

Abstract
Glucose-induced β-cell action potential (AP) repolarization is regulated by potassium efflux through voltage gated (Kv) and calcium activated (KCa) potassium channels. Thus, ablation of the primary Kv channel of the β-cell, Kv2.1, causes increased AP duration. However, Kv2.1−/− islet electrical activity still remains sensitive to the potassium channel inhibitor tetraethylammonium. Therefore, we utilized Kv2.1−/− islets to characterize Kv and KCa channels and their respective roles in modulating the β-cell AP. The remaining Kv current present in Kv2.1−/−β-cells is inhibited with 5 μm CP 339818. Inhibition of the remaining Kv current in Kv2.1−/− mouse β-cells increased AP firing frequency by 39.6% but did not significantly enhance glucose stimulated insulin secretion (GSIS). The modest regulation of islet AP frequency by CP 339818 implicates other K+ channels, possibly KCa channels, in regulating AP repolarization. Blockade of the KCa channel BK with slotoxin increased β-cell AP amplitude by 28.2%, whereas activation of BK channels with isopimaric acid decreased β-cell AP amplitude by 30.6%. Interestingly, the KCa channel SK significantly contributes to Kv2.1−/− mouse islet AP repolarization. Inhibition of SK channels decreased AP firing frequency by 66% and increased AP duration by 67% only when Kv2.1 is ablated or inhibited and enhanced GSIS by 2.7-fold. Human islets also express SK3 channels and their β-cell AP frequency is significantly accelerated by 4.8-fold with apamin. These results uncover important repolarizing roles for both Kv and KCa channels and identify distinct roles for SK channel activity in regulating calcium- versus sodium-dependent AP firing.
Introduction
Coordinated electrical activity allows pancreatic β-cells to respond to secretagogues with Ca2+ entry via specific Ca2+ channels followed by insulin secretion. In the case of glucose stimulation of mouse islets, this activity is organized into slow depolarizing waves with a plateau from which action potentials rapidly fire. The resulting depolarization activates voltage-gated channels, including the L-type Ca2+ channel, which leads to the rising phase of the AP in rodent islets (Dean & Matthews, 1968; Satin & Cook, 1985). Continued depolarization activates voltage-gated K+ (Kv) channels, in particular Kv2.1, allowing membrane repolarization and the falling phase of the action potential via K+ efflux from the β-cell (Roe et al. 1996). Calcium entry and elevated cytoplasmic Ca2+ additionally regulate Ca2+-activated K+ (KCa) channels, which affects AP repolarization and the duration of the slow wave (Ammala et al. 1993; Gopel et al. 1999). Both Kv and KCa channels influence rodent and human β-cell APs and thereby influence insulin secretion.
The Kv2.1 channel gives rise to the predominant Kv conductance in β-cells (Roe et al. 1996; MacDonald et al. 2001; Herrington et al. 2005; Jacobson et al. 2007; Braun et al. 2008). Thus, islets from a mouse model with an ablated Kv2.1 gene show increased glucose-induced action potential duration and reduced firing frequency compared with controls (Jacobson et al. 2007). However, Kv2.1−/− islet APs show a significantly increased duration and amplitude with reduced frequency when treated with tetraethylammonium (TEA), a broad-spectrum blocker of potassium channels at low millimolar concentrations (Armstrong & Binstock, 1965; Jacobson et al. 2007). Thus, additional repolarizing K+ channels modulate AP shape and firing frequency together with Kv2.1. As glucose stimulated insulin secretion (GSIS) can be modulated by TEA application during inhibition or ablation of Kv2.1, it is important to conclusively characterize the non-Kv2.1 β-cell K+ channels involved in this response (Herrington et al. 2006; D. A. Jacobson, unpublished observation). One possibility is that remaining Kv channels of the Kv2.1−/−β-cell play an important role in regulating islet AP frequency and shape. Another possibility may be that the TEA responses of Kv2.1−/− result from the inhibition of non-Kv K+ channels such as KCa. Since Kv2.1 does not play a significant role in human islet β-cell electrical activity during glucose stimulation, a better understanding of the repolarizing currents in Kv2.1−/− islets may also help to understand this aspect of human islet function (Braun et al. 2008).
Here we investigated the role of KCa and non-Kv2.1 Kv channels in regulating secretagogue induced action potential firing in intact islets. We show important roles for non-Kv2.1 Kv, BK and SK channels and their influences on membrane potential, AP amplitude, AP duration and insulin secretion. Our results also provide evidence that SK channels are expressed in and regulate human islet glucose induced β-cell electrical activity.
Methods
Ethical approval
The experiments using mice in this paper involve the isolation of pancreatic islets. Mice were killed following inhalation of isoflurane, until general anaesthesia was obtained, by cervical dislocation. This method is consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association and has received approval of the University of Chicago Animal Care and Use Committee.
Mouse islet and β-cell isolation
Islets were isolated from pancreata of 1- to 5-month-old C57BL/6 wild-type (The Jackson Laboratory, Bar Harbor, ME, USA), MIP-GFP (Hara et al. 2003), Kv2.1−/− (Jacobson et al. 2007), or Kv1.4−/− mice (London et al. 1998) using collagenase digestion and Ficoll gradients as previously described (Philipson et al. 1994). Human cadaveric islets were a generous gift from Drs M. Garfinkel, M. Nothias and P. Witkowsi (University of Chicago). Islets were plated or dissociated in 0.005% trypsin, placed on glass coverslips, and cultured for 16 h in RPMI-1640 medium supplemented with 10% fetal calf serum (FCS), concentrations of glucose specified, 100 IU ml−1 penicillin, and 100 mg ml−1 streptomycin. Dissociated primary β-cells were specifically used in all voltage clamp experiments recording K+ currents. β-Cells on the periphery of intact islets were recorded in current clamp mode in all of the membrane potential recordings. In some islet preparations a brief 5 min treatment with 0.005% trypsin without titration was used to promote coverslip attachment. Cells and islets were maintained in a humidified incubator at 37°C under an atmosphere of 95% air–5% CO2.
Perforated-patch electrophysiology
Patch electrodes (2–4 MΩ) loaded with solution containing (in mmol l−1) 140 KCl, 1 MgCl2[H2O]6, 10 EGTA, 10 Hepes (pH 7.25 with KOH) and the pore-forming antibiotic amphotericin B (Sigma) (Rae et al. 1991) were used to record islet attached β-cells. Islets were perfused with Krebs–Ringer-HEPES buffer containing (in mmol l−1) 119 NaCl, 2 CaCl2, 4.7 KCl, 10 Hepes, 1.2 MgSO4, 1.2 KH2PO4, adjusted to pH 7.35 with NaOH, with the indicated concentrations of glucose toxins and compounds. Cells on the periphery of islets on glass coverslips were sealed in voltage clamp at −80 mV, and good access was obtained over several minutes through perforations by amphotericin (Rae et al. 1991). After being switched to current clamp, cells that had a resting membrane voltage near −65 mV in 2 mm glucose were assumed to be β-cells. Solution temperatures were maintained between 34 and 37°C. To confirm that the human recordings were performed in β-cells, an adenovirus that allows expression of a fluorescent protein only in endocrine cells that express insulin was employed. The adenoviral construct was generated by placing a fragment of the rat insulin promoter 2, equivalent to the region previously described (Hanahan, 1985), upstream of the FRET indicator TN-XL (a gift from Oliver Griesbeck; Mank et al. 2006). Construction and purification of the resulting adenovirus was performed by Vector BioLabs (Philadelphia, PA, USA). Transduction of mouse and human endocrine cells with the RIP-TN-XL virus results in expression of the TN-XL fluorescent protein only in insulin positive cells (authors’ unpublished observation).
