease in Th and recruitment scheme disorganization of the injured motor pool result solely from up-regulation of nNOS in HMNs and require cGMP-mediated NO signalling.
threshold (see supplementary Results and Fig. S4). Together, these results suggest that the decrease in Th and recruitment scheme disorganization of the injured motor pool result solely from up-regulation of nNOS in HMNs and require cGMP-mediated NO signalling.
Figure 7. Viral expression of nNOS in HMNs mimics changes in the recruitment distribution of HMNs induced by XIIth nerve crushing.
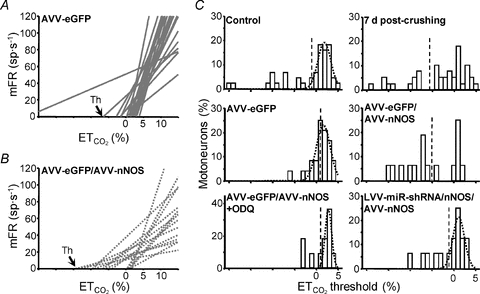
A and B, collection of linear regression lines of mean firing rate (mFR) versus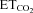 including the whole analysed pools in AVV-eGFP (A) and AVV-eGFP/AVV-nNOS (B) treated animals. The abscissa intercepts of the regression lines indicate the theoretical recruitment threshold (Th) for each motor unit. C, distribution histograms of Ths obtained in HMNs from the indicated conditions. All distributions except those from crushed non-treated, AVV-eGFP/AVV-nNOS and LVV-miR-shRNA/nNOS/crush+Dox (not shown) conditions were well fitted to Gaussian peak equations (r > 0.85, P < 0.0001). This indicates that recruitment distribution disruption after nerve injury is a nNOS-dependent process. Dashed lines point to the mean of each group. The number of analysed HMNs per condition is indicated in Table 2.
including the whole analysed pools in AVV-eGFP (A) and AVV-eGFP/AVV-nNOS (B) treated animals. The abscissa intercepts of the regression lines indicate the theoretical recruitment threshold (Th) for each motor unit. C, distribution histograms of Ths obtained in HMNs from the indicated conditions. All distributions except those from crushed non-treated, AVV-eGFP/AVV-nNOS and LVV-miR-shRNA/nNOS/crush+Dox (not shown) conditions were well fitted to Gaussian peak equations (r > 0.85, P < 0.0001). This indicates that recruitment distribution disruption after nerve injury is a nNOS-dependent process. Dashed lines point to the mean of each group. The number of analysed HMNs per condition is indicated in Table 2.
Transgenic nNOS expression reduces the synaptic coverage of hypoglossal neurons
Axonal injury of HMNs reduces responses of HMNs to chemoreceptor-modulated inspiratory drive underpinned, at least in part, by NO-mediated synaptic stripping of motoneurons (Gonzalez-Forero et al. 2004; Sunico et al. 2005, 2010). Additionally, we have recently reported that de novo synthesis of NO was sufficient to induce the withdrawal of synaptic boutons on HMNs (Sunico et al. 2010). This was accompanied by a strong decrease in the evoked excitatory postsynaptic potential on motoneurons in vitro (Sunico et al. 2010). Subsequently, we investigated whether the effect of nNOS transgene on the discharge pattern of HMNs could be explained, at least in part, by alterations in the synaptic arrangement of motoneurons. Given that the HN consists of 90% motoneurons and 10% interneurons (Sturrock, 1991), we assume some contamination of the results by data from plausible interneurons, which we minimized by focusing our studies on the larger neurons (>65 μm cell body perimeter).
We performed immunohistochemistry for synaptophysin (Syn), a synaptic marker, in sections obtained from animals after intranuclear administration of different combinations of viral vectors. The action of nNOS transgene expression on the synaptic array of HMNs was investigated using confocal microscopy. We measured the linear density of Syn-immunoreactive (Syn-ir) puncta (Syn-ir puncta/100 μm of membrane perimeter) opposed to eGFP-positive neurons. Importantly, the linear density of Syn-ir puncta was significantly reduced (−42.1 ± 2.8%; P < 0.05; one-way ANOVA; post hoc Tukey's test) on HMNs from AVV-eGFP/AVV-nNOS relative to AVV-eGFP injected animals (23.3 ± 0.7 Syn-ir puncta/100 μm; Fig. 8A, B and F). This effect was fully prevented by preceding injection into the HN of LVV-miR-shRNA/nNOS (−1.0 ± 3.0%; Fig. 8C and F).
Figure 8. AVV-nNOS injection into the HN induces reduction in the frequency of Syn-ir puncta opposed to HMN.
