

Abstract
BK channels are large unitary conductance K+ channels cooperatively activated by intracellular calcium and membrane depolarisation. We show that BK channels regulate electrical activity in β-cells of mouse pancreatic islets exposed to elevated glucose. In 11.1 mm glucose, the non-peptidyl BK channel blocker paxilline increased the height of β-cell action potentials (APs) by 21 mV without affecting burst- or silent-period durations. In isolated β-cells, paxilline increased AP height by 16 mV without affecting resting membrane potential. In voltage clamp, paxilline blocked a transient component of outward current activated by a short depolarisation, which accounted for at least 90% of the initial outward K+ current. This BK current (IBK) was blocked by the Ca2+ channel blockers Cd2+ (200 μm) or nimodipine (1 μm), and potentiated by FPL-64176 (1 μm). IBK was also 56% blocked by the BK channel blocker iberiotoxin (100 nm). IBK activated more than 10-fold faster than the delayed rectifier IKv over the physiological voltage range, and partially inactivated. An AP-like command revealed that IBK activated and deactivated faster than IKv and accounted for 86% of peak IK, explaining why IBK block increased AP height. A higher amplitude AP-like command, patterned on an AP recorded in 11.1 mm glucose plus paxilline, activated 4-fold more IKv and significantly increased Ca2+ entry. Paxilline increased insulin secretion in islets exposed to 11.1 mm glucose by 67%, but did not affect basal secretion in 2.8 mm glucose. These data suggest a modified model of β-cell AP generation where IBK and IKv coordinate the AP repolarisation.
Introduction
Elevated ambient glucose triggers action potentials (APs) in β-cells of murine pancreatic islets (Dean & Matthews, 1968). It is thought that voltage-dependent Ca2+ current (ICav) underlies the AP upstroke, and delayed rectifier K+ current (IKv, mediated mostly by Kv2.1 channels (Roe et al. 1996; MacDonald & Wheeler, 2003; Herrington et al. 2006)) drives the downstroke (Ashcroft & Rorsman, 1989; Houamed et al. 2004).
BK channels are K+ channels of large unitary conductance that are activated cooperatively by elevated [Ca2+]i and membrane depolarisation (Marty, 1981; Kaczorowski et al. 1996; Vergara et al. 1998). This dual sensitivity allows BK channels to subserve disparate physiological functions in many cell types (Orio et al. 2002). BK channels vary widely in their biophysical and pharmacological properties as a result of multiple splicing of the ion-conducting α-subunit mRNA (Adelman et al. 1992; Butler et al. 1993), differential incorporation of four modulatory β-subunits (Orio et al. 2002), and multiple phosphorylation patterns (Yan et al. 2008).
Rodent β-cells and certain insulinomas express BK channels (Cook et al. 1984; Findlay et al. 1985; Satin et al. 1989; Tabcharani & Misler, 1989; Bokvist et al. 1990; Mancilla & Rojas, 1990; Smith et al. 1990b; Kukuljan et al. 1991; Li et al. 1999), but the role of these channels in β-cell physiology has been controversial. Early studies and theoretical models proposed that IBK regulated glucose-dependent electrical excitability in β-cells (Atwater et al. 1979, 1983; Chay, 1986). However, Smith et al. (1990b) observed that IBK constituted only a minor and variable component of the depolarisation-activated IK, concluding that it was thus unlikely to play a major role in shaping APs. Similarly, Kukuljan et al. (1991) reported that the IBK blocker charybdotoxin (ChTx) did not affect glucose-induced APs or islet bursting, and concluded that IBK did not participate in glucose-induced electrical activity. Thus, there emerged a consensus that BK channels do not significantly participate in the glucose-mediated islet electrical activity or in glucose-stimulated insulin secretion (GSIS) in mouse. However, β-cells from a Kv2.1 knockout mouse lacking IKv are still capable of AP repolarisation, and moreover, express a transient outward K+ current of unknown molecular identity (Jacobson et al. 2007). Similarly, toxin block of β-cell Kv2.1 channels slowed, but did not inhibit, AP repolarisation. Further, human β-cells have recently been shown to express a robust IBK whose pharmacological modulation affects both AP shape and GSIS (Braun et al. 2008).
The present study examines whether IBK is activated in primary mouse β-cells under physiological conditions, and investigates its possible role in the control of islet electrical activity and GSIS. Preliminary results have been described previously in abstract form (Houamed & Satin, 2009).
Methods
Isolation of islets and single β-cells
Islets were isolated from adult male Swiss Webster mice as previously described (Goforth et al. 2002; Zhang et al. 2005), using a protocol approved by the University of Michigan University Committee on the Use and Care of Animals, and following published standards (Drummond, 2009). Animals were killed by neck dislocation. The pancreas was perfused through the bile duct with cold Krebs solution containing 1 mg ml−1 collagenase P (Roche Diagnostics, Indianapolis, IN, USA) and 0.1% bovine serum albumin (BSA), then rapidly excised, incubated at 37°C for 15 min, triturated and washed. Krebs solution contained (in mm): 137 NaCl, 5 KCl, 1.2 MgCl2, 1 CaCl2, 5 NaHCO3, 5 glucose, 10 Hepes; pH 7.35. Single β-cells and islets were dispersed in Spinner's salts, washed with culture medium and plated onto glass coverslips. Islets and single β-cells were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum and antibiotics (Invitrogen, Carlsbad, CA, USA) in 5% CO2 at 37°C. Most islets and single cells were used within 24 h and 72 h, respectively. As we have noted previously (Zhang et al. 2003), these culture periods did not appear to affect the electrical properties or the glucose sensitivity of islets and individual β-cells.
Electrophysiology
Single islets or coverslips with attached β-cells were placed in a ∼300 μl recording chamber constantly superfused, at 2 ml min−1 and 33.5°C, with a saline solution containing (in mm): 140 NaCl, 10 Hepes, 1 MgCl2, 2.5 CaCl2, 3.6 KCl, pH 7.35. Drugs and glucose were dissolved directly into the saline solution; when drugs were diluted from DMSO stocks, the highest final concentration of the solvent was <0.1%. Iberiotoxin solutions contained 0.1% BSA to prevent the toxin from sticking to the perfusion system. Glucose (11.1 mm) was added for membrane potential recording; for voltage-clamp recordings, 5 mm glucose and 200 μm tolbutamide were present. The two glucose concentrations, which have been used in β-cell studies involving measurement of membrane potential and current, Ca2+i dynamics, and insulin secretion (Gopel et al. 1999b; Zhang et al. 2005), were used to facilitate comparison with previous studies. For ICav measurements, 30 mm TEA was added to block IK, and NaCl reduced accordingly to maintain the osmolarity of the saline solution.
Recordings utilized the perforated patch technique, using the pore-forming compounds nystatin or amphotericin-B (Falke et al. 1989). Pipette resistance was 1.6–4.4 MΩ when filled with a solution composed of (in mm): 76 K2SO4, 5 NaCl, 10 KCl, 1 MgCl2, 10 Hepes, pH 7.35. For ICav measurements, Cs2SO4 replaced K2SO4. The pore formers were dissolved in DMSO at 50 μg ml−1 then diluted in pipette solution to a final concentration of 250 μg ml−1. Pipette tips were dipped in pipette solution, then back-filled with pipette solution containing the pore former. Recordings commenced when the series resistance (Rs) was <40 MΩ; most of the remaining Rs was compensated electronically so that the voltage error generated by the largest currents was <6 mV. All salts and drugs were from Sigma (St Louis, MO, USA), except paxilline, which was from Enzo Life Sciences (Plymouth Meeting, PA, USA), and iberiotoxin, which was from AnaSpec (Fremont, CA, USA).