Insulin secretion measurements
Mouse islets were allowed to recover following isolation overnight. For insulin measurements, 20 islets/animal (n = 6 animals) were incubated with Krebs–Ringer buffer containing 2 mm glucose for 10 min followed by Krebs–Ringer buffer with 14 mm glucose for 10 min with the indicated potassium channel toxins for the remainder of the experiment and analysed for insulin content using an ELISA-based detection kit (ALPCO Diagnostics, Salem, NH, USA), and data are presented as means ±s.d.
Photorelease of caged calcium
Dispersed single β-cells loaded with caged calcium o-nitrophenyl (NP)-EGTA AM (4 μm; Invitrogen/Molecular Probes, Eugene, OR, USA) and Fluo-4 (10 μm, Invitrogen/Molecular Probes) for 30 min washed and incubated in Krebs–Ringer buffer with 2 mm glucose. The dishes were transferred to a heated stage and perfused with Krebs–Ringer buffer with 2 mm glucose and held at −80 mV in the perforated patch voltage clamp configuration. The cells were stepped to −60 mV and switched to Krebs–Ringer buffer with 2 mm glucose and 200 μm tolbutamide while recording the changes in fluorescence of the calcium indicator fluo-4 with a photomultiplier tube (Photon Technology International, Birmingham, NJ, USA). A brief light pulse at 360 nm was applied to the cell through the monochrometer at full intensity to release a small quantity of calcium in the cytoplasm as described (Voets et al. 2001).
High-speed intracellular calcium imaging
Mouse islets attached to glass-bottom Matek tissue-culture dishes were loaded with 5 μm fluo-4-AM loaded in Krebs–Ringer buffer for 30 min at 37°C, as described (Bao et al. 2008). Islets were excited at 488 nm from a monochrometer (Till Photonics, Gräfelfing, Germany), and the emitted light was filtered with a 535/40 filter (Chroma Technology Corp., Bellows Falls, VT, USA) and recorded with a photomultiplier tube (Photon Technology International). Acquisition was controlled with pCLAMP 9 software (Molecular Devices, Sunnyvale, CA, USA). For the experiments utilizing caged calcium, the primary β-cells were imaged for calcium as described above with an excitation wavelength of 360 nm. All experiments were acquired at 10 kHz, to obtain data with sufficiently high temporal resolution. Data analysis was performed with pCLAMP 9, Microsoft Excel, and Graph Pad (GraphPad Prism Software Inc., La Jolla, CA, USA). For the experiments utilizing caged calcium, the primary β-cells were imaged for calcium as described above except the excitation wavelength was 360 nm.
Western blot analysis
Protein extracts were prepared from human islets by extraction with SDS loading buffer (1% SDS, 30 mmol l−1 Tris-HCl, pH 6.8, 5%β-mercaptoethanol, 5% glycerol, and 0.1% bromophenol blue) and heating at 70°C for 10 min. Proteins were prepared as a Western blot on a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA). After electrophoresis through a 12% denaturing polyacrylamide gel, SK3 antibody (Alomone Laboratories, Jerusalem, Israel) was used to probe the membrane at 1:250 dilution in phosphate buffered saline (PBS), 0.1% Tween, and 3% powdered dried milk, followed by goat anti-rabbit horseradish peroxidase (HRP)-coupled secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) at 1:5,000 in the same solution. The membranes were washed in PBS containing 0.1% Tween between and after antibody incubations; HRP was illuminated using Pico Signal (Thermo Scientific) and exposed on Kodak X-omat film (Kodak).
Results
Voltage gated K+ channel regulation of the β-cell action potential
Ablation of the primary β-cell Kv channel, Kv2.1, increases AP duration and reduces their frequency (Jacobson et al. 2007). However, Kv2.1−/− islet APs also show a significantly increased duration and amplitude with reduced frequency when treated with the K+ channel blocker TEA, which correlates precisely with fast intracellular Ca2+ concentration changes (online Supplemental Material, Fig. S1; Jacobson et al. 2007). The TEA response may result from its inhibitory action on the remaining Kv current in the Kv2.1−/−β-cells. We therefore examined the pharmacology of β-cell non-Kv2.1 Kv currents, recorded from Kv2.1−/−β-cells. The remaining Kv current present in Kv2.1−/−β-cells is insensitive to inhibitors of Kv1.1, Kv1.2, Kv1.3, Kv1.6, Kv3.2, Kv3.4 and Kv11.1 channels whereas the remaining Kv current is completely inhibited by 10 μm of the dihydroquinoline CP 33818 (Fig. 1). CP 33818 has been shown to block Kv1.4 and Kv1.3 channels at concentrations below 10 μm (Nguyen et al. 1996), and thus the remaining current may be Kv1.4 as Kv1.3 specific toxins do not inhibit the remaining Kv current from Kv2.1−/−β-cells (Fig. 1). The CP 339818 sensitive Kv-current observed in Kv2.1−/−β-cells is not due to compensation from the loss of Kv2.1 as primary mouse β-cells also have a CP 33818 sensitive Kv current (Figs S2 and S3). Kv1.4 protein is expressed in rodent islets and the mouse insulinoma cell line Min6, and has been shown to be expressed in rhesus pancreatic β-cells (Fig. 2, and see MacDonald et al. 2001; Yan et al. 2004). However, one previous report indicates no expression of Kv1.4 in mouse islets and primary mouse Kv1.4−/−β-cells still show the CP 339818 sensitive Kv current while Kv2.1 is inhibited with stromatoxin (ScTX) (Gopel et al. 2000, Fig. 2A and B). To determine if Kv1.4 is expressed in mouse β-cells, pancreatic sections were stained for Kv1.4 together with insulin or glucagon and revealed that Kv1.4 is not expressed in islet β-cells (Fig. 2C). Therefore, CP 339818 may have different specificities for the endogenous β-cell Kv-channels; unless blockade of Kv1.4 or Kv1.3 channels by CP 339818 in non-β-cells can result in the β-cells also changing their electrical activity, exactly which channels are targets of CP 339818 remains undetermined. The CP 339818 sensitive Kv-channel was assessed for its role in regulating islet β-cell electrical activity and insulin secretion. CP 339818 treatment of Kv2.1−/− islets undergoing AP firing, induced by tolbutamide, resulted in a depolarization of the membrane potential and an increase in AP firing frequency by 39.6% (n = 5, Fig. 3A, Table 1). Similarly, C57BL/6 mouse islet fast Ca2+ fluctuations recorded following depolarization, with tolbutamide to block KATP and ScTX to block Kv2.1, respond to 5 μm CP 339818 with an increase in the Ca2+ fluctuation frequency (38.5%± 21%, n = 3, Fig. 3C). The increase in AP firing frequency in response to CP 339818 when Kv2.1 is inhibited does not result in a significant change in GSIS (Fig. 3D). Interestingly, C57BL/6 islets treated with glucose (without Kv2.1 inhibition) show only a depolarization in membrane potential and no change in AP firing frequency when treated with 5 μm CP 339818, which is reversible (Fig. 3B, Fig. S3 and Table 1, frequency measured 5 min post-treatment). Thus the non-Kv2.1 Kv channel plays a minimal role in regulating mouse islet β-cell electrical activity and insulin secretion. Furthermore the TEA induced changes in Kv2.1−/−β-cell electrical activity are not mediated by inhibition of the remaining Kv currents. These results implicate an important role for another family (or families) of potassium channels in modulating β-cell AP firing frequency, amplitude and duration.