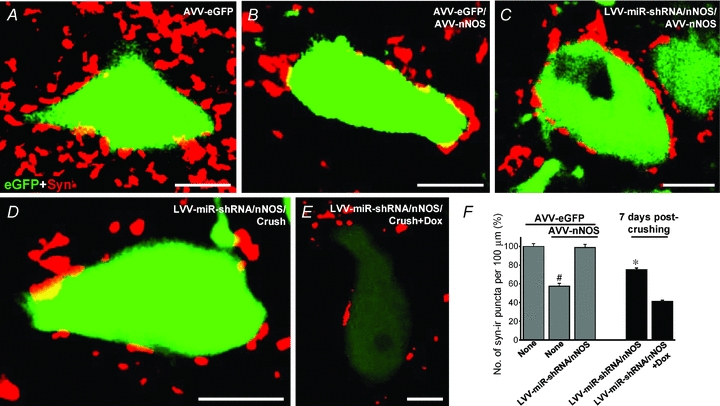
A–E, Syn-ir puncta around eGFP-identified HMNs obtained from animals receiving the indicated viral vectors. Scale bars, 10 μm. Secondary antibody was labelled with Cy5 for immunolabelling of Syn. F, average number of Syn-ir puncta per 100 μm of eGFP-identified HMN perimeter at the indicated conditions. Note that AVV-nNOS induces a reduction in the number of Syn-ir puncta opposed to HMNs. Besides, LVV-miR-shRNA/nNOS partially prevented crushing-induced Syn-ir punta reduction. The number of analysed HMNs per condition was as follows: AVV-eGFP, n = 53 neurons; AVV-eGFP/AVV-nNOS, n = 64 neurons; AVV-eGFP/AVV-nNOS+LVV-miR-shRNA/nNOS, n = 50 neurons; 7 days post-crushing+LVV-miR-shRNA/nNOS, n = 119 neurons; 7 days post-crushing+LVV-miR-shRNA/nNOS+Dox, n = 108 neurons. Significant differences (P < 0.05; one-way ANOVA; post hoc Tukey's test) relative to AVV-eGFP- (#) or 7 days post-crushing+LVV-miR-shRNA/nNOS+Dox-treated (*) groups.
Preceding injection of LVV-miR-shRNA/nNOS in the HN strongly attenuated reduction in Syn-ir induced by XIIth nerve crushing (−24.9 ± 1.7%) as compared to Dox-treated injured animals (−58.8 ± 1.3%; Fig. 8D–F). The last result is essentially identical to the previously reported reduction in the synaptic coverage after XIIth nerve injury (−57.2 ± 2.9%) previously reported by our group (Sunico et al. 2005). These observations suggest that relevant functional alterations in motoneurons induced by nNOS transgene expression or by axotomy involve NO-triggered synaptic rearrangements.
Discussion
We report here that de novo nNOS expression is sufficient to provoke axotomy-like functional disturbances in motoneurons which, at least partially, result from NO-induced synaptic loss. HMNs retrogradely transfected with nNOS express a functional enzyme that synthesizes NO in response to glutamatergic stimulation. nNOS transduction altered conduction properties of motor axons, decreased baseline discharge activity of HMNs, reduced motoneuron sensitivity to chemoreceptor-modulated inspiratory drive and modified the recruitment scheme of the motor units. These functional outcomes paralleled the impairment in the synaptic coverage of transfected motoneurons. Electrophysiological alterations were all prevented by systemic administration of a relatively specific nNOS inhibitor, a sGC inhibitor or by a preceding injection into the nucleus of a neuron-specific lentivirus expressing a shRNA targeting nNOS. In addition, synaptic stripping induced by nNOS was fully avoided by lentiviral injection. This pattern of changes is indistinguishable from a set of effects previously documented after a mechanical trauma to HMN axons. Moreover, LVV-mediated nNOS knock-down effectively prevented pivotal pathophysiological changes induced by XIIth nerve trauma. Together, these results identify expressed nNOS as a key factor responsible for an array of pathological processes triggered by axonal damage.
We first set out to model the pathological expression of nNOS in HMNs using AVV. It was important to demonstrate that the nNOS expressed in that way is functional. Therefore, we retrogradely transduced small numbers of HMNs using AVV injections into the tongue. Next, we performed ratiometric real-time imaging in brainstem slices obtained from transfected pups with the NO-specific fluorescent probe DAA (Chen et al. 2001). To offset time dependent changes in the tissue concentrations of the dye, we mixed it with a reference dye, Alexa 633, and monitored the DAA/Alexa 633 ratio. DAA fluorescence linearly rose in response to bath addition of a NO donor, proportional to the donor concentration. The slope of DAA fluorescence increased close to the membrane of eGFP/nNOS-transfected motoneurons in response to glutamate, and this could be blocked by adding the NOS inhibitor l-NAME to the incubation solution. We found that NO released by the HMNs formed a clear gradient in the brain tissue with an estimated space constant of 12.3 μm. Signalling distances for NO reported in previous studies range from few micrometres to up to 200 μm (Wood & Garthwaite, 1994; Hall & Garthwaite, 2006; Tornieri & Rehder, 2007; Steinert et al. 2008; Artinian et al. 2010). Here it was estimated using fluorescence of a NO-sensitive dye, but not a NO-mediated functional response in target structures. We assume that differences could be mainly explained by the sensitivity of the assay or, perhaps differences in NO degradation speed under different experimental conditions and in different tissues. In addition, nNOS transduction induced a reduction in the linear density of Syn-ir puncta opposed to transfected motoneurons (current results), which correlated with actual bouton detachment (Sunico et al. 2010). As these gradients have a clear vector and spread from the nNOS expressing neurons towards incoming axonal terminals, we suggest that they may represent a repulsive signal leading to the withdrawal of these inputs.
Disconnection of motoneurons from their target myocytes disrupts reciprocal trophic interactions, leading to profound alterations in the structural and physiological properties of both motoneurons and muscle fibres. Axotomy induces changes in axonal, synaptic and intrinsic membrane properties. It comprises enhanced somato-dendritic excitability, decreased axonal conduction velocity, massive loss of afferent synaptic inputs (Titmus & Faber, 1990) and disturbances in the firing properties and recruitment order of motor units (Gonzalez-Forero et al. 2004, 2007). All these changes were concomitant with nNOS up-regulation in motoneurons (Yu, 1997; Sunico et al. 2005). To model the situation observed after the nerve injury, we injected AVV-nNOS into the HN and studied changes in the physiological properties of HMNs.