Functional criteria used to identify β-cells in situ on the surface of an islet were presence of AP bursts in 11.1 mm glucose, membrane capacitances >5.7 pF, and the absence of an early Na+ current that remains activatable at physiological membrane potentials (Gopel et al. 1999a). Similar criteria were used to identify dissociated β-cells; in addition, we visually selected for larger cells, which were more likely to be β-cells (Leung et al. 2005). Data reported in the present study are based on recordings obtained from 51 isolated β-cells whose average membrane electrical capacitance was 7.86 ± 0.32 pF. The average capacitance of β-cells in situ in islets was 8.34 ± 0.39 pF (n = 18).
An EPC9 amplifier and PULSE software (HEKA, Lambrecht/Pfalz, Germany) provided stimulation and data acquisition. This study utilized four pulse protocols (P1, AP1, AP2 and IV1). P1 consisted of 40 ms depolarising pulse from −70 mV to −10 mV. AP1 and AP2 were synthetic APs patterned on β-cell APs recorded in 11.1 mm glucose without and with 1 μm paxilline, respectively (Fig. 1D). Each AP command consisted of a −65 mV holding potential segment followed by 14 ramps (Fig. 5A and B). IV1 consisted of 500 ms depolarising test pulses from −70 mV holding potential; test potentials were −60 mV to +10 mV in 10 mV increments. Linear leak and capacitive components of membrane current were subtracted by a combination of analog and digital (p/n) techniques; data traces shown in Figs 2, 5 and 6 are averages of several consecutive recordings. Data were analysed using PULSEFIT (HEKA), Igor (Wavemetrics, Lake Oswego, OR, USA), Excel (Microsoft, Redmond, WA, USA) and Prism (Graphpad, La Jolla, CA, USA). APs were analysed using Synaptosoft (Decatur, GA, USA). Recordings were corrected for a +10.8 to +12.3 mV calculated junction potential (Neher, 1992).
Figure 1. Effects of BK channel blocker paxilline on the electrical activity of glucose-stimulated islets and single β-cells.
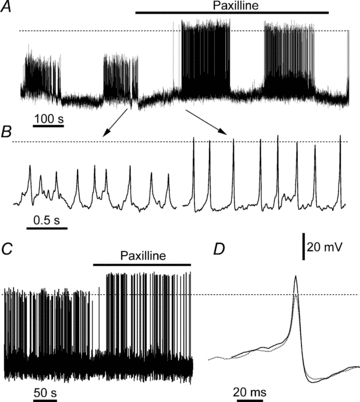
A, membrane potential recording in an islet exposed to 11.1 mm glucose, showing typical bursting. Paxilline (1 μm; horizontal bar) dramatically increased AP height. B, data from A on an expanded time scale. The arrows show approximate location of traces in A. C, effect of paxilline on APs of a single β-cell exposed to 1 μm paxilline in 11.1 mm glucose (denoted by the horizontal bar). D, two APs extracted from the trace in C are shown superimposed; dotted and continuous traces were recorded in the absence and presence of paxilline, respectively. Dashed lines denote 0 mV level; all panels have the same voltage calibration.
Figure 5. Activation of IBK and IKv by waveform commands patterned on β-cell APs.
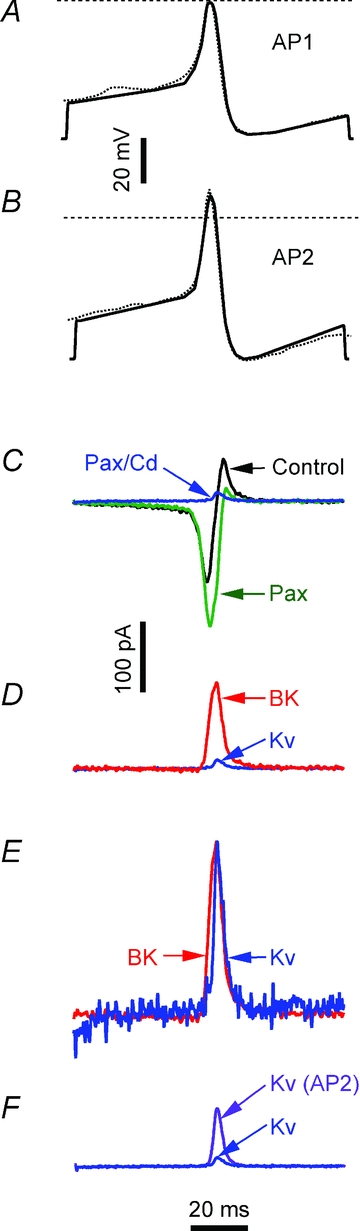
The waveform in A: low amplitude AP1 command (continuous line) patterned on the AP (dotted line), recorded in 11.1 mm glucose, shown in Fig. 1D. B, high amplitude AP2 command (continuous line) patterned on the high amplitude AP, recorded in 11.1 mm glucose and 1 μm paxilline, shown in Fig. 1B. Voltage calibration bar refers to A and B. C, membrane currents activated by AP1. ‘Control’ (black) was recorded in 5 mm glucose and 200 μm tolbutamide. ‘Pax’ (green) was recorded after 1 μm paxilline. ‘Pax/Cd’ (blue) was recorded in 1 μm paxilline and 200 μm Cd2+. D, comparison of IBK and IKv activated by AP1. ‘BK’ (red) is the paxilline-sensitive IBK component, obtained by subtracting ‘Pax’ from ‘Control’ in A. ‘Kv’ (blue) is IKv, the current remaining after paxilline and Cd2+ have blocked IBK and ICav; it is identical to the ‘Pax/Cd’ trace in C. E, comparison of IBK and IKv activation and deactivation time courses. IBK (‘BK’; red) and IKv (‘Kv’; blue) have been normalized to their respective peaks (arbitrary amplitude scale). F, AP2 command enhances IKv activation. ‘Kv’ (blue) is the same IKv trace marked ‘Pax/Cd’ and ‘Kv’, in C and D, respectively. ‘Kv AP2’ (purple) is IKv elicited by AP2 command. Current calibration bar refer to C, D and F; vertical calibration of E is arbitrary; the time calibration bar refers to all the panels.
Figure 2. Identification of IBK in voltage-clamped β-cells.
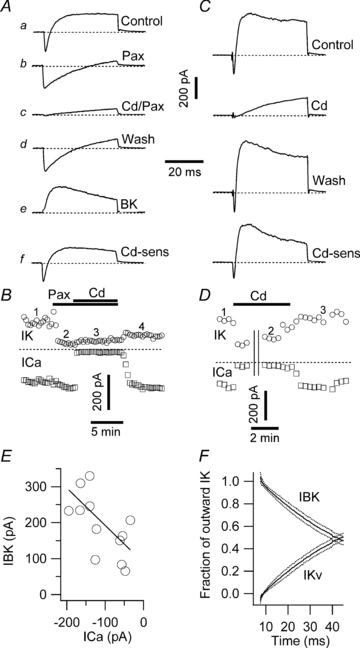
A, membrane currents recorded in response to P1 command. a was recorded in 5 mm glucose and 200 μm tolbutamide. b was recorded in the added presence of 1 μm paxilline; c was recorded in the added presence of 200 μm Cd2+. d shows full recovery of inward current and partial recovery of the outward current upon return to ‘Control’ solution. e shows the paxilline-sensitive IBK, obtained by subtraction of b from a. f shows Cd2+-sensitive current, obtained by subtraction of c from a. B, time course of the experiment illustrated in A. Circles and squares denote the peak outward IK and inward ICav currents, respectively. The numbers above the graph depict the approximate positions for a, b, c and d shown in A. The horizontal bars indicate periods of paxilline and Cd2+application. C, membrane currents, recorded in a different β-cell. ‘Control’ trace was recorded in solution containing 5 mm glucose and 200 μm tolbutamide. ‘Cd’ trace was recorded in the presence of 200 μm Cd2+ to block ICav and Ca2+-dependent currents. ‘Wash’ trace shows full recovery of inward and outward current components upon return to Control solution. ‘Cd-sens’ trace shows Cd2+-sensitive current (IBK, ICav and potentially other Ca2+-sensitive currents) obtained by subtraction of Cd from Control traces. The calibration bar is the same as in A. D, time course of experiment in C. Circles and squares denote peak outward and inward currents, respectively. The numbers above the graph depict positions of the traces shown in C; the horizontal bar indicates duration of Cd2+ application. Both inward and outward currents were blocked by Cd2+ and recovered rapidly upon washout. Parallel vertical lines depict a 255 s break in the recording while other protocols were executed. Dashed lines in A–D indicate zero current level. E, correlation between ICav and IBK in individual β-cells. The continuous line shown is a least-square fit to the data with slope −0.9763. F, dynamics of the contributions of IBK and IKv to current elicited by a short depolarizing pulse. The proportion of total outward IK carried by IBK and IKv, respectively, are shown as a function of time after the pulse commenced at 5 ms. The continuous lines represent the mean of 12 cells; the dotted lines represent the s.e.m.