Figure 1. The Kv current remaining in Kv2.1−/−β-cells is inhibited by CP 339818.
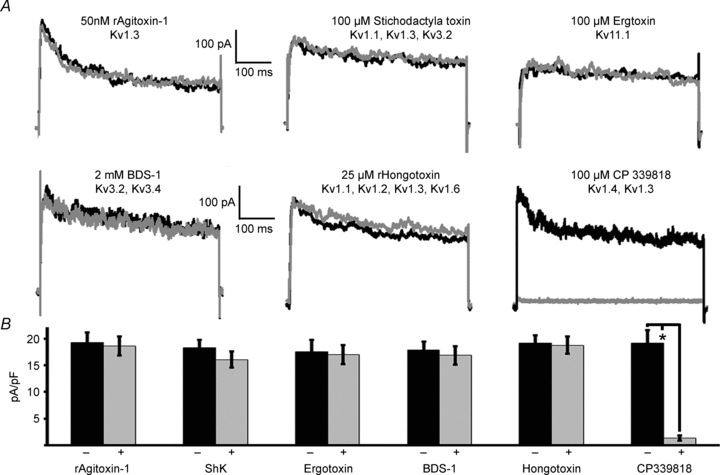
A, Kv current traces recorded from Kv2.1−/− in response to a voltage step from −80 mV to 30 mV before (black trace) and after (grey trace) the indicated treatment. B, analysis of the recordings shown in A before (black bars) and after (grey bars) the indicated treatment (+s.d., *P < 0.05).
Figure 2. Kv1.4 is not the remaining β-cell Kv-current when Kv2.1 is inhibited.
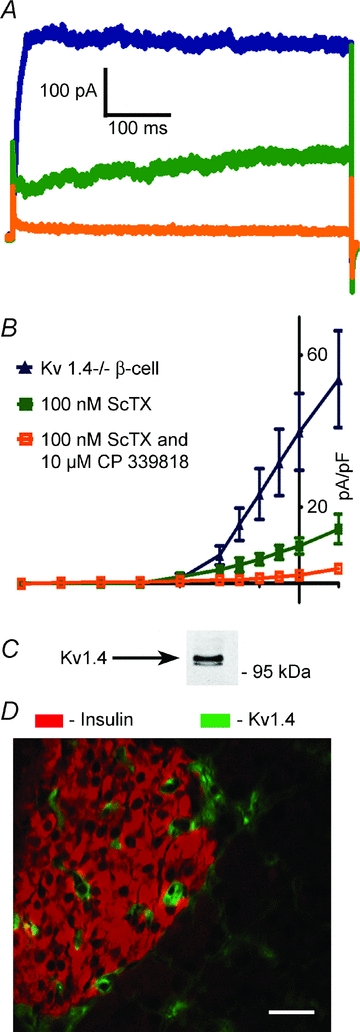
A, Kv-current traces from Kv1.4−/−β-cells in response to a voltage step from −80 mV to 30 mV (blue trace) with 100 nm ScTX (green trace) and 5 μm CP 339818 (orange trace). B, current density vs. voltage plots for β-cells recorded in steps from −80 mV to the indicated voltage (n = 5 for each condition). C, Western blot analysis of mouse islet protein preparation probed with a Kv1.4 specific antibody. Mouse pancreatic section probed with an antibody specific to Kv1.4 (green) and insulin (red) showing non-overlap of the two signals); scale bar 10 μm.
Figure 3. The non Kv2.1 Kv current modulates β-cell membrane potential.
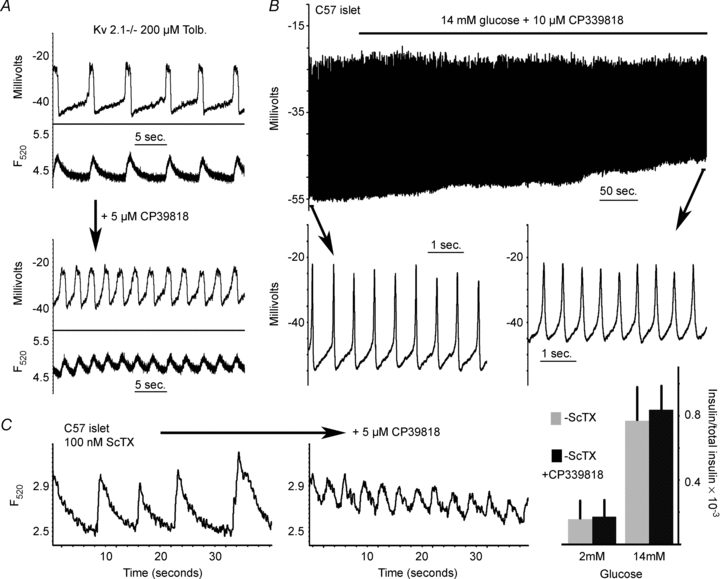
A, Kv2.1−/− tolbutamide induced islet electrical activity (top trace) and Ca2+ flux (lower trace) from the same islet with or without 5 μm CP 339818. B, C57BL/6 glucose induced depolarization in response to 10 μm CP 339818 (black bar; the recording represents the initial depolarization induced by glucose). Inset shows the APs from the segment of activity indicated by the black arrow. C, C57BL/6 islet tolbutamide induced fast Ca2+ fluctuations from the same islet 1 min post each indicated treatment. D, glucose stimulated insulin secretion in response to Kv2 channel inhibition with 100 nm ScTX and in combination with 10 μm CP 339818 (+s.d.).
Table 1.