This study provides strong evidence for the causative role of nNOS dysregulation and electrophysiological changes occurring in injured motoneurons. In the current experiments, the intranuclear AVV-nNOS administration evoked changes very similar to those observed after XIIth nerve crushing (Gonzalez-Forero et al. 2004). Viral de novo expression of nNOS led to a distortion in the recruitment scheme of motor units, with a reduction in Th of the motor pool similar to that of crushing. This is consistent with an enhancement in motoneuron excitability. Recruitment order in a motor pool is primarily determined by intrinsic membrane properties that could eventually modify Th range or recruitment gain (Gustafsson & Pinter, 1985; Heckman & Binder, 1993; Cope & Sokoloff, 1999). Nerve injury has been shown to induce motoneuron hyperexcitability by a drastic NO/cGMP-mediated enhancement in the input resistance (Gonzalez-Forero et al. 2007). This effect on intrinsic membrane properties could be mimicked by AVV-nNOS-mediated transduction of HMNs. Furthermore, nerve injury reduces response of HMNs to baseline and chemoreceptor-modulated inspiratory drive in the adult rat (Gonzalez-Forero et al. 2004), which was mimicked by nNOS transgene expression into the HN. Injury-induced alterations of the chemosensory-modulated responsiveness of HMNs are accompanied by a nNOS/sGC-mediated loss of afferent inputs (Sunico et al. 2005). Strikingly, nNOS transduction of HMNs induced a reduction in their synaptic coverage. Interestingly, de novo synthesis of NO is sufficient to induce excitatory, but not inhibitory, synapse detachment from motoneurons in a cGMP-dependent way (Sunico et al. 2010). AVV-nNOS-induced loss of excitatory synapses on the transfected HMNs is the most probable explanation for their almost complete loss of responsiveness to baseline and chemoreceptor-modulated inspiratory drive.
It can be argued that nNOS from AVV-transduced glia or up-regulated nNOS and iNOS by the surgical trauma caused by intracerebral injection (Rao et al. 1999; Petrov et al. 2000) could be the actual sources of the NO that induces functional alterations in motoneurons. However, intranuclear injection of LVV-miR-shRNA/nNOS, which is highly selective for neurons (Liu et al. 2008), prevented all disturbances induced by AVV-nNOS, making this possibility highly unlikely. All functional changes induced in HMNs by nNOS transgene expression into the HN were also prevented by the chronic administration of the broad spectrum NOS inhibitor l-NAME or 7-NI, a relatively specific inhibitor of nNOS in vivo that lacks the vascular effects of l-NAME (Moreno-Lopez et al. 2004). Because 7-NI has nearly identical affinity for nNOS and eNOS in vitro (Wolff et al. 1994; Bland-Ward & Moore, 1995; Moore & Bland-Ward, 1996), it could be possible that its protective effects were actually due to block of eNOS. We think that this is highly unlikely. 7-NI did not inhibit acetylcholine-induced endothelium-dependent relaxation of the isolated rabbit aorta preparation and was without effect on arterial blood pressure in a range of species including mouse (Moore et al. 1993), rat (Beierwaltes, 1995) and pigeon (Zagvazdin et al. 1996). In addition, at the dose used here, 7-NI did not accelerate axonal regeneration and the beginning of neuromuscular reconnection after XIIth nerve crushing, where eNOS plays a key role because these processes can be accelerated by a relatively specific eNOS inhibitor or by local administration of a dominant negative for eNOS (Sunico et al. 2008). These findings, together with the fact that neuronally targeted LVV-miR-shRNA/nNOS fully prevented AVV-nNOS-induced effects on motoneuron physiology, make highly unlikely that another isoform of NOS was involved in alterations induced in HMNs after adenoviral administration. Thus, transgenic nNOS expression in HMNs is sufficient to induce axotomy-like alterations in several electrophysiological characteristics of HMNs. Furthermore, since it could be prevented by ODQ, this is a sGC-dependent effect. These findings indicate that nNOS is a key molecule which sets off a range of functional alterations in response to trauma and suggest that the same may happen in certain degenerative processes underpinning motor neuropathies.
The functional significance of these NO-mediated physiological changes in injured motoneurons remains uncertain. Electrical alterations would imply a functional de-differentiation of axotomized motoneurons to a postnatal-like stage (Kuno et al. 1974). In the same context, during postnatal development nNOS is transitorily expressed in motoneurons (Vazquez et al. 1999; Gao et al. 2008), suggesting a role for NO in their postnatal maturation. Therefore, induced NO could be the trigger to activate re-growth programmes in injured adult motor axons that would involve reversion to an immature electrical phenotype. Another, but not mutually exclusive, possibility is that NO-induced hyperexcitability in axotomized motoneurons constitutes a compensatory response to counteract the deficiency in incoming excitatory drive (Eccles et al. 1958; Kuno & Llinas, 1970; Sumner, 1975) which, paradoxically, was also NO dependent (Sunico et al. 2005). In this scenario, NO-induced membrane hyperexcitability could maintain minimal levels of electrical activity necessary to prevent neuronal death and support axonal growth, synaptogenesis and differentiation in both immature and regenerating motoneurons.
Finally, we have tested whether knock-down of nNOS is able to prevent some electrophysiological and synaptic alterations induced by nerve crushing, a model of acquired peripheral neuropathy. We found that nNOS knock-down in motoneurons prevents reduction in the chemosensory-modulated responsiveness of HMNs, distortion in the recruitment scheme of motor units and, at least in part, synapse loss induced by nerve damage. This agrees with our previous assumption about the causative role of de novo expression of nNOS in the reduction of the excitatory synaptic coverage (Sunico et al. 2005, 2010) and hyperexcitability (Gonzalez-Forero et al. 2007) occurring in motoneurons after axonal injury by a sGC-dependent mechanism. The present results support the model whereby a NO gradient created around nNOS-expressing motoneurons could be repulsive for sGC-containing excitatory synapses in an activity-dependent way. This might be a protective mechanism delaying degeneration of NO-mediated hyperexcitable motoneurons in the face of excitotoxic stimuli. In animal models of ALS, NO causes hyperexcitability and sensitizes motoneurons to death (Raoul et al. 2002; Kuo et al. 2004, 2005).