Figure 6. ICav activation by AP-waveform commands.
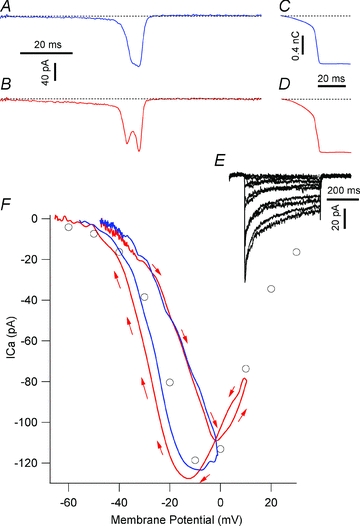
ICav activated by AP1 (A) and AP2 (B) commands. C and D show the corresponding cumulative Ca2+influx (QCav). The dashed lines indicate the zero ICav or QCav. E, a family of ICav traces activated by IV1 protocol, recorded in the same cell as panels A and C. F, comparison of ICav waveforms activated by AP commands and step depolarisations. Circles represent the voltage dependence of peak ICav traces activated by the depolarizing steps in E. Blue and red curves represent the ICavs activated by AP1 and AP2, plotted as a function of their voltage waveforms. The red arrows illustrate the direction of the current progression with time in response to AP2 command. Both currents loop clockwise, beginning and ending at the top left corner of the panel; the current activated by AP2 (red trace) has an additional anticlockwise loop, covering ∼−5 to +15 mV potential range, nested within the main current loop.
Insulin secretion measurements
Insulin secretion measurements were as previously described (Jung et al. 2009). Islets were preincubated for 60 min in Krebs–Ringer bicarbonate solution composed of (in mm): 98.5 NaCl, 4.9 KCl, 2.6 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25.9 NaHCO3, 20 Hepes, 2.8 glucose and 0.1% BSA. Batches of 10 islets were incubated for 60 min in 200 μl of RPMI 1640 media containing either 2.8 or 11.1 mm glucose, in the presence or absence of 1 μm paxilline; supernatant insulin was determined by radioimmunoassay (RIA; Linco Research Inc., St Charles, MO, USA). Determinations were in triplicate, and the experiments were repeated on three different islet preparations.
Statistics
Error bars denote s.e.m.; Student's t test was used to determine statistical significance.
Results
The effect of BK channel block on glucose-dependent electrical activity in islets
Islets exposed to elevated glucose generate AP bursts separated by silent intervals (Fig. 1A) (Henquin & Meissner, 1984; Gopel et al. 1999b; Zhang et al. 2003, 2005). Slow-, fast- and mixed-bursting patterns, ranging in frequency from ∼2 to ∼0.2 min−1, have been observed in individual islets (Zhang et al. 2003). We investigated the effect of 1 μm paxilline on the membrane potential of superficial β-cells (n = 22; average recording per cell ∼25 min) on intact islets stimulated by 11.1 mm glucose (Fig. 1A). Paxilline is a non-peptidyl tremourogenic BK channel inhibitor with nanomolar potency (Knaus et al. 1994; Sanchez & McManus, 1996). As the effects of paxilline were poorly reversible, each islet or single β-cell was exposed to the drug only once.
Analysis of burst- and silent-interval durations in pooled data revealed that the pattern of alternating bursts and silent intervals was unaffected by addition of paxilline, while AP amplitude was increased dramatically. The mean burst duration was 178.29 ± 39.80 s (n = 18 islets) in 11.1 mm glucose, and 162.37 ± 32.14 s (n = 17) after paxilline addition; these values were not significantly different (P = 0.76). Likewise, the mean silent interval duration in 11.1 mm glucose was 106.31 ± 18.85 s (n = 16), and 94.80 ± 14.55 s (n = 16) in paxilline; this difference was also not statistically significant (P = 0.63). In terms of duty cycle, islets spent on average 62.7% of the time bursting in 11.1 mm glucose alone, and 63.1% when paxilline was present.
Paxilline changed the baseline potential immediately preceding each AP by a statistically insignificant −0.5 ± 0.9 mV, but significantly potentiated the AP peak by 20.3 ± 3.0 mV (n = 6; P < 0.0001; Fig. 1B). Consequently, BK channel blockade in islets exposed to 11.1 mm glucose resulted in increased AP height, but did not alter burst dynamics.
The effect of BK channel block on glucose-dependent APs in single β-cells
In contrast to the bursting behaviour seen in intact islets, elevated glucose triggers continuous AP firing in most single β-cells, with a minority of cells showing a variety of bursting behaviours (Smith et al. 1990a; Ammala et al. 1991; Kinard et al. 1999). To test whether the effects of paxilline on APs in whole islets were recapitulated in isolated single β-cells, membrane potential was recorded in 11.1 mm glucose in the absence and presence of 1 μm paxilline (Fig. 1C). Analysis of 751 APs in 11.1 mm glucose and 352 APs in the added presence of paxilline showed that paxilline enhanced AP peak height by an average of 15.6 ± 0.5 mV, a difference which was highly significant (P < 0.0001) in each of the four β-cells tested. In contrast, paxilline had no significant effect on baseline potential (0.1 ± 1.0 mV).
Figure 1D shows two exemplar APs, selected from the trace in Fig. 1C, plotted on an expanded time scale and manually aligned so that their peaks coincided temporally. The dotted-line AP was recorded in 11.1 mm glucose, and the continuous-line AP was recorded after 1 μm paxilline. Paxilline potentiated AP height, resulting in a positive overshoot, did not affect baseline potential, and did not appear to affect AP width, nor did it inhibit the afterhyperpolarisation (AHP) phase that often follows the AP.
IBK mediates a pharmacologically and kinetically distinct component of the outward current activated by a brief depolarisation
Membrane current recorded in single mouse β-cells elicited by a P1 protocol consisted of an early inward component that relaxed outwards (Fig. 2Aa). The outward component has been considered to be mainly delayed rectifier IKv (Smith et al. 1990b; Roe et al. 1996). However, paxilline block revealed that IBK constituted a substantial component of the outward IK that was larger than the paxilline-insensitive IKv component (Fig. 2Ab).
The early inward current was blocked by 200 μm Cd2+ (Fig. 2Ac), confirming its identity as ICav (Satin & Cook, 1985; Rorsman & Trube, 1986; Smith et al. 1993; Jing et al. 2005). Addition of Cd2+ in the presence of paxilline did not decrease IKv further (compare Fig. 2Ab and 2Ac). Upon removal of paxilline and Cd2+, ICav recovered rapidly and fully, whereas IBK recovered slowly and partially (Fig. 2Ad). The difference current traces in Fig. 2Ae and 2Af (Fig. 2Ab minus 2Aa, Fig. 2Ac minus 2Aa, respectively) represent IBK and the paxilline- and Cd2+-sensitive current.