| Islets and treatments | A. AP freq. (Hz) | s.d. | B. Membrane potential (mV) | s.d. | t test (samples; P value) | |
|---|---|---|---|---|---|---|
| 1. Kv2.1−/− 200 mm tolb., 2 mm glucose | 0.201 | 0.035 | −46.9 | 3.15 | n = 5 | 1B, 2B; P = 0.0496 |
| 2. Kv2.1−/− 200 mm tolb., 2 mm glucose, 5 μm CP 339818 | 0.333 | 0.045 | −40.8 | 1.92 | n = 5 | 1A, 2A; P = 0.0002 |
| 3. C57 14 mm glucose* | 1.928 | 0.341 | −54.25 | 6.19 | n = 6 | 3B, 4B; P = 0.0241 |
| 4. C57 14 mm glucose, 10 μm CP 339818* | 1.642 | 0.259 | −45.542 | 5.12 | n = 6 | 3A, 4A; P = 0.132 |
| 5. Kv1.4−/− 14 mm glucose* | 1.791 | 0.132 | −51.667 | 3.21 | n = 4 | 5B, 6B; P = 0.0302 |
| 6. Kv1.4−/− 14 mm glucose, 10 μm CP 339818* | 1.533 | 0.104 | −40.583 | 4.87 | n = 4 | 5A, 6A; P = 0.0516 |
| B. Time to peak (ms) | ||||||
| 7. Kv2.1−/− 200 mm tolb., 2 mm glucose | 0.216 | 0.052 | 295.492 | 126.9 | n = 3 | 7B, 8B; P = .0139 |
| 8. Kv2.1−/− 200 mm tolb., 2 mm glucose, 100 nm apamin | 0.072 | 0.03 | 905.192 | 107.3 | n = 3 | 7A, 8A; P = 0.0031 |
| 9. C57 200 mm tolb., 100 nm ScTX | 0.233 | 0.056 | 452.963 | 104.3 | n = 3 | 9B, 10B; P = 0.0183 |
| 10. C57 200 mm tolb., 100 nm ScTX, 100 nm apamin | 0.078 | 0.042 | 1364.388 | 305.5 | n = 3 | 9A, 10A; P = 0.00001 |
| 11. C57 +tolb | 0.841 | 0.306 | 129.85 | 4.738 | n = 3 | 11B, 12B, P = 0.91 |
| 12. C57 +tolb+ UCL 1684 | 1.098 | 0.429 | 133.15 | 37.68 | n = 3 | 11A, 12A; P = 0.44 |
| 13. C57+ tolb + ScTX | 0.251 | 0.071 | 422.6 | 83.27 | n = 4 | 13B, 14B; P = 0012 |
| 14. C57 200 mm tolb., 100 nm ScTX, 10 nm UCL 1684 | 0.043 | 0.017 | 1785.631 | 215.9 | n = 4 | 13A, 14A; P = 0.00002 |
Frequency taken 5 min post-treatment.
Large-conductance Ca2+-activated K+ channels regulate β-cell action potential amplitude
Recently the TEA sensitive Ca2+ activated potassium channel, BK, has been implicated in regulating human β-cell AP amplitude (Braun et al. 2008). Slotoxin, a BK specific inhibitor, and isopimaric acid, a BK specific activator, were utilized to assess the role of BK channels in regulating mouse islet AP dynamics (Garcia-Valdes et al. 2001; Imaizumi et al. 2002). Islet attached β-cells undergoing depolarization induced APs, induced with tolbutamide, show a modest but significant increase in AP amplitude when treated with 200 nm slotoxin by 28.2 ± 9.5% independent of Kv2.1 activity (n = 3, P < 0.01), when compared to vehicle treated islet APs (Fig. 4A). Similarly glucose induced mouse islet AP amplitude is increased with BK channel inhibition from a peak of −20 ± 2.5 mV to −11 ± 3.6 mV (n = 3, Fig. 4B). Whereas, activating BK channels with isopimaric acid (10 μm) causes a significant reduction in islet attached β-cell tolbutamide induced AP amplitude by 30.6 ± 7.8% (n = 3, P < 0.01, Fig. 4C). Fast calcium fluctuations respond with similar changes in amplitude while treated with the BK channel pharmacological inhibition or activation (data not shown). However, TEA still significantly increases the islet AP calcium fluctuation duration and amplitude when BK channels are inhibited (Fig. S4). These data demonstrate that BK channels can modulate islet AP amplitude and resulting calcium influx in mouse islets, identifying a modest role for their associated currents that have been recorded in primary rodent β-cells (Tabcharani & Misler, 1989). Interestingly BK channel inhibition during glucose stimulation results in increased oscillations in membrane potential. This may result from an increase in repolarization from Kslow channel activation due to the increased calcium influx during each AP, which in combination with the remaining KATP current that is not inhibited during glucose stimulation could cause membrane repolarization (Gopel et al. 1999). The changes in AP amplitude induced by BK inhibition or activation, however, are significantly less than the changes in AP amplitude resulting from TEA treatment and have no effect on AP frequency or duration.
Figure 4. BK channels regulate mouse islet AP amplitude.
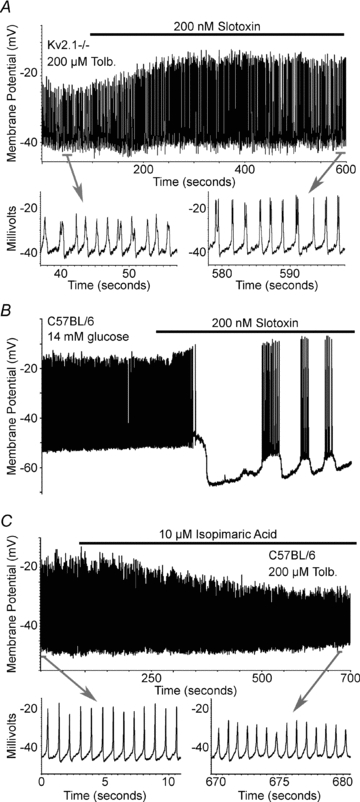
A, Kv2.1−/− tolbutamide induced islet electrical activity in response to 200 nm slotoxin (black bar). Inset shows the APs from the segment of activity indicated by the grey bar. B, C57BL/6 glucose induced islet electrical activity in response to 200 nm slotoxin (black bar). C, C57BL/6 tolbutamide induced islet electrical activity in response to 10 μm isopimaric acid (black bar). Inset shows the APs from the segment of activity indicated by the grey bar.
Small conductance Ca2+-activated K+ channels regulate mouse and human islet β-cell action potential shape and firing frequency
Another calcium activated K+ channel, the SK channel, is also expressed in β-cells (Tamarina et al. 2003). Apamin sensitive SK channels play important roles in regulating AP firing in many types of cells including neurons; however, a clear role for these channels during islet β-cell AP firing has not been determined (Lebrun et al. 1983; Bond et al. 1999; Zhang et al. 2005). The SK channels are sensitive to TEA, with IC50 values ranging from 3 to 14 mm (Monaghan et al. 2004). Thus, concentrations of 15–25 mm TEA, which induce the observed changes in β-cell AP firing from Kv2.1−/− islets (Fig. S1), may result from SK current inhibition. To examine the role of SK channels in islet β-cell AP firing islet membrane potential and calcium fluctuations were recorded during administration of the selective SK channel inhibitor apamin or the non-peptide inhibitor UCL 1684 (Fig. 5 and Fig. S5, Table 1; Rosa et al. 1998). When Kv2.1−/−β-cells undergoing tolbutamide induced APs were treated with 100 nm apamin their associated AP duration was increased by 67.4% while the firing frequency was decreased by 66.6% (n = 3, Table 1); these changes were also observed with the corresponding islet calcium fluctuations (Fig. 5A). This response might be due to compensation for the loss of Kv2.1 channels in the Kv2.1−/− islets by altered expression of SK channels. Thus, C57BL/6 islet responses were also measured in response to apamin. When C57BL/6 islets were treated with tolbutamide and ScTX, to induce APs and inhibit Kv2.1, the APs were modified similarly to the Kv2.1−/− islets when treated with 100 nm apamin (Fig. 5B) with a significant slowing in frequency by 66.5% and increase in duration by 66.8% (n = 3, Table 1). Another SK channel inhibitor, UCL 1684, causes a similar affect as apamin on tolbutamide induced fast calcium fluctuations, which is reversible upon washout of the compound (Fig. 5C, Fig. S5 and Table 1). Islet APs and calcium fluctuations are modestly increased in frequency with 10 nm UCL 1684 (by 20.1%, n = 3, Fig. S5, Table 1, with tolbutamide induced depolarization). However, when islet Kv2.1 channels are inhibited, the addition of 10 nm UCL 1684 causes a significant reduction in fast calcium fluctuation frequency (by 80.6%, n = 4, Fig. S5 and Table 1, with tolbutamide induced depolarization) with an increase in their time to peak (by 76.2%, n = 4, Fig. S5 and Table 1). Similarly glucose induced mouse C57BL/6 islet AP duration is increased with a corresponding decrease in frequency in response to apamin only when Kv2.1 is inhibited with ScTX (Fig. S6). Thus, apamin and UCL 1684 sensitive SK channels play a significant role in regulating mouse islet action potentials only when Kv2.1 currents are inhibited. This associated increase in calcium observed during each AP when SK channels are inhibited under conditions where Kv2.1 activity is blocked results in an increase in GSIS by 2.4-fold (n = 7, P < 0.05) compared to islets treated with 100 nm ScTX (Fig. 5D).