LVV-miR-shRNA/nNOS was unable to protect against changes induced by nerve injury in axonal conduction properties or firing rate at basal conditions of  . It is unlikely that the possibility of other NO-synthesizing sources, not targeted by LVV-miR-shRNA/nNOS, such as iNOS and/or eNOS, is responsible for the persistence of the above-mentioned effects. First, if that were the case, then we would expect similar alterations in non-crushed animals which received AVV and LVV central injections, but that did not occur. In addition, iNOS up-regulation was not detected by immunohistochemistry in the HN after XIIth nerve crushing (Sunico et al. 2005). Besides, eNOS was not up-regulated in lumbar neurons after nerve injury (Rogerio et al. 2006) and was not involved in motoneuron degeneration after nerve avulsion (Martin et al. 2005). On the other hand, this could be the consequence of a residual nNOS expression, as this system results in ∼50% reduction of the nNOS protein (Fig. S1) as measured by Western blotting. It could also explain partial preventive action against synapse reduction. It is well established that NO can mediate physiological or pathological actions across a wide range of concentrations from the nanomolar to the micromolar range (Malinski et al. 1993; Bellamy et al. 2002; Wang et al. 2006). It is possible that pathological upregulation of nNOS after nerve injury leads to generation of micromolar concentrations of NO which affect NO-sensitive synapses (Sunico et al. 2010) and, therefore, the chemoreceptor-modulated inspiratory drive. But when nNOS is downregulated by LVV-miR-shRNA/nNOS injection, only NO effects on more sensitive ionic channels (Ahern et al. 2002; Montero et al. 2008) and/or synapse neurotransmission (Wang et al. 2007; Sunico et al. 2010) may persist. Interestingly, differential sensitivity in the potentiation of glutamatergic EPSPs and GABAergic IPSPs to the NO/cGMP pathway has been described in the nucleus tractus solitarii at physiological concentrations of NO (Wang et al. 2007).
. It is unlikely that the possibility of other NO-synthesizing sources, not targeted by LVV-miR-shRNA/nNOS, such as iNOS and/or eNOS, is responsible for the persistence of the above-mentioned effects. First, if that were the case, then we would expect similar alterations in non-crushed animals which received AVV and LVV central injections, but that did not occur. In addition, iNOS up-regulation was not detected by immunohistochemistry in the HN after XIIth nerve crushing (Sunico et al. 2005). Besides, eNOS was not up-regulated in lumbar neurons after nerve injury (Rogerio et al. 2006) and was not involved in motoneuron degeneration after nerve avulsion (Martin et al. 2005). On the other hand, this could be the consequence of a residual nNOS expression, as this system results in ∼50% reduction of the nNOS protein (Fig. S1) as measured by Western blotting. It could also explain partial preventive action against synapse reduction. It is well established that NO can mediate physiological or pathological actions across a wide range of concentrations from the nanomolar to the micromolar range (Malinski et al. 1993; Bellamy et al. 2002; Wang et al. 2006). It is possible that pathological upregulation of nNOS after nerve injury leads to generation of micromolar concentrations of NO which affect NO-sensitive synapses (Sunico et al. 2010) and, therefore, the chemoreceptor-modulated inspiratory drive. But when nNOS is downregulated by LVV-miR-shRNA/nNOS injection, only NO effects on more sensitive ionic channels (Ahern et al. 2002; Montero et al. 2008) and/or synapse neurotransmission (Wang et al. 2007; Sunico et al. 2010) may persist. Interestingly, differential sensitivity in the potentiation of glutamatergic EPSPs and GABAergic IPSPs to the NO/cGMP pathway has been described in the nucleus tractus solitarii at physiological concentrations of NO (Wang et al. 2007).
It is known that after a nerve injury, NO production is up-regulated in endothelial cells, recruited macrophages and Schwann cells within the damaged nerve (Moreno-Lopez, 2010). Clearly, these sources could not have been affected by the LVV used in this study for nNOS knock-down. In addition, the amplitude and duration of the action potential afterhyperpolarization are increased in neonatal spinal motoneurons after sciatic nerve crushing (Mentis et al. 2007). Yet, incubation of slices containing HMNs with DEA/NO failed to alter afterhyperpolarization in motoneurons (F. Portillo, S. Kasparov & B. Moreno-López, unpublished results) even though the NO donor induced membrane potential depolarization (Gonzalez-Forero et al. 2007). Thus, NO-independent alterations of afterhyperpolarization induced by nerve injury might explain the ineffectiveness of LVV-miR-shRNA/nNOS in preventing decrease in the firing rate at basal conditions of injured HMNs.
A comparison of the time courses of pathophysiological changes and nNOS expression in HMNs after XIIth nerve crushing supports the idea that alterations in the baseline inspiratory drive and in antidromic latency are independent of NO-mediated mechanisms. Expression of nNOS in injured motoneurons begins at day 3 post-injury, and then rises to its peak at day 7, after which it gradually returns to control values (Moreno-Lopez, 2010). This time course was similar to that observed in synaptic alterations (Sunico et al. 2005) and chemoreceptor-modulated inspiratory drive (Gonzalez-Forero et al. 2004) of injured HMNs. However, changes in the inspiratory discharge pattern at baseline and in antidromic latency reached their maximum at day 15 after XIIth nerve injury (Gonzalez-Forero et al. 2004), suggesting that these alterations do not have a close temporal relationship with nNOS up-regulation.