The time course of the experiment shown in Fig. 2A is plotted in Fig. 2B, illustrating that paxilline blocks the IBK component of outward current in a slowly reversible manner, without affecting the inward, Cd2+-sensitive, ICav. In this experiment, the poor reversibility of paxilline block did not allow us to distinguish whether Cd2+ and paxilline were blocking the same IK component. In an independent set of experiments Cd2+, applied alone, blocked a current grossly indistinguishable from that seen when Cd2+ was combined with paxilline (Fig. 2C). However, unlike with paxilline, Cd2+ block recovered readily upon drug washout (Fig. 2D). In pooled data, Cd2+ alone blocked 60.0 ± 11.6% of the peak total outward current (n = 11), and in combination with paxilline blocked 60.2 ± 3.6% (n = 17); the two values were not significantly different. IKv accounted for 38.3 ± 3.7% of the peak total outward current (n = 17). Therefore, 1 μm paxilline blocks the β-cell IBK selectively and completely.
Figure 2E depicts peak IBK as a function of the corresponding peak ICav, determined in12 separate β-cells, spanning ∼5.7- and ∼5.0-fold ranges of ICav and IBK, respectively. The continuous line fit to the data has a slope of −0.9763; ICav and IBK are significantly correlated (P = 0.0267).
Additional pharmacological evidence for a Ca2+-activated IK was provided by the L-type ICav blocker nimodipine (1 μm) and the enhancer FPL-64176 (1 μm; McDonough et al. 2005), which reduced and enhanced outward current by 15.6 ± 3.3% (P = 0.020; n = 3) and 72.2 ± 38.1% (P = 0.099; n = 3), respectively.
The scorpion-toxin peptide iberiotoxin (IbTx) is a ChTx analogue that is equally potent at blocking BK channels; unlike ChTx, which was used in an earlier study of BK channels in β-cells (Kukuljan et al. 1991), IbTx does not block Kv channels (Galvez et al. 1990). In the current study, IbTx blocked 56.2 ± 12.6% of the paxilline-sensitive IBK current (n = 4; individual-cell block ranged from 22.3 to 81.2%).
To compare their activation time courses quantitatively, IBK and IKv in 12 cells were normalized to the total outward IK (i.e. IBK+IKv; Fig. 2F). IBK carries most of the early repolarizing current (∼100%, 2.6 ms into the pulse). Concurrent IBK inactivation and IKv activation result in either current approaching 50% by the end of the 40 ms pulse.
Taken together, these observations show that IBK constitutes a sizeable component of the outward IK activated by depolarizing pulses in mouse β-cells, in agreement with our observation that paxilline increased the amplitude of β-cell APs.
Membrane potential- and time-dependence of IBK in β-cells
Time- and voltage-dependence of IBK activation and inactivation were characterized using the IV1 protocol. The multiphasic and noisy currents in Fig. 3A were recorded in 5 mm glucose and 200 μm tolbutamide. Paxilline blocked IBK, resulting in outward currents with slower activation, less inactivation and less noise (Fig. 3B). IKv recorded in paxilline and Cd2+ activated exponentially (continuous lines superimposed on the current traces; Fig. 3C). Subtracting traces in Fig. 3B from those in Fig. 3A revealed IBK (Fig. 3D), characterized by rapid activation, partial inactivation and prominent fluctuations; IBK inactivated monoexponentially (continuous lines superimposed on current traces). Figure 3E shows the paxilline- and Cd2+-sensitive membrane current component obtained by subtracting traces in Fig. 3C from those in Fig. 3A; addition of Cd2+ following paxilline had little effect on outward currents but, as expected, blocked ICav, so that the subtracted current shown in Fig. 3E closely resembles the current in Fig. 3D, with an added early inward ICav component. This early ICav is shown on an expanded timescale in Fig. 3G.
Figure 3. Voltage- and time-dependence of IBK and IKv.
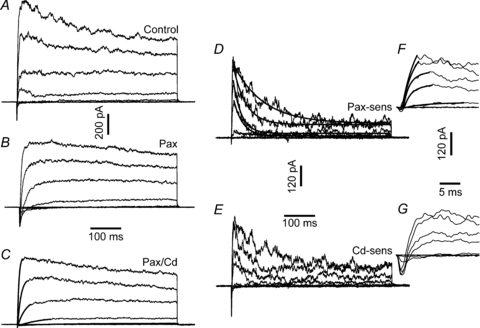
Family of membrane currents activated by IV1 protocol. A, control (5 mm glucose + 200 μm tolbutamide). B, in 1 μm paxilline. C, IKv recorded in the presence of Cd2+ and paxilline. The continuous lines are single exponential fits to the rising phase of the current. D, paxilline-sensitive IBK, obtained by subtracting the currents in B from A. The continuous lines are monoexponential fits of IBK decay. E, Cd2++ paxilline-sensitive current, obtained by subtracting traces in C from A. F, IBK activation. The early phase of IBK, shown in D, is plotted on an expanded time scale. The continuous lines are double exponential fits to IBK rising phase. G, early inward ICav current. The traces were obtained by plotting the early stages of the traces in E on an expanded time scale. The leftmost calibration bars refer to A, B and C; the middle bars refer to D and E, and the rightmost bars refer to F and G.
Normalized conductance–voltage (G–V) curves for IBK and IKv show that both currents activate over a similar membrane potential range (Fig. 4A). The dual time constants (τs) for IBK activation and its inactivation τ, have U-shaped membrane potential dependence (Fig. 4B); IBK activates at least an order of magnitude faster than IKv over all but the most depolarised potentials. Figure 4C shows the ratio of steady-state IBK, recorded at the end of the 500 ms pulse, to peak IBK. The line fit through the data in the range of −20 to +30 mV has a slope of 7.8 × 10−3 mV−1, suggesting that this parameter is slightly voltage dependent.
Figure 4. Voltage dependence of amplitude and kinetics of IBK and IKv.
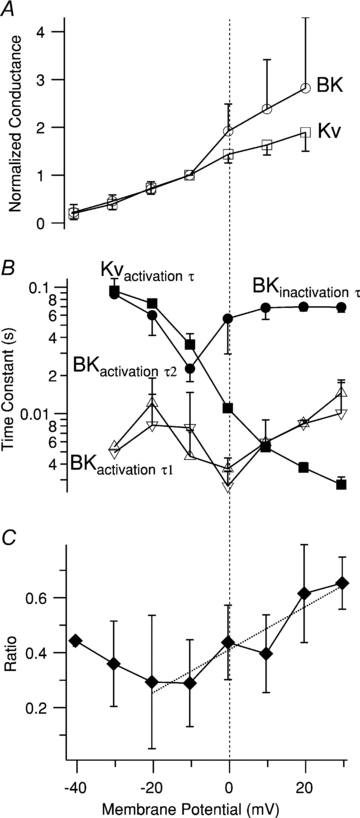
A, normalized conductance–voltage (G–V) curves for IBK (open circles) and IKv (open squares). Each point is the mean ±s.e.m. of 3–4 determinations in different cells normalised to the value at −10 mV. B, voltage dependence of the time constant of IKv activation (Kvactivation τ; filled squares), the time constant of IBK inactivation (BKinactivation τ; filled circles), the first time constant of IBK activation (BKactivation τ1; upright open triangles), and the second time constant of IBK activation (BKactivation τ2; inverted open triangles). Each point is mean ±s.e.m. of 3–4 determinations. Note logarithmic axis for τs. C, voltage dependence of ratio of steady-state IBK current (average of IBK over the last 100 ms of the 500 ms pulse) to peak IBK. Each point is the average of 3–4 measurements in separate cells, except the leftmost point, which is a single determination. The dotted line is a linear fit to the data between −20 and +30 mV; its slope is 0.0078 mV−1. The membrane potential axis is the same for all panels; the vertical dashed line denotes 0 mV.
Therefore, IBK activates with the same voltage dependence, but ∼10-fold faster, as IKv, and inactivates rapidly to a steady-state component that is ∼30 to ∼60% of the peak.