Figure 5. SK channels limit islet AP duration and frequency and regulate GSIS in the absence of Kv2.1 activity.
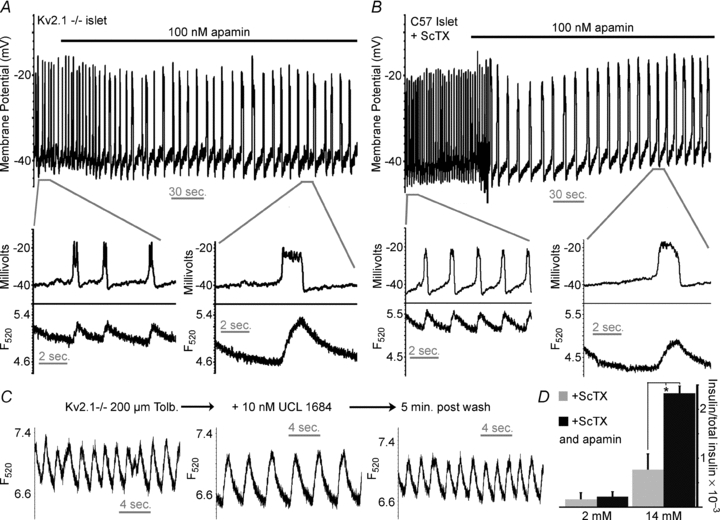
A, Kv2.1−/− islet tolbutamide induced electrical activity in response to 100 nm apamin (black bar). Inset shows the APs (top trace) and Ca2+ change (bottom trace) from the segment of activity indicated by the grey bar. B, C57BL/6 islet tolbutamide induced electrical activity, in the presence of 100 nm ScTX, in response to 100 nm apamin (black bar). Inset shows the APs (top trace) and Ca2+ change (bottom trace) from the segment of activity indicated by the grey bar. C, Kv2.1−/− islet tolbutamide induced Ca2+ flux in response to the indicated treatment; the traces were taken 5 min post-treatment. D, glucose stimulated insulin secretion in response to Kv2 channel inhibition with 100 nm ScTX and in combination with SK channel inhibition with 100 nm apamin (n = 7, +s.d., *P < 0.05).
Apamin sensitive currents have not been observed in primary pancreatic β-cells (Ammala et al. 1993; Zhang et al. 2005). However, SK currents have been recorded from the rat insulinoma cell line, INS-1 (Andres et al. 2009). Therefore, we measured SK currents from primary mouse β-cells to ensure the specificity of the pharmacological approaches utilized. The dissociated primary β-cells were loaded with o-nitrophenyl EGTA (caged Ca2+) and currents were recorded at a holding potential of −60 mV in the perforated patch configuration with tolbutamide present to inhibit KATP channels. Holding at −60 mV also ensures no contaminating current from BK channels, which require greater depolarization (>−40 mV) for their activation (Braun et al. 2008). Photolysis of the caged Ca2+ resulted in a small conductance that persisted during the light pulse and then slowly returned to baseline (Fig. 6A); this current closely resembles heterologously expressed SK currents (Kohler et al. 1996). The current could be reactivated by an additional light pulse to release more of the caged Ca2+, providing that Ca2+ release was detectable as recorded from the β-cells with the calcium indicator Fluo-4 (Fig. 6A). The resulting calcium-activated current was significantly inhibited by 82.4% after treatment of the cells with 500 nm apamin (P = 0.003, n = 4, Fig. 6C). Therefore, apamin sensitive SK channel currents are expressed and can be activated in β-cells by calcium.
Figure 6. Calcium activates mouse β-cell SK currents which significantly regulate β-cell membrane potential.
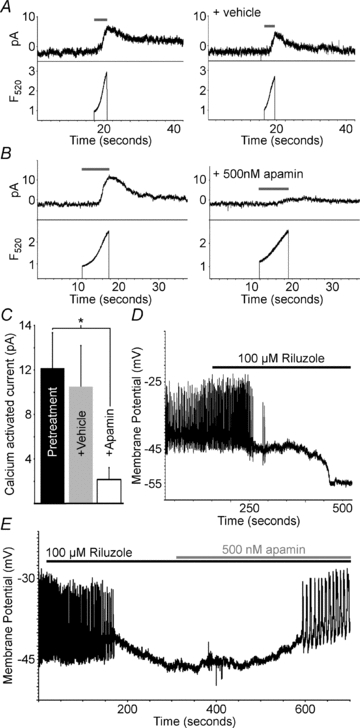
A, C57BL/6 mouse β-cell current recorded at −60 mV (top trace) and calcium change (lower trace) recorded during photolysis of caged calcium with the indicated pulse of 360 nm light (grey line); the right set of traces is from the same cell 5 min following vehicle treatment. B, C57BL/6 mouse β-cell current recorded at −60 mV (top trace) and calcium change (lower trace) recorded during photolysis of caged calcium with the indicated pulse of 360 nm light (grey line); the right set of traces is from the same cell 5 min following 100 nm apamin treatment. C, analysis of SK currents in response to vehicle or apamin (500 nm) (n = 4, +s.d., *P < 0.05). D, representative C57BL/6 islet electrical activity, induced with tolbutamide, in response to 100 μm riluzole (black bar). E, representative C57BL/6 islet electrical activity, induced with tolbutamide, in response to 100 μm riluzole (black bar) and 500 nm apamin (grey bar).
Specific activators of SK channels were utilized to assess if SK channel activity can regulate β-cell membrane potential in the presence of Kv2.1 channel activity. When C57BL/6 islets undergoing tolbutamide induced APs were treated with the SK channel activator riluzole (100 μm; Grunnet et al. 2001) the APs increased briefly in some islets followed by a polarization of the resting membrane potential, which results in inhibition of AP firing in all islets (n = 4, Fig. 6D). Islets treated with tolbutamide and riluzole until the membrane potential stabilized without AP firing were then treated with apamin (500 nm), which caused a depolarization of the membrane potential and a return of AP firing (n = 4, Fig. 6E). Therefore, activation of SK channels can significantly regulate β-cell electrical activity.