In summary, we report here that de novo expression of nNOS creates a gradient of NO around the motoneurons in an activity-dependent manner that can affect their intrinsic membrane properties and synaptic coverage. These results point to nNOS as a pivotal target for the development of therapeutic tools for the treatment of peripheral neuropathies and neurodegenerative disorders characteristically accompanied by nNOS up-regulation.
Acknowledgments
This work was supported by grants from the Ministerio de Ciencia e Innovación, (SAF2008-01415) and the Consejería de Innovación, Ciencia y Empresa from the Junta de Andalucía, (PAI2007-CTS-02606), both co-financed with FEDER funds and the Mútua Madrileña Foundation, Spain (B.M.-L.). B.L. and S.K. were supported by the British Heart Foundation (RG/07/006). C.R.S. was supported by a personal fellowship (AP2003-3548) from the Ministerio de Educación y Ciencia. F.M. received a fellowship (AP2006-03925) from the Ministerio de Ciencia e Innovación, Spain. We thank Elaine Lilly, PhD (Writer's First Aid), for English language revision of this manuscript.
Glossary
Abbreviations
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- AVV
adenoviral vector
- AVV-eGFP
adenoviral vector directing the expression of the enhanced green fluorescent protein
- AVV-mRFP
adenoviral vector directing the expression of the monomeric red fluorescent protein
- AVV-nNOS
adenoviral vector directing the expression of the neuronal nitric oxide synthase
- BR
burst rate
- eNOS
endothelial nitric oxide synthase
end-tidal CO2
- FR
instantaneous firing rate
- hCMV
human cytomegalovirus
- HD
Huntington's disease
- HMN
hypoglossal motoneuron
- HN
hypoglossal nucleus
- iNOS
inducible nitric oxide synthase
- LVV
lentiviral vector
- LVV-miR-shRNA/nNOS
lentiviral vector harbors an expression cassette for a miRNA30 (miR30)-based short hairpin (shRNA) interference system for neuronal nitric oxide synthase
- mFR
mean firing rate
- 7-NI
7-nitroindazole
- nNOS
neuronal nitric oxide synthase
- NO
nitric oxide
- pfu
plaque forming unit
- PD
Parkinson's disease
- ROIs
regions of interest
- SB
number of spikes per burst
- sGC
soluble guanylyl cyclase
- Syn-ir
synaptophysin immunoreactive
- Tet
tetracycline
- Th
threshold
- TU
transducing unit
- XIIth
hypoglossal nerve
Author contributions
B.M.L. was responsible for conception of the study. B.M.L. and S.K. contributed in the design of the experiments. F.M. carried out electrophysiological recordings and histological/immunohistochemical studies. C.R.S. performed real-time imaging experiments. B.L., J.F.R.P. and S.K. developed viral vectors and performed western blotting procedures. F.M., C.R.S. and B.L. analysed the data. B.M.L. and S.K. were involved in the interpretation of the data. All authors approved the final version of the manuscript. Viral vectors were developed and real-time imaging experiments were performed in the Department of Physiology, School of Medical Sciences, University of Bristol, Bristol, UK. Electrophysiological recordings and histological/immunohistochemical studies were conducted in the Área de Fisiología, Facultad de Medicina, Universidad de Cádiz, Cádiz, Spain.
Author's present address
C. R. Sunico: Burnham Institute for Medical Research, 10901 North Torrey Pines Road, La Jolla, CA 92037, USA.
Supplemental material
MATERIAL AND METHODS
Figure S1
Figure S2
Figure S3
Figure S4
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Ahern GP, Klyachko VA, Jackson MB. cGMP and S-nitrosylation: two routes for modulation of neuronal excitability by NO. Trends Neurosci. 2002;25:510–517. doi: 10.1016/s0166-2236(02)02254-3. [DOI] [PubMed] [Google Scholar]
- Artinian L, Tornieri K, Zhong L, Baro D, Rehder V. Nitric oxide acts as a volume transmitter to modulate electrical properties of spontaneously firing neurons via apamin-sensitive potassium channels. J Neurosci. 2010;30:1699–1711. doi: 10.1523/JNEUROSCI.4511-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierwaltes WH. Selective neuronal nitric oxide synthase inhibition blocks furosemide-stimulated renin secretion in vivo. Am J Physiol Renal Physiol. 1995;269:F134–139. doi: 10.1152/ajprenal.1995.269.1.F134. [DOI] [PubMed] [Google Scholar]
- Bellamy TC, Griffiths C, Garthwaite J. Differential sensitivity of guanylyl cyclase and mitochondrial respiration to nitric oxide measured using clamped concentrations. J Biol Chem. 2002;277:31801–31807. doi: 10.1074/jbc.M205936200. [DOI] [PubMed] [Google Scholar]
- Bland-Ward PA, Moore PK. 7-Nitro indazole derivatives are potent inhibitors of brain, endothelium and inducible isoforms of nitric oxide synthase. Life Sci. 1995;57:PL131–135. doi: 10.1016/0024-3205(95)02046-l. [DOI] [PubMed] [Google Scholar]
- Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A, Musella A, D’Amelio M, Cavallucci V, Martorana A, Bergamaschi A, Cencioni MT, Diamantini A, Butti E, Comi G, Bernardi G, Cecconi F, Battistini L, Furlan R, Martino G. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci. 2009;29:3442–3452. doi: 10.1523/JNEUROSCI.5804-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS. The corticostriatal pathway in Huntington's disease. Prog Neurobiol. 2007;81:253–271. doi: 10.1016/j.pneurobio.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Aston-Jones G. Cerebellar injury induces NADPH diaphorase in Purkinje and inferior olivary neurons in the rat. Exp Neurol. 1994;126:270–276. doi: 10.1006/exnr.1994.1064. [DOI] [PubMed] [Google Scholar]