K+ currents activated by voltage waveforms patterned on APs
To characterize the roles of IBK and IKv in AP repolarisation, currents were evoked by two voltage commands (AP1 and AP2) patterned on APs recorded in 11.1 mm glucose alone and with paxilline, respectively (Figs 1D, and 5A and B). AP1 evoked a biphasic current (Fig. 5C, black trace) comprising an early transient inward component, followed by a transient outward component. Paxilline (green trace) enhanced the inward component and reduced the outward component. Subsequent Cd2+ addition blocked the inward ICav component, revealing a small transient outward IKv (blue trace). A transient outward paxilline-sensitive IBK component was obtained by subtraction of the traces. The AP1-activated IBK component was substantially larger than IKv (red and blue traces in Fig. 5D). In five cells, peak IBK was 7.01 ± 2.29-fold larger than IKv. Normalized IKv and IBK (Fig. 5E) show that IKv activated ∼2 ms slower than IBK, a delay much shorter than observed with the P1 step voltage command (Figs 2–4).
The AP2 command was applied in the presence of paxilline and Cd2+ to investigate the IKv-dependent repolarizing mechanisms in the higher amplitude APs observed in paxilline (Fig. 5B). AP2 activated a dramatically larger (4.11 ± 0.76-fold; n = 5) IKv than AP1 (Fig. 5F). In a paired comparison, peak amplitude of IKv activated by AP2 was not significantly different from the peak amplitude of IBK activated by AP1 (n = 5; P = 0.1440), indicating that the IKv increase observed with AP2 suffices in repolarizing the AP in the absence of IBK.
AP2 peak is 13 mV more positive than AP1. To determine the contribution of increased conductance (GKv) or K+ electromotive driving force in the IKv increase, a point-by-point subtraction of the K+ reversal potential (determined from IKv tail current protocol; data not shown) from AP commands resulted in driving-voltage (ΔV) waveforms. IKv was divided by ΔV to produce the GKv waveform. Comparison of the peak amplitudes of the GKv and IKv waveforms activated by AP1 and AP2 revealed that increased GKv underlay 91.28 ± 2.17% (n = 8) of the IKv increase, with the remaining ∼8% increase due to increased ΔV. This is considerably larger than the ∼20% expected from the steady-state GKv calculated from the step activation experiments illustrated in Fig. 4A.
Thus, because the change in peak AP height observed in paxilline occurs over a potential range where the GKv is steeply voltage dependent, the enhanced activation of IKv compensates for the loss of IBK as a repolarizing mechanism.
Ca2+ currents activated by voltage waveforms patterned on APs
AP1 and AP2 were applied in the presence of K+ channel blockers to characterize the waveform of ICav underlying the AP upstroke and to investigate whether the increase in AP height seen in paxilline was associated with changes in Ca2+ influx. AP1 and AP2 evoked ICavs with multiple peaks (Figs 6A and B, and 7) that had similar maximum amplitudes. ICav activated by AP1 had a broad staircase-like peak; AP2-evoked ICav had twin peaks separated by a prominent trough. AP2 elicited more Ca2+ influx, calculated as QCav, the time integral of ICav (Fig. 6C and D). In paired recordings, AP2 significantly increased QCav (11.91 ± 4.29%; n = 4; P = 0.037). Additionally, the traces in Fig. 6C and D show that a substantial fraction of ICav activated during the slow depolarisation preceding the AP threshold. This slow ICav component carried 27.03 ± 5.64% and 33.78 ± 6.02% of the total QCav activated by AP1 and AP2, respectively (n = 4). Thus, inhibition of IBK enhances Ca2+ influx per AP, which may subsequently raise [Ca2+]i and potentiate insulin exocytosis.
Figure 7. Temporal alignment of membrane voltage and normalized currents activated by AP1 (A) and AP2 (B).
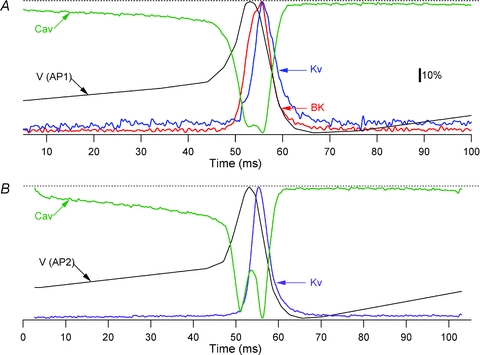
Colour coding for the IK traces is as in Fig. 5. In both panels, black traces marked V (AP1) and V (AP2) represent membrane voltage and green traces marked Cav represent ICav. Dashed lines show zero level for ICav and maximum level for IKv, IBK and membrane voltage. The calibration bar refers to both panels.
Figure 6E shows a set of ICavs activated by the IV1 step protocol recorded in the same cell as panels A and C. ICavs activated by IV1 were biophysically similar to those previously reported for β-cells (Satin & Cook, 1985; Rorsman & Trube, 1986). Figure 6F compares trajectory of ICavs evoked by AP1 and AP2 plotted as a function of voltage to the I–V relation of peak ICavs obtained from the IV1 protocol. ICavs activated by AP commands show more complex time- and voltage-dependence than expected from the I–V relation. In all four cells examined, both AP1 and AP2 evoked ICavs that displayed prominent hysteresis; thus, at any membrane potential, more ICav was activated on the AP downstroke than the upstroke.
BK channel block enhances glucose-stimulated insulin secretion in islets
As IBK enhanced Ca2+ entry during APs, we determined whether paxilline enhanced GSIS. Basal insulin secretion rate, measured in 2.8 mm glucose, was not significantly affected by paxilline (0.028 ± 0.005 and 0.032 ± 0.006 ng (100 islets)−1 min−1 in the absence and presence of 1 μm paxilline, respectively; P > 0.05). In contrast, in the presence of a stimulatory glucose concentration (11.1 mm), 1 μm paxilline significantly enhanced insulin secretion by approximately 67%, from 0.576 ± 0.110 to 0.960 ± 0.211 ng (100 islets)−1 min−1 (P = 0.0148).
Discussion
We report that BK channels activate under physiological conditions and regulate AP amplitude, Ca2+ entry and GSIS in mouse pancreatic β-cells. IBK activates over a similar membrane potential range as IKv; however, it activates more rapidly and inactivates partially; thus, it carries most of the outward current during the initial phases of the AP. Compared to previous studies (Smith et al. 1990b; Kukuljan et al. 1991), our results suggest a more important role for IBK, and a reappraisal of the role of IKv, in β-cell function, and posit that IBK serves a similar role in modulating AP shape and GSIS in mouse as it does in human β-cells (Braun et al. 2008).
While the IBKs in the present study are kinetically similar to previous reports (Smith et al. 1990b; Kukuljan et al. 1991), our observation that IBK constituted >50% and >85% of outward IK activated by P1 and AP1, does not support the previous conclusion that BK channels play at most a minor role in AP regulation (Smith et al. 1990b). Similarly, our observation that paxilline enhanced AP height by ∼21 mV contrasts with the report that ChTx did not affect glucose-stimulated electrical activity in individual β-cells and whole islets (Kukuljan et al. 1991).
IBKs and APs recorded in the present study are somewhat larger than reported previously (Smith et al. 1990b; Kukuljan et al. 1991). Mouse strain-dependent differences in BK- and Cav-channel expression, or the elevated recording temperature and intracellular milieu-preserving perforated patch technique, may have facilitated ICav and contributed to enhanced IBK amplitudes in the present study (Fig. 2E). In addition, use of ChTx by Kukuljan et al. (1991) may have led these authors to underestimate the amplitude and role of IBK. ChTx and IbTx are large charged peptides whereas paxilline is a small lipid-soluble molecule. Therefore, paxilline may have accessed the islet interior better and exerted a bigger effect on APs and GSIS, as has been reported for other tissues (Hu et al. 2001; Imlach et al. 2010). Moreover, use of ChTx and IbTx in previous studies may have led to underestimation of IBK, as we found that IbTx only partially blocked the paxilline-sensitive IBK in mouse β-cells. Parenthetically, the variable (22% to 81%) IbTx-mediated blockade of β-cell IBK, as well as BK inactivation, may reflect the heterogeneous stoichiometry of β-subunits of the underlying BK channels. Inclusion of β1-, β2- and β4-subunits have been shown to reduce ChTx/IbTx potency by ∼11-, ∼50- and >1000-fold, respectively (Wallner et al. 1999; Lippiat et al. 2003), whereas paxilline appears able to equally block all known BK channel stoichiometries (Hu et al. 2001; Imlach et al. 2010). BK channels containing β2- or β3-subunits also inactivate rapidly (Xia et al. 1999, 2003). Therefore, if a β-cell expresses multiple β-subunits, the resulting macroscopic IBK may exhibit both inactivating and IbTx-insensitive components (Ding et al. 1998). The BK channel β-subunit expression profile of mouse primary β-cells is unknown; however, an insulinoma expresses mRNA of the β3 subunit (Xia et al. 1999), and human β-cells express mRNAs for both β2 and β3 (Braun et al. 2008). In summary, quantitative technical issues may help account for the discrepancies between earlier studies and the conclusions of the present study.