Human β-cells differ from mouse β-cells in their electrical responses to glucose, so we went on to determine if SK channels can also regulate human islet glucose stimulated electrical activity. Human islets express SK3 as determined by Western blot analysis from three independent islet preparations (Fig. 7A). SK-like currents were also recorded from dissociated human β-cells by photolysis of caged calcium while recording membrane currents at −60 mV (Fig. 7B). To test the role of SK channels on human β-cell electrical activity, intact islet β-cells were recorded during glucose stimulation, 14 mm, with and without apamin, 100 nm. Human islet cells with hyperpolarized membrane potentials, below −60 mV, in 2 mm glucose were switched to 14 mm glucose resulting in AP firing in many cells that were likely to be β-cells (Fig. 7C). To confirm β-cell identity, cells were transduced with a RIP-TN-XL adenovirus, which fluorescently labels insulin expressing human endocrine cells, and the APs were recorded from the fluorescent cells. Human β-cell glucose stimulated APs were significantly increased in frequency with the addition of apamin, 100 nm, in 4 out of 5 cells (Fig. 7C). Of the four cells that respond to apamin, the frequency of AP firing significantly increases by 4.84-fold (±2.42 fold) after SK channel inhibition (P = 0.03 when compared to vehicle treated cells). These results implicate SK channels in regulating human β-cell glucose stimulated electrical activity; however, this regulation differs from the role of SK channels during mouse β-cell AP firing. These differences are presumably due to variations in the ion channels activated in mouse versus human β-cells during glucose induced electrical activity.
Figure 7. Human islet SK channels regulate glucose stimulated electrical activity.
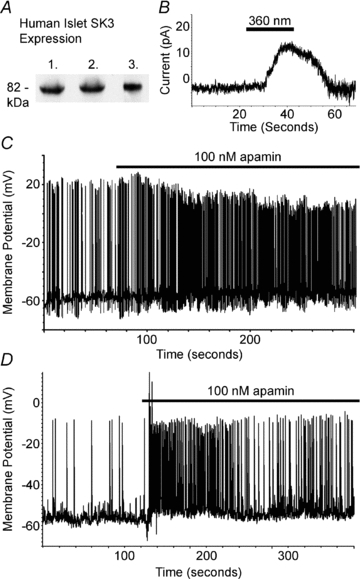
A, Western blot analysis of three independent islet preparations with an SK3 specific antibody. B, human β-cell current recorded at −60 mV (top trace) during photolysis of caged calcium with the indicated pulse of 360 nm light (grey line). C, two representative human islet glucose induced β-cell electrical responses to 100 nm apamin (black line).
Discussion
Following glucose stimulation pancreatic islets respond with insulin secretion resulting from changes in β-cell ion flux through transporters, pumps and ion channels. The activities of a diverse group of ion channels regulate the β-cell membrane potential during glucose stimulation, resulting in action potential firing and allowing physiological insulin secretion (Ashcroft & Rorsman, 1989). The results presented here help further this understanding by conclusively demonstrating the roles of KCa and non-Kv2.1 Kv channels in intact islet β-cell AP dynamics and calcium flux. The results suggest three independent regulatory roles for these K+ channels on AP firing frequency and/or AP shape. Furthermore these results elucidate a functional role of SK channels in regulating human β-cell AP firing frequency.
The rodent β-cell utilizes Kv2.1 channels to limit AP duration and sustain a robust AP firing frequency in response to glucose. Therefore when Kv2.1 is inhibited and/or ablated, glucose-induced AP duration is prolonged while AP firing frequency is attenuated (Jacobson et al. 2007). However, although Kv2.1 is expressed as a functional channel in human β-cells it has been shown to play an insignificant role during glucose-dependent AP firing (Yan et al. 2004; Herrington et al. 2005; Braun et al. 2008). This may result in part from the differences in APs observed between mouse and human islet β-cells (Pressel & Misler, 1990). Human β-cells exhibit a primarily sodium-dependent AP upstroke mediated by voltage-gated sodium channels whereas mouse β-cells exhibit a primarily calcium-dependent AP upstroke mediated by voltage-dependent calcium channels (CaV) (Dean & Matthews, 1970; Hellman et al. 1974; Donatsch et al. 1977; Pressel & Misler, 1990). The kinetic differences between voltage-dependent calcium currents versus voltage-gated sodium currents causes a much longer AP duration in mouse β-cells compared to the human β-cells, which will activate a substantially greater amount of Kv2.1 current. However, TEA still induces a significant increase in AP amplitude while reducing their frequency both in Kv2.1 null mouse β-cells and human β-cells (Fig. 1; Jacobson et al. 2007; Braun et al. 2008). This may result from the inhibition of the remaining Kv current observed in Kv2.1−/−β-cells (Jacobson et al. 2007). Our results show that the remaining Kv current from mouse Kv2.1−/−β-cells is inhibited by the dihydroquinoline CP 339818 (Nguyen et al. 1996). Interestingly, instead of causing TEA-like patterns on β-cell APs, CP 339818 causes an increase in AP firing frequency in Kv2.1−/− islets and C57BL/6 islets treated with ScTX. This is most likely due to a modest decrease in the β-cell membrane potential upon treatment with CP 339818 that leads to increased activation of CaV channels resulting in an increased AP firing frequency. The depolarization induced by CP 339818 is observed in islets with and without Kv2.1 activity; however, it only increases AP firing frequency when Kv2.1 is inhibited. This may result from the significantly increased mouse islet AP firing frequency observed when Kv2.1 is active. The small change in membrane potential induced with CP 339818 does not result in a significant alteration of rapidly firing action potentials and may be compensated for by the Kv2.1 repolarizing current induced during each AP. Therefore Kv2.1 activity is the primary voltage gated K+ current that regulates AP frequency and insulin secretion from mouse pancreatic islets.
The human β-cell utilizes the calcium activated and voltage-dependent channel, BK, to regulate AP firing in response to glucose (Braun et al. 2008). Thus when BK channels are inhibited, the glucose induced AP amplitude is increased or in some cells their firing is inhibited (Braun et al. 2008). The TEA sensitive BK channels may, therefore, also play a role in the TEA induced change in APs observed in mouse islets (Reinhart et al. 1989). To test this idea, BK channels were inhibited in islets, which resulted in a modest increase in tolbutamide or glucose induced AP amplitude without significantly altering AP firing frequency, whereas when BK channels were activated with isopimaric acid, the amplitude of the APs was also modestly reduced. However, the changes seen with inhibition of BK channels are still small compared to the changes in AP dynamics observed with TEA. Therefore the changes in islet β-cell APs by TEA may result from a coordinated inhibition of many channels including BK, Kv2.1, and presumably other K+ channels.