- Chen X, Sheng C, Zheng X. Direct nitric oxide imaging in cultured hippocampal neurons with diaminoanthraquinone and confocal microscopy. Cell Biol Int. 2001;25:593–598. doi: 10.1006/cbir.2001.0734. [DOI] [PubMed] [Google Scholar]
- Coleman P, Federoff H, Kurlan R. A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology. 2004;63:1155–1162. doi: 10.1212/01.wnl.0000140626.48118.0a. [DOI] [PubMed] [Google Scholar]
- Cope TC, Sokoloff AJ. Orderly recruitment among motoneurons supplying different muscles. J Physiol (Paris) 1999;93:81–85. doi: 10.1016/s0928-4257(99)80138-7. [DOI] [PubMed] [Google Scholar]
- Deckel AW, Tang V, Nuttal D, Gary K, Elder R. Altered neuronal nitric oxide synthase expression contributes to disease progression in Huntington's disease transgenic mice. Brain Res. 2002;939:76–86. doi: 10.1016/s0006-8993(02)02550-7. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Libet B, Young RR. The behaviour of chromatolysed motoneurones studied by intracellular recording. J Physiol. 1958;143:11–40. doi: 10.1113/jphysiol.1958.sp006041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emre M. Dementia associated with Parkinson's disease. Lancet Neurol. 2003;2:229–237. doi: 10.1016/s1474-4422(03)00351-x. [DOI] [PubMed] [Google Scholar]
- Eve DJ, Nisbet AP, Kingsbury AE, Hewson EL, Daniel SE, Lees AJ, Marsden CD, Foster OJ. Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson's disease. Brain Res Mol Brain Res. 1998;63:62–71. doi: 10.1016/s0169-328x(98)00259-9. [DOI] [PubMed] [Google Scholar]
- Fernandez-Vizarra P, Fernandez AP, Castro-Blanco S, Encinas JM, Serrano J, Bentura ML, Munoz P, Martinez-Murillo R, Rodrigo J. Expression of nitric oxide system in clinically evaluated cases of Alzheimer's disease. Neurobiol Dis. 2004;15:287–305. doi: 10.1016/j.nbd.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Gao S, Cheng C, Zhao J, Chen M, Li X, Shi S, Niu S, Qin J, Lu M, Shen A. Developmental regulation of PSD-95 and nNOS expression in lumbar spinal cord of rats. Neurochem Int. 2008;52:495–501. doi: 10.1016/j.neuint.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Forero D, Portillo F, Gomez L, Montero F, Kasparov S, Moreno-Lopez B. Inhibition of resting potassium conductances by long-term activation of the NO/cGMP/protein kinase G pathway: A new mechanism regulating neuronal excitability. J Neurosci. 2007;27:6302–6312. doi: 10.1523/JNEUROSCI.1019-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Forero D, Portillo F, Sunico CR, Moreno-Lopez B. Nerve injury reduces responses of hypoglossal motoneurones to baseline and chemoreceptor-modulated inspiratory drive in the adult rat. J Physiol. 2004;557:991–1011. doi: 10.1113/jphysiol.2003.059972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson B, Pinter MJ. Factors determining the variation of the afterhyperpolarization duration in cat lumbar α-motoneurones. Brain Res. 1985;326:392–395. doi: 10.1016/0006-8993(85)90053-8. [DOI] [PubMed] [Google Scholar]
- Hall CN, Garthwaite J. Inactivation of nitric oxide by rat cerebellar slices. J Physiol. 2006;577:549–567. doi: 10.1113/jphysiol.2006.118380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman CJ, Binder MD. Computer simulations of the effects of different synaptic input systems on motor unit recruitment. J Neurophysiol. 1993;70:1827–1840. doi: 10.1152/jn.1993.70.5.1827. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Brecht S, Mayer B, Leah J, Kummer W, Bravo R, Zimmermann M. Long-lasting expression of JUN and KROX transcription factors and nitric oxide synthase in intrinsic neurons of the rat brain following axotomy. J Neurosci. 1993;13:4130–4145. doi: 10.1523/JNEUROSCI.13-10-04130.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto A, Kawanami T, Llena JF, Hirano A. Immunocytochemical studies on synaptophysin in the anterior horn of lower motor neuron disease. J Neuropathol Exp Neurol. 1994;53:196–201. doi: 10.1097/00005072-199403000-00011. [DOI] [PubMed] [Google Scholar]
- Ince PG, Slade J, Chinnery RM, McKenzie J, Royston C, Roberts GW, Shaw PJ. Quantitative study of synaptophysin immunoreactivity of cerebral cortex and spinal cord in motor neuron disease. J Neuropathol Exp Neurol. 1995;54:673–679. doi: 10.1097/00005072-199509000-00009. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Martemyanov KA, Thayer SA. Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci. 2008;28:12604–12613. doi: 10.1523/JNEUROSCI.2958-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno M, Llinas R. Alterations of synaptic action in chromatolysed motoneurones of the cat. J Physiol. 1970;210:823–838. doi: 10.1113/jphysiol.1970.sp009244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno M, Miyata Y, Munoz-Martinez EJ. Differential reaction of fast and slow α-motoneurones to axotomy. J Physiol. 1974;240:725–739. doi: 10.1113/jphysiol.1974.sp010631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo JJ, Schonewille M, Siddique T, Schults AN, Fu R, Bar PR, Anelli R, Heckman CJ, Kroese AB. Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol. 2004;91:571–575. doi: 10.1152/jn.00665.2003. [DOI] [PubMed] [Google Scholar]