The multiple peaks in ICavs activated by AP commands (Figs 6 and 7) are qualitatively similar to those reported in an insulinoma cell line (Li et al. 1999). Differences in activation and deactivation rates of the underlying Cav channel subtypes may have caused time-dependent changes in the composition of the macroscopic ICavs, resulting in the hysteresis shown in Fig. 6F (Chan et al. 2005).
BK channel block increased Ca2+ influx into the β-cells by ∼12% and enhanced GSIS by ∼67%. A single AP may raise [Ca2+] at the secretory apparatus sufficiently to trigger exocytosis (Ammala et al. 1993). Insulin secretion is steeply dependent on [Ca2+]i in the physiological range (Hill coefficient = 3.4; Renstrom et al. 1997). This is also reflected in the observation that secretion rate increased 3-fold from −20 to 0 mV (Gopel et al. 2004), a similar voltage range over which the AP peaks are enhanced by paxilline. Thus, the increase in QCav could in principle account for the ∼67% increase in GSIS associated with IBK block. Moreover, the substantial Ca2+ influx that precedes AP threshold may stimulate insulin vesicle priming or exocytosis from the highly Ca2+-sensitive pool (Gromada et al. 1999; Neher & Sakaba, 2008).
The results of the present study suggest a modification of the current model of AP generation in the β-cell. Figure 7A shows normalized IBK, IKv and ICav, activated by AP1, averaged from four to five β-cells and aligned with voltage trace. Figure 7B shows the corresponding IKv and ICav activated by AP2 and aligned with voltage trace.
In 11.1 mm glucose, a depolarisation beyond the AP threshold triggers a sharp increase in ICav that depolarizes the membrane rapidly. The depolarisation and Ca2+ influx activate IBK, beginning ∼4 ms after the AP threshold and coinciding with the last ∼3 ms of the AP upstroke (Fig. 7A). IBK activation slows the depolarization rate and truncates the AP. The slower IKv activates with a further ∼2 ms delay, coinciding with the last ∼1 ms of the AP upstroke. As IKv is slower and 7-fold smaller than IBK (Fig. 5D), it minimally affects the AP upstroke and peak amplitude. IBK and IKv peak ∼3 ms after the AP peak and underlie the downstroke. The larger IBK dominates early repolarisation; however, IBK turns off faster than IKv, which is consistent with the latter contributing preferentially to the later stages of downstroke and the AHP.
In paxilline (Fig. 7B), IBK block eliminates a major part of the early repolarising drive, allowing ICav to depolarise the membrane further, and resulting in a taller AP. However, increased depolarisation increases GKv and K+ driving force, and accelerates IKv activation, resulting in a 4-fold increase in peak IKv than that activated by AP1 command. Along with ICav inactivation and decreased Ca2+ driving force (Fig. 6E), this larger IKv serves to repolarise the AP and underlies the AHP.
Our results and model for the distinct roles of IBK and IKv in AP repolarisation and AHP generation are consistent with previous studies of IK in β-cells. Thus, while the IKv in β-cells is mostly carried by Kv2.1 channels (Roe et al. 1996; MacDonald et al. 2002; Herrington et al. 2006; Herrington, 2007; Jacobson et al. 2007) absence of this current does not prevent AP repolarisation (Jacobson et al. 2007). Instead, a specific Kv2.1 blocker broadens (>2-fold) the AP and inhibits the AHP, while only marginally affecting the AP amplitude (Herrington et al. 2006; Herrington, 2007). Similarly, genetic ablation of Kv2.1 reveals a transient outward current that is remarkably similar in its amplitude and kinetics to the IBK described in the present study (see Figs 2 and 3 of Jacobson et al. (2007)). Thus, it is conceivable that the remaining outward current in Kv2.1-knockout β-cells is carried by BK type channels. This remains to be determined in future studies.
In conclusion, this study demonstrates that BK channels in the mouse β-cell activate under physiological conditions, and modulate glucose-dependent electrical activity and insulin secretion, raising the possibility that inherited BK channel defects (Du et al. 2005) may have metabolic consequences. As BK channels are modulated by multiple signalling mechanisms (Hou et al. 2009), this mechanism has the potential to dynamically integrate, on an AP-by-AP basis, multiple cellular signalling pathways, such as those triggered by neurotransmitters or incretin hormones, to modulate GSIS by up to 67%. In addition, the wide variety of drugs that modulate BK channel function (Nardi et al. 2003; Nardi & Olesen, 2008) could provide a novel approach for modulating GSIS in diabetic and hyperinsulinaemic patients.
Acknowledgments
The research was supported by National Institutes of Health (USA) grant RO1 DK46409 to L.S.S., and was supported in part by the Islet Core of the University of Washington Diabetes Education and Research Center (NIDDK grant P30DK017047). We thank Mary Clark for providing the islets. We also thank Mary Clark, Matt Merrins, Paula Goforth, Jim Ren, Artie Sherman and Richard Bertram for helpful comments and discussions.
Glossary
Abbreviations
- AHP
afterhyperpolarisation
- AP
action potential
- BK
large-conductance Ca2+-activated K+
- ChTx
charybdotoxin
- GKv
voltage-activated K+ conductance
- GSIS
glucose-stimulated insulin secretion
- IBK
large-conductance Ca2+-activated K+ current
- IbTx
iberiotoxin
- ICav
voltage-activated Ca2+ current
- IK
total outward K+ current
- IKv
voltage-activated K+ current
- QCav
voltage-activated Ca2+ charge influx
- RIA
radioimmunoassay
- Rs
series resistance
Author contributions
All authors contributed to the conception, design, analysis and/or interpretation of the data presented in this paper. The electrophysiological experiments were designed by K.M.H. and L.S.S. and carried out by K.M.H. Analysis of the electrophysiological experiments was done by K.M.H. The insulin secretion experiment was designed by K.M.H., L.S.S. and I.R.S., and carried out and analysed by I.R.S. The paper was written by K.M.H., initially in collaboration with L.S.S., and revised with the critical appraisal of I.R.S. All authors approved the final version of the paper for publication.