- Kuo JJ, Siddique T, Fu R, Heckman CJ. Increased persistent Na+ current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol. 2005;563:843–854. doi: 10.1113/jphysiol.2004.074138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Paton JF, Kasparov S. Viral vectors based on bidirectional cell-specific mammalian promoters and transcriptional amplification strategy for use in vitro and in vivo. BMC Biotechnol. 2008;8:49. doi: 10.1186/1472-6750-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luth HJ, Holzer M, Gertz HJ, Arendt T. Aberrant expression of nNOS in pyramidal neurons in Alzheimer's disease is highly co-localized with p21ras and p16INK4a. Brain Res. 2000;852:45–55. doi: 10.1016/s0006-8993(99)02178-2. [DOI] [PubMed] [Google Scholar]
- Malinski T, Bailey F, Zhang ZG, Chopp M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1993;13:355–358. doi: 10.1038/jcbfm.1993.48. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Chen K, Liu Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J Neurosci. 2005;25:6449–6459. doi: 10.1523/JNEUROSCI.0911-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Diaz E, Moran LB, Navarrete R. Early alterations in the electrophysiological properties of rat spinal motoneurones following neonatal axotomy. J Physiol. 2007;582:1141–1161. doi: 10.1113/jphysiol.2007.133488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero F, Portillo F, Gonzalez-Forero D, Moreno-Lopez B. The nitric oxide/cyclic guanosine monophosphate pathway modulates the inspiratory-related activity of hypoglossal motoneurons in the adult rat. Eur J Neurosci. 2008;28:107–116. doi: 10.1111/j.1460-9568.2008.06312.x. [DOI] [PubMed] [Google Scholar]
- Moore PK, Babbedge RC, Wallace P, Gaffen ZA, Hart SL. 7-Nitro indazole, an inhibitor of nitric oxide synthase, exhibits anti-nociceptive activity in the mouse without increasing blood pressure. Br J Pharmacol. 1993;108:296–297. doi: 10.1111/j.1476-5381.1993.tb12798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PK, Bland-Ward PA. 7-Nitroindazole: an inhibitor of nitric oxide synthase. Methods Enzymol. 1996;268:393–398. doi: 10.1016/s0076-6879(96)68041-0. [DOI] [PubMed] [Google Scholar]
- Moreno-Lopez B. Local isoform-specific NOS inhibition: a promising approach to promote motor function recovery after nerve injury. J Neurosci Res. 2010;88:1846–1857. doi: 10.1002/jnr.22353. [DOI] [PubMed] [Google Scholar]
- Moreno-Lopez B, Gonzalez-Forero D. Nitric oxide and synaptic dynamics in the adult brain: Physiopathological aspects. Rev Neurosci. 2006;17:309–357. doi: 10.1515/revneuro.2006.17.3.309. [DOI] [PubMed] [Google Scholar]
- Moreno-Lopez B, Romero-Grimaldi C, Noval JA, Murillo-Carretero M, Matarredona ER, Estrada C. Nitric oxide is a physiological inhibitor of neurogenesis in the adult mouse subventricular zone and olfactory bulb. J Neurosci. 2004;24:85–95. doi: 10.1523/JNEUROSCI.1574-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Ishiwata Y, Inaba N, Kuroda T, Nakamura Y. Hypoglossal premotor neurons with rhythmical inspiratory-related activity in the cat: localization and projection to the phrenic nucleus. Exp Brain Res. 1994;98:1–12. doi: 10.1007/BF00229103. [DOI] [PubMed] [Google Scholar]
- Peever JH, Shen L, Duffin J. Respiratory pre-motor control of hypoglossal motoneurons in the rat. Neuroscience. 2002;110:711–722. doi: 10.1016/s0306-4522(01)00594-2. [DOI] [PubMed] [Google Scholar]
- Perez-Severiano F, Escalante B, Vergara P, Rios C, Segovia J. Age-dependent changes in nitric oxide synthase activity and protein expression in striata of mice transgenic for the Huntington's disease mutation. Brain Res. 2002;951:36–42. doi: 10.1016/s0006-8993(02)03102-5. [DOI] [PubMed] [Google Scholar]
- Petrov T, Page AB, Owen CR, Rafols JA. Expression of the inducible nitric oxide synthase in distinct cellular types after traumatic brain injury: an in situ hybridization and immunocytochemical study. Acta Neuropathol. 2000;100:196–204. doi: 10.1007/s004019900167. [DOI] [PubMed] [Google Scholar]
- Rao VL, Dogan A, Bowen KK, Dempsey RJ. Traumatic injury to rat brain upregulates neuronal nitric oxide synthase expression and L-[3H]nitroarginine binding. J Neurotrauma. 1999;16:865–877. doi: 10.1089/neu.1999.16.865. [DOI] [PubMed] [Google Scholar]
- Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G, Pettmann B. Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS-linked SOD1 mutations. Neuron. 2002;35:1067–1083. doi: 10.1016/s0896-6273(02)00905-4. [DOI] [PubMed] [Google Scholar]
- Rogerio F, Teixeira SA, Junior HJ, Maria CC, Vieira AS, de Rezende AC, Pereira GA, Muscara MN, Langone F. mRNA and protein expression and activities of nitric oxide synthases in the lumbar spinal cord of neonatal rats after sciatic nerve transection and melatonin administration. Neurosci Lett. 2006;407:182–187. doi: 10.1016/j.neulet.2006.08.035. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Maruyama S. Synapse loss in anterior horn neurons in amyotrophic lateral sclerosis. Acta Neuropathol. 1994;88:222–227. doi: 10.1007/BF00293397. [DOI] [PubMed] [Google Scholar]
- Saxon DW, Beitz AJ. Cerebellar injury induces NOS in Purkinje cells and cerebellar afferent neurons. Neuroreport. 1994;5:809–812. doi: 10.1097/00001756-199403000-00018. [DOI] [PubMed] [Google Scholar]
- Simic G, Lucassen PJ, Krsnik Z, Kruslin B, Kostovic I, Winblad B, Bogdanovi nNOS expression in reactive astrocytes correlates with increased cell death related DNA damage in the hippocampus and entorhinal cortex in Alzheimer's disease. Exp Neurol. 2000;165:12–26. doi: 10.1006/exnr.2000.7448. [DOI] [PubMed] [Google Scholar]
- Singh OV, Yaster M, Xu JT, Guan Y, Guan X, Dharmarajan AM, Raja SN, Zeitlin PL, Tao YX. Proteome of synaptosome-associated proteins in spinal cord dorsal horn after peripheral nerve injury. Proteomics. 2009;9:1241–1253. doi: 10.1002/pmic.200800636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small DH. Network dysfunction in Alzheimer's disease: does synaptic scaling drive disease progression? Trends Mol Med. 2008;14:103–108. doi: 10.1016/j.molmed.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2005;102:13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert JR, Kopp-Scheinpflug C, Baker C, Challiss RA, Mistry R, Haustein MD, Griffin SJ, Tong H, Graham BP, Forsythe ID. Nitric oxide is a volume transmitter regulating postsynaptic excitability at a glutamatergic synapse. Neuron. 2008;60:642–656. doi: 10.1016/j.neuron.2008.08.025. [DOI] [PubMed] [Google Scholar]
- Sturrock RR. Stability of motor neuron and interneuron number in the hypoglossal nucleus of the ageing mouse brain. Anat Anz. 1991;173:113–116. [PubMed] [Google Scholar]
- Sumner BE. A quantitative analysis of boutons with different types of synapse in normal and injured hypoglossal nuclei. Exp Neurol. 1975;49:406–417. doi: 10.1016/0014-4886(75)90097-7. [DOI] [PubMed] [Google Scholar]
- Sunico CR, Gonzalez-Forero D, Dominguez G, Garcia-Verdugo JM, Moreno-Lopez B. Nitric oxide induces pathological synapse loss by a protein kinase G-, Rho kinase-dependent mechanism preceded by myosin light chain phosphorylation. J Neurosci. 2010;30:973–984. doi: 10.1523/JNEUROSCI.3911-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunico CR, Portillo F, Gonzalez-Forero D, Kasparov S, Moreno-Lopez B. Evidence for a detrimental role of nitric oxide synthesized by endothelial nitric oxide synthase after peripheral nerve injury. Neuroscience. 2008;157:40–51. doi: 10.1016/j.neuroscience.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Sunico CR, Portillo F, Gonzalez-Forero D, Moreno-Lopez B. Nitric oxide-directed synaptic remodeling in the adult mammal CNS. J Neurosci. 2005;25:1448–1458. doi: 10.1523/JNEUROSCI.4600-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titmus MJ, Faber DS. Axotomy-induced alterations in the electrophysiological characteristics of neurons. Prog Neurobiol. 1990;35:1–51. doi: 10.1016/0301-0082(90)90039-j. [DOI] [PubMed] [Google Scholar]
- Tornieri K, Rehder V. Nitric oxide release from a single cell affects filopodial motility on growth cones of neighboring neurons. Dev Neurobiol. 2007;67:1932–1943. doi: 10.1002/dneu.20572. [DOI] [PubMed] [Google Scholar]
- Vazquez C, Anesetti G, Martinez Palma L. Transient expression of nitric oxide synthase in the hypoglossal nucleus of the rat during early postnatal development. Neurosci Lett. 1999;275:5–8. doi: 10.1016/s0304-3940(99)00686-2. [DOI] [PubMed] [Google Scholar]
- Wang S, Paton JF, Kasparov S. Differential sensitivity of excitatory and inhibitory synaptic transmission to modulation by nitric oxide in rat nucleus tractus solitarii. Exp Physiol. 2007;92:371–382. doi: 10.1113/expphysiol.2006.036103. [DOI] [PubMed] [Google Scholar]
- Wang S, Teschemacher AG, Paton JF, Kasparov S. Mechanism of nitric oxide action on inhibitory GABAergic signaling within the nucleus tractus solitarii. FASEB J. 2006;20:1537–1539. doi: 10.1096/fj.05-5547fje. [DOI] [PubMed] [Google Scholar]
- Wolff DJ, Lubeskie A, Umansky S. The inhibition of the constitutive bovine endothelial nitric oxide synthase by imidazole and indazole agents. Arch Biochem Biophys. 1994;314:360–366. doi: 10.1006/abbi.1994.1454. [DOI] [PubMed] [Google Scholar]
- Wood J, Garthwaite J. Models of the diffusional spread of nitric oxide: implications for neural nitric oxide signalling and its pharmacological properties. Neuropharmacology. 1994;33:1235–1244. doi: 10.1016/0028-3908(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Yu WH. Regulation of nitric oxide synthase expression in motoneurons following nerve injury. Dev Neurosci. 1997;19:247–254. doi: 10.1159/000111213. [DOI] [PubMed] [Google Scholar]
- Zagvazdin YS, Fitzgerald ME, Sancesario G, Reiner A. Neural nitric oxide mediates Edinger-Westphal nucleus evoked increase in choroidal blood flow in the pigeon. Invest Ophthalmol Vis Sci. 1996;37:666–672. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.