References
- Adelman JP, Shen KZ, Kavanaugh MP, Warren RA, Wu YN, Lagrutta A, Bond CT, North RA. Calcium-activated potassium channels expressed from cloned complementary DNAs. Neuron. 1992;9:209–216. doi: 10.1016/0896-6273(92)90160-f. [DOI] [PubMed] [Google Scholar]
- Ammala C, Eliasson L, Bokvist K, Larsson O, Ashcroft FM, Rorsman P. Exocytosis elicited by action potentials and voltage-clamp calcium currents in individual mouse pancreatic B-cells. J Physiol. 1993;472:665–688. doi: 10.1113/jphysiol.1993.sp019966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammala C, Larsson O, Berggren PO, Bokvist K, Juntti-Berggren L, Kindmark H, Rorsman P. Inositol trisphosphate-dependent periodic activation of a Ca2+-activated K+ conductance in glucose-stimulated pancreatic beta-cells. Nature. 1991;353:849–852. doi: 10.1038/353849a0. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- Atwater I, Dawson CM, Ribalet B, Rojas E. Potassium permeability activated by intracellular calcium ion concentration in the pancreatic beta-cell. J Physiol. 1979;288:575–588. [PMC free article] [PubMed] [Google Scholar]
- Atwater I, Rosario L, Rojas E. Properties of the Ca-activated K+ channel in pancreatic beta-cells. Cell Calcium. 1983;4:451–461. doi: 10.1016/0143-4160(83)90021-0. [DOI] [PubMed] [Google Scholar]
- Bokvist K, Rorsman P, Smith PA. Block of ATP-regulated and Ca2+-activated K+ channels in mouse pancreatic beta-cells by external tetraethylammonium and quinine. J Physiol. 1990;423:327–342. doi: 10.1113/jphysiol.1990.sp018025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR, Rorsman P. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes. 2008;57:1618–1628. doi: 10.2337/db07-0991. [DOI] [PubMed] [Google Scholar]
- Butler A, Tsunoda S, McCobb DP, Wei A, Salkoff L. mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channels. Science. 1993;261:221–224. doi: 10.1126/science.7687074. [DOI] [PubMed] [Google Scholar]
- Chan SA, Polo-Parada L, Smith C. Action potential stimulation reveals an increased role for P/Q-calcium channel-dependent exocytosis in mouse adrenal tissue slices. Arch Biochem Biophys. 2005;435:65–73. doi: 10.1016/j.abb.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Chay TR. On the effect of the intracellular calcium-sensitive K+ channel in the bursting pancreatic beta-cell. Biophys J. 1986;50:765–777. doi: 10.1016/S0006-3495(86)83517-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DL, Ikeuchi M, Fujimoto WY. Lowering of pHi inhibits Ca2+-activated K+ channels in pancreatic B-cells. Nature. 1984;311:269–271. doi: 10.1038/311269a0. [DOI] [PubMed] [Google Scholar]
- Dean PM, Matthews EK. Electrical activity in pancreatic islet cells. Nature. 1968;219:389–390. doi: 10.1038/219389a0. [DOI] [PubMed] [Google Scholar]
- Ding JP, Li ZW, Lingle CJ. Inactivating BK channels in rat chromaffin cells may arise from heteromultimeric assembly of distinct inactivation-competent and noninactivating subunits. Biophys J. 1998;74:268–289. doi: 10.1016/S0006-3495(98)77785-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, Kotagal P, Luders HO, Shi J, Cui J, Richerson GB, Wang QK. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733–738. doi: 10.1038/ng1585. [DOI] [PubMed] [Google Scholar]
- Falke LC, Gillis KD, Pressel DM, Misler S. ‘Perforated patch recording’ allows long-term monitoring of metabolite-induced electrical activity and voltage-dependent Ca2+ currents in pancreatic islet B cells. FEBS Lett. 1989;251:167–172. doi: 10.1016/0014-5793(89)81448-6. [DOI] [PubMed] [Google Scholar]
- Findlay I, Dunne MJ, Petersen OH. High-conductance K+ channel in pancreatic islet cells can be activated and inactivated by internal calcium. J Membr Biol. 1985;83:169–175. doi: 10.1007/BF01868748. [DOI] [PubMed] [Google Scholar]
- Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J Biol Chem. 1990;265:11083–11090. [PubMed] [Google Scholar]
- Goforth PB, Bertram R, Khan FA, Zhang M, Sherman A, Satin LS. Calcium-activated K+ channels of mouse beta-cells are controlled by both store and cytoplasmic Ca2+: experimental and theoretical studies. J Gen Physiol. 2002;120:307–322. doi: 10.1085/jgp.20028581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. Voltage-gated and resting membrane currents recorded from B-cells in intact mouse pancreatic islets. J Physiol. 1999a;521:717–728. doi: 10.1111/j.1469-7793.1999.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopel S, Zhang Q, Eliasson L, Ma XS, Galvanovskis J, Kanno T, Salehi A, Rorsman P. Capacitance measurements of exocytosis in mouse pancreatic α-, β- and δ-cells within intact islets of Langerhans. J Physiol. 2004;556:711–726. doi: 10.1113/jphysiol.2003.059675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopel SO, Kanno T, Barg S, Eliasson L, Galvanovskis J, Renstrom E, Rorsman P. Activation of Ca2+-dependent K+ channels contributes to rhythmic firing of action potentials in mouse pancreatic beta cells. J Gen Physiol. 1999b;114:759–770. doi: 10.1085/jgp.114.6.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromada J, Hoy M, Renstrom E, Bokvist K, Eliasson L, Gopel S, Rorsman P. CaM kinase II-dependent mobilization of secretory granules underlies acetylcholine-induced stimulation of exocytosis in mouse pancreatic B-cells. J Physiol. 1999;518:745–759. doi: 10.1111/j.1469-7793.1999.0745p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC, Meissner HP. Significance of ionic fluxes and changes in membrane potential for stimulus-secretion coupling in pancreatic B-cells. Experientia. 1984;40:1043–1052. doi: 10.1007/BF01971450. [DOI] [PubMed] [Google Scholar]
- Herrington J. Gating modifier peptides as probes of pancreatic beta-cell physiology. Toxicon. 2007;49:231–238. doi: 10.1016/j.toxicon.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Herrington J, Zhou YP, Bugianesi RM, Dulski PM, Feng Y, Warren VA, et al. Blockers of the delayed-rectifier potassium current in pancreatic beta-cells enhance glucose-dependent insulin secretion. Diabetes. 2006;55:1034–1042. doi: 10.2337/diabetes.55.04.06.db05-0788. [DOI] [PubMed] [Google Scholar]
- Hou S, Heinemann SH, Hoshi T. Modulation of BKCa channel gating by endogenous signaling molecules. Physiology (Bethesda) 2009;24:26–35. doi: 10.1152/physiol.00032.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houamed KM, Fu J, Roe MW, Philipson LH. Electrophysiology of the pancreatic beta cell. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus. Philadelphia: Lippincott Williams and Wilkins; 2004. pp. 51–68. [Google Scholar]
- Houamed KM, Satin LS. Multiple components of Ca-activated K currents in mouse pancreatic beta cells. Biophys J. 2009;96:473a. [Google Scholar]
- Hu H, Shao LR, Chavoshy S, Gu N, Trieb M, Behrens R, Laake P, Pongs O, Knaus HG, Ottersen OP, Storm JF. Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci. 2001;21:9585–9597. doi: 10.1523/JNEUROSCI.21-24-09585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlach WL, Finch SC, Miller JH, Meredith AL, Dalziel JE. A role for BK channels in heart rate regulation in rodents. PLoS One. 2010;5:e8698. doi: 10.1371/journal.pone.0008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson DA, Kuznetsov A, Lopez JP, Kash S, Ammala CE, Philipson LH. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab. 2007;6:229–235. doi: 10.1016/j.cmet.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing X, Li DQ, Olofsson CS, Salehi A, Surve VV, Caballero J, et al. CaV2.3 calcium channels control second-phase insulin release. J Clin Invest. 2005;115:146–154. doi: 10.1172/JCI22518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SR, Reed BJ, Sweet IR. A highly energetic process couples calcium influx through L-type calcium channels to insulin secretion in pancreatic beta-cells. Am J Physiol Endocrinol Metab. 2009;297:E717–E727. doi: 10.1152/ajpendo.00282.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski GJ, Knaus HG, Leonard RJ, McManus OB, Garcia ML. High-conductance calcium-activated potassium channels; structure, pharmacology, and function. J Bioenerg Biomembr. 1996;28:255–267. doi: 10.1007/BF02110699. [DOI] [PubMed] [Google Scholar]
- Kinard TA, de Vries G, Sherman A, Satin LS. Modulation of the bursting properties of single mouse pancreatic beta-cells by artificial conductances. Biophys J. 1999;76:1423–1435. doi: 10.1016/S0006-3495(99)77303-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus HG, McManus OB, Lee SH, Schmalhofer WA, Garcia-Calvo M, Helms LM, et al. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry. 1994;33:5819–5828. doi: 10.1021/bi00185a021. [DOI] [PubMed] [Google Scholar]
- Kukuljan M, Goncalves AA, Atwater I. Charybdotoxin-sensitive KCa channel is not involved in glucose-induced electrical activity in pancreatic beta-cells. J Membr Biol. 1991;119:187–195. doi: 10.1007/BF01871418. [DOI] [PubMed] [Google Scholar]
- Leung YM, Ahmed I, Sheu L, Tsushima RG, Diamant NE, Hara M, Gaisano HY. Electrophysiological characterization of pancreatic islet cells in the mouse insulin promoter-green fluorescent protein mouse. Endocrinology. 2005;146:4766–4775. doi: 10.1210/en.2005-0803. [DOI] [PubMed] [Google Scholar]
- Li ZW, Ding JP, Kalyanaraman V, Lingle CJ. RINm5f cells express inactivating BK channels whereas HIT cells express noninactivating BK channels. J Neurophysiol. 1999;81:611–624. doi: 10.1152/jn.1999.81.2.611. [DOI] [PubMed] [Google Scholar]
- Lippiat JD, Standen NB, Harrow ID, Phillips SC, Davies NW. Properties of BK(Ca) channels formed by bicistronic expression of hSloα and β1–4 subunits in HEK293 cells. J Membr Biol. 2003;192:141–148. doi: 10.1007/s00232-002-1070-0. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Sewing S, Wang J, Joseph JW, Smukler SR, Sakellaropoulos G, et al. Inhibition of Kv2.1 voltage-dependent K+ channels in pancreatic beta-cells enhances glucose-dependent insulin secretion. J Biol Chem. 2002;277:44938–44945. doi: 10.1074/jbc.M205532200. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Wheeler MB. Voltage-dependent K+ channels in pancreatic beta cells: role, regulation and potential as therapeutic targets. Diabetologia. 2003;46:1046–1062. doi: 10.1007/s00125-003-1159-8. [DOI] [PubMed] [Google Scholar]
- McDonough SI, Mori Y, Bean BP. FPL 64176 modification of CaV1.2 L-type calcium channels: dissociation of effects on ionic current and gating current. Biophys J. 2005;88:211–223. doi: 10.1529/biophysj.104.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancilla E, Rojas E. Quinine blocks the high conductance, calcium-activated potassium channel in rat pancreatic beta-cells. FEBS Lett. 1990;260:105–108. doi: 10.1016/0014-5793(90)80078-w. [DOI] [PubMed] [Google Scholar]
- Marty A. Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature. 1981;291:497–500. doi: 10.1038/291497a0. [DOI] [PubMed] [Google Scholar]
- Nardi A, Calderone V, Chericoni S, Morelli I. Natural modulators of large-conductance calcium-activated potassium channels. Planta Med. 2003;69:885–892. doi: 10.1055/s-2003-45095. [DOI] [PubMed] [Google Scholar]
- Nardi A, Olesen SP. BK channel modulators: a comprehensive overview. Curr Med Chem. 2008;15:1126–1146. doi: 10.2174/092986708784221412. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Orio P, Rojas P, Ferreira G, Latorre R. New disguises for an old channel: MaxiK channel beta-subunits. News Physiol Sci. 2002;17:156–161. doi: 10.1152/nips.01387.2002. [DOI] [PubMed] [Google Scholar]
- Renstrom E, Eliasson L, Rorsman P. Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic B-cells. J Physiol. 1997;502:105–118. doi: 10.1111/j.1469-7793.1997.105bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe MW, Worley JF, 3rd, Mittal AA, Kuznetsov A, DasGupta S, Mertz RJ, et al. Expression and function of pancreatic beta-cell delayed rectifier K+ channels. Role in stimulus-secretion coupling. J Biol Chem. 1996;271:32241–32246. doi: 10.1074/jbc.271.50.32241. [DOI] [PubMed] [Google Scholar]
- Rorsman P, Trube G. Calcium and delayed potassium currents in mouse pancreatic beta-cells under voltage-clamp conditions. J Physiol. 1986;374:531–550. doi: 10.1113/jphysiol.1986.sp016096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez M, McManus OB. Paxilline inhibition of the α-subunit of the high-conductance calcium-activated potassium channel. Neuropharmacology. 1996;35:963–968. doi: 10.1016/0028-3908(96)00137-2. [DOI] [PubMed] [Google Scholar]
- Satin LS, Cook DL. Voltage-gated Ca2+ current in pancreatic B-cells. Pflugers Arch. 1985;404:385–387. doi: 10.1007/BF00585354. [DOI] [PubMed] [Google Scholar]
- Satin LS, Hopkins WF, Fatherazi S, Cook DL. Expression of a rapid, low-voltage threshold K current in insulin-secreting cells is dependent on intracellular calcium buffering. J Membr Biol. 1989;112:213–222. doi: 10.1007/BF01870952. [DOI] [PubMed] [Google Scholar]
- Smith PA, Aschroft FM, Fewtrell CM. Permeation and gating properties of the L-type calcium channel in mouse pancreatic beta cells. J Gen Physiol. 1993;101:767–797. doi: 10.1085/jgp.101.5.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PA, Ashcroft FM, Rorsman P. Simultaneous recordings of glucose dependent electrical activity and ATP-regulated K+-currents in isolated mouse pancreatic beta-cells. FEBS Lett. 1990a;261:187–190. doi: 10.1016/0014-5793(90)80667-8. [DOI] [PubMed] [Google Scholar]
- Smith PA, Bokvist K, Arkhammar P, Berggren PO, Rorsman P. Delayed rectifying and calcium-activated K+ channels and their significance for action potential repolarization in mouse pancreatic beta-cells. J Gen Physiol. 1990b;95:1041–1059. doi: 10.1085/jgp.95.6.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabcharani JA, Misler S. Ca2+-activated K+ channel in rat pancreatic islet B cells: permeation, gating and blockade by cations. Biochim Biophys Acta. 1989;982:62–72. doi: 10.1016/0005-2736(89)90174-0. [DOI] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- Wallner M, Meera P, Toro L. Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: a transmembrane β-subunit homolog. Proc Natl Acad Sci U S A. 1999;96:4137–4142. doi: 10.1073/pnas.96.7.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Ding JP, Lingle CJ. Molecular basis for the inactivation of Ca2+- and voltage-dependent BK channels in adrenal chromaffin cells and rat insulinoma tumor cells. J Neurosci. 1999;19:5255–5264. doi: 10.1523/JNEUROSCI.19-13-05255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Ding JP, Lingle CJ. Inactivation of BK channels by the NH2 terminus of the β2 auxiliary subunit: an essential role of a terminal peptide segment of three hydrophobic residues. J Gen Physiol. 2003;121:125–148. doi: 10.1085/jgp.20028667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Olsen JV, Park KS, Li W, Bildl W, Schulte U, Aldrich RW, Fakler B, Trimmer JS. Profiling the phospho-status of the BKCa channel α subunit in rat brain reveals unexpected patterns and complexity. Mol Cell Proteomics. 2008;7:2188–2198. doi: 10.1074/mcp.M800063-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Goforth P, Bertram R, Sherman A, Satin L. The Ca2+ dynamics of isolated mouse beta-cells and islets: implications for mathematical models. Biophys J. 2003;84:2852–2870. doi: 10.1016/S0006-3495(03)70014-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Houamed K, Kupershmidt S, Roden D, Satin LS. Pharmacological properties and functional role of Kslow current in mouse pancreatic beta-cells: SK channels contribute to Kslow tail current and modulate insulin secretion. J Gen Physiol. 2005;126:353–363. doi: 10.1085/jgp.200509312. [DOI] [PMC free article] [PubMed] [Google Scholar]