Another type of KCa channel expressed in mouse pancreatic β-cells is the SK channel, although its role in regulating electrical activity is not well understood (Lebrun et al. 1983; Tamarina et al. 2003; Zhang et al. 2005). Glucose-stimulated islet APs are modestly regulated by the SK channel inhibitor apamin (Lebrun et al. 1983). Similarly, we find that inhibition of SK channels only modestly regulates mouse islet AP firing frequency and duration. These results corroborate the idea that SK channels may not play a dominant role in regulating glucose stimulated rodent β-cell electrical activity (Lebrun et al. 1983). However, under certain conditions the calcium influx measured during each AP is increased which may activate more SK channel activity. One such condition occurs in Kv2.1−/−β-cells where glucose results in the firing of APs with increased duration. Interestingly when Kv2.1 is ablated and or inhibited, SK channel inhibition with apamin substantially increases β-cell AP duration while significantly reducing their frequency and amplifying glucose stimulated insulin secretion. Thus, the increase in calcium entry observed during each AP when Kv2.1 is ablated or inhibited may allow for activation of SK channels. This implicates a role for SK channels in regulating β-cell electrical activity during conditions where calcium currents are augmented. Therefore, it would be interesting to address the activation of SK channels during incretin stimulation of islets when the amplitude of calcium currents has been shown to significantly increase (Yada et al. 1993; Gromada et al. 1995; Suga et al. 1997).
SK-like currents can also be recorded from human β-cells and the SK3 channel protein is expressed in human islets (Fig. 7). Human β-cell glucose induced APs involve voltage-gated sodium channels and thus have significantly reduced duration compared with mouse β-cells (Fig. 7; Pressel & Misler, 1990; Braun et al. 2008). As SK channels play an important role in regulating sodium channel-dependent AP firing in neurons they may also play an important role in regulating human β-cell electrical activity (Bond et al. 1999). Indeed human islet β-cell glucose stimulated AP firing is significantly increased in frequency in response to apamin. As calcium influx occurs during the AP an increase in the frequency of APs would be predicted to increase calcium influx into the human β-cell. This important observation leads to the question: Do SK channels regulate human islet GSIS, and if so to what extent? Future studies are needed to comprehensively address the role of SK channels in human islet insulin secretion. Interestingly, in the absence of Kv2.1 activity, SK channel inhibition leads to an increase in AP firing frequency in human β-cells but a reduction in AP firing frequency in mouse β-cells. This difference may be directly linked to SK channel activation by calcium entry during the AP. During the calcium-dependent mouse β-cell AP, SK channel activity limits the duration as its activity increases proportionally to the calcium influx during the AP, which eventually results in repolarization, whereas in the human β-cell sodium-dependent AP the kinetics of the sodium current result in a very rapid depolarization and repolarization resulting in calcium influx but providing a minimal role for the slowly activating and inactivating SK current during the repolarization. Therefore, instead of regulating repolarization the SK current activated during the rapid sodium-dependent spike may result in a delay before the next AP can fire (Bond et al. 1999). These results uncover distinct roles for SK channels in regulating sodium-dependent versus calcium-dependent APs that would be predicted to limit both human and mouse β-cell calcium influx.
The β-cell action potential results in calcium influx that stimulates insulin secretion. Extending the mouse AP duration with Kv2.1 inhibition and furthermore with SK inhibition both result in increased glucose stimulated insulin secretion (Fig. 4; Jacobson et al. 2007). These results implicate a significant role for the duration of each calcium fluctuation during the AP on insulin secretion. An increase in the duration of the AP and the associated augmentation of calcium influx results in a significant increase in insulin secretion. Future studies are required to record the calcium fluctuations of human β-cells during the AP and assess how they influence insulin secretion and if their duration is dynamically regulated in response to secretagogues.
The results presented here implicate important and dynamic roles for Kv, SK and BK channels in regulating islet AP shape, firing frequency and insulin secretion. The duration of the AP is clearly linked to insulin secretion by these observations. The data also illuminate the complexity of the repolarization process in β-cells, which shows the existence of overlapping and in some cases redundant roles for specific K+ channels and illustrates the importance of understanding species specificity when interpreting pharmacological effects.
Acknowledgments
We thank Jeanne Nerbonne and Richard Wilson for the Kv1.4−/− animals. We also thank John Adelman and Paco Herson for helpful discussions and suggestions. This work was supported by NIH grant DK48494 (L.P.), the University of Chicago DRTC DK020595 and in part by NIH grant DK081666-02 (D.J.).
Glossary
Abbreviations
- AP
action potential
- BK
large conductance calcium activated potassium channel
- Ca2+
calcium
- CaV
voltage-dependent calcium channel
- GSIS
glucose stimulated insulin secretion.
- K+
potassium
- KCa
calcium activated potassium channel
- Kv
voltage gated potassium channel
- SD
standard deviation
- SK
small conductance calcium activated potassium channel
- TEA
tetraethylammonium
Author contributions
Conception and design of the experiments was performed by D.J. and L.P. Collection, analysis and interpretation of the data were performed by D.J., F.M., M.T., J.T. and O.C. The article was drafted and revised by D.J. and L.P. The experiments were performed at the University of Chicago and all authors approved this manuscript for publication.
Supplemental material
Supplemental Material, Fig. S1
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Ammala C, Bokvist K, Larsson O, Berggren PO, Rorsman P. Demonstration of a novel apamin-insensitive calcium-activated K+ channel in mouse pancreatic B cells. Pflugers Arch. 1993;422:443–448. doi: 10.1007/BF00375069. [DOI] [PubMed] [Google Scholar]
- Andres MA, Baptista NC, Efird JT, Ogata KK, Bellinger FP, Zeyda T. Depletion of SK1 channel subunits leads to constitutive insulin secretion. FEBS Lett. 2009;583:369–376. doi: 10.1016/j.febslet.2008.12.024. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, Binstock L. Anomalous rectification in the squid giant axon injected with tetraethylammonium chloride. J Gen Physiol. 1965;48:859–872. doi: 10.1085/jgp.48.5.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic β-cell. Prog Biophys Mol Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- Bao S, Jacobson DA, Wohltmann M, Bohrer A, Jin W, Philipson LH, Turk J. Glucose homeostasis, insulin secretion, and islet phospholipids in mice that overexpress iPLA2β in pancreatic β-cells and in iPLA2β-null mice. Am J Physiol Endocrinol Metab. 2008;294:E217–229. doi: 10.1152/ajpendo.00474.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond CT, Maylie J, Adelman JP. Small-conductance calcium-activated potassium channels. Ann N Y Acad Sci. 1999;868:370–378. doi: 10.1111/j.1749-6632.1999.tb11298.x. [DOI] [PubMed] [Google Scholar]
- Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR, Rorsman P. Voltage-gated ion channels in human pancreatic β-cells: electrophysiological characterization and role in insulin secretion. Diabetes. 2008;57:1618–1628. doi: 10.2337/db07-0991. [DOI] [PubMed] [Google Scholar]
- Dean PM, Matthews EK. Electrical activity in pancreatic islet cells. Nature. 1968;219:389–390. doi: 10.1038/219389a0. [DOI] [PubMed] [Google Scholar]
- Dean PM, Matthews EK. Electrical activity in pancreatic islet cells: effect of ions. J Physiol. 1970;210:265–275. doi: 10.1113/jphysiol.1970.sp009208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donatsch P, Lowe DA, Richardson BP, Taylor P. The functional significance of sodium channels in pancreatic β-cell membranes. J Physiol. 1977;267:357–376. doi: 10.1113/jphysiol.1977.sp011817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Valdes J, Zamudio FZ, Toro L, Possani LD. Slotoxin, αKTx1.11, a new scorpion peptide blocker of MaxiK channels that differentiates between α and α+β (β1 or β4) complexes. FEBS Lett. 2001;505:369–373. doi: 10.1016/s0014-5793(01)02791-0. [DOI] [PubMed] [Google Scholar]
- Gopel SO, Kanno T, Barg S, Eliasson L, Galvanovskis J, Renstrom E, Rorsman P. Activation of Ca2+-dependent K+ channels contributes to rhythmic firing of action potentials in mouse pancreatic β cells. J Gen Physiol. 1999;114:759–770. doi: 10.1085/jgp.114.6.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopel SO, Kanno T, Barg S, Rorsman P. Patch-clamp characterisation of somatostatin-secreting δ-cells in intact mouse pancreatic islets. J Physiol. 2000;528:497–507. doi: 10.1111/j.1469-7793.2000.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromada J, Dissing S, Bokvist K, Renstrom E, Frokjaer-Jensen J, Wulff BS, Rorsman P. Glucagon-like peptide I increases cytoplasmic calcium in insulin-secreting beta TC3-cells by enhancement of intracellular calcium mobilization. Diabetes. 1995;44:767–774. doi: 10.2337/diab.44.7.767. [DOI] [PubMed] [Google Scholar]
- Grunnet M, Jespersen T, Angelo K, Frokjaer-Jensen C, Klaerke DA, Olesen SP, Jensen BS. Pharmacological modulation of SK3 channels. Neuropharmacology. 2001;40:879–887. doi: 10.1016/s0028-3908(01)00028-4. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Heritable formation of pancreatic β-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115–122. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- Hara M, Wang X, Kawamura T, Bindokas VP, Dizon RF, Alcoser SY, Magnuson MA, Bell GI. Transgenic mice with green fluorescent protein-labeled pancreatic β-cells. Am J Physiol Endocrinol Metab. 2003;284:E177–183. doi: 10.1152/ajpendo.00321.2002. [DOI] [PubMed] [Google Scholar]
- Hellman B, Idahl LA, Lernmark A, Sehlin J, Taljedal IB. The pancreatic β-cell recognition of insulin secretagogues. Effects of calcium and sodium on glucose metabolism and insulin release. Biochem J. 1974;138:33–45. doi: 10.1042/bj1380033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington J, Sanchez M, Wunderler D, Yan L, Bugianesi RM, Dick IE, Clark SA, Brochu RM, Priest BT, Kohler MG, McManus OB. Biophysical and pharmacological properties of the voltage-gated potassium current of human pancreatic β-cells. J Physiol. 2005;567:159–175. doi: 10.1113/jphysiol.2005.089375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington J, Zhou YP, Bugianesi RM, Dulski PM, Feng Y, Warren VA, Smith MM, Kohler MG, Garsky VM, Sanchez M, Wagner M, Raphaelli K, Banerjee P, Ahaghotu C, Wunderler D, Priest BT, Mehl JT, Garcia ML, McManus OB, Kaczorowski GJ, Slaughter RS. Blockers of the delayed-rectifier potassium current in pancreatic β-cells enhance glucose-dependent insulin secretion. Diabetes. 2006;55:1034–1042. doi: 10.2337/diabetes.55.04.06.db05-0788. [DOI] [PubMed] [Google Scholar]
- Imaizumi Y, Sakamoto K, Yamada A, Hotta A, Ohya S, Muraki K, Uchiyama M, Ohwada T. Molecular basis of pimarane compounds as novel activators of large-conductance Ca2+-activated K+ channel α-subunit. Mol Pharmacol. 2002;62:836–846. doi: 10.1124/mol.62.4.836. [DOI] [PubMed] [Google Scholar]
- Jacobson DA, Kuznetsov A, Lopez JP, Kash S, Ammala CE, Philipson LH. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab. 2007;6:229–235. doi: 10.1016/j.cmet.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- Lebrun P, Atwater I, Claret M, Malaisse WJ, Herchuelz A. Resistance to apamin of the Ca2+-activated K+ permeability in pancreatic B-cells. FEBS Lett. 1983;161:41–44. doi: 10.1016/0014-5793(83)80726-1. [DOI] [PubMed] [Google Scholar]
- London B, Wang DW, Hill JA, Bennett PB. The transient outward current in mice lacking the potassium channel gene Kv1.4. J Physiol. 1998;509:171–182. doi: 10.1111/j.1469-7793.1998.171bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald PE, Ha XF, Wang J, Smukler SR, Sun AM, Gaisano HY, Salapatek AM, Backx PH, Wheeler MB. Members of the Kv1 and Kv2 voltage-dependent K+ channel families regulate insulin secretion. Mol Endocrinol. 2001;15:1423–1435. doi: 10.1210/mend.15.8.0685. [DOI] [PubMed] [Google Scholar]
- Mank M, Reiff DF, Heim N, Friedrich MW, Borst A, Griesbeck O. A FRET-based calcium biosensor with fast signal kinetics and high fluorescence change. Biophys J. 2006;90:1790–1796. doi: 10.1529/biophysj.105.073536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan AS, Benton DC, Bahia PK, Hosseini R, Shah YA, Haylett DG, Moss GW. The SK3 subunit of small conductance Ca2+-activated K+ channels interacts with both SK1 and SK2 subunits in a heterologous expression system. J Biol Chem. 2004;279:1003–1009. doi: 10.1074/jbc.M308070200. [DOI] [PubMed] [Google Scholar]
- Nguyen A, Kath JC, Hanson DC, Biggers MS, Canniff PC, Donovan CB, Mather RJ, Bruns MJ, Rauer H, Aiyar J, Lepple-Wienhues A, Gutman GA, Grissmer S, Cahalan MD, Chandy KG. Novel nonpeptide agents potently block the C-type inactivated conformation of Kv1.3 and suppress T cell activation. Mol Pharmacol. 1996;50:1672–1679. [PubMed] [Google Scholar]
- Philipson LH, Rosenberg MP, Kuznetsov A, Lancaster ME, Worley JF, 3rd, Roe MW, Dukes ID. Delayed rectifier K+ channel overexpression in transgenic islets and β-cells associated with impaired glucose responsiveness. J Biol Chem. 1994;269:27787–27790. [PubMed] [Google Scholar]
- Pressel DM, Misler S. Sodium channels contribute to action potential generation in canine and human pancreatic islet B cells. J Membr Biol. 1990;116:273–280. doi: 10.1007/BF01868466. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Reinhart PH, Chung S, Levitan IB. A family of calcium-dependent potassium channels from rat brain. Neuron. 1989;2:1031–1041. doi: 10.1016/0896-6273(89)90227-4. [DOI] [PubMed] [Google Scholar]
- Roe MW, Worley JF, 3rd, Mittal AA, Kuznetsov A, DasGupta S, Mertz RJ, Witherspoon SM, 3rd, Blair N, Lancaster ME, McIntyre MS, Shehee WR, Dukes ID, Philipson LH. Expression and function of pancreatic β-cell delayed rectifier K+ channelsRole in stimulus-secretion coupling. J Biol Chem. 1996;271:32241–32246. doi: 10.1074/jbc.271.50.32241. [DOI] [PubMed] [Google Scholar]
- Rosa JC, Galanakis D, Ganellin CR, Dunn PM, Jenkinson DH. Bis-quinolinium cyclophanes: 6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)- diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non-peptidic blocker of the apamin-sensitive Ca2+-activated K+ channel. J Med Chem. 1998;41:2–5. doi: 10.1021/jm970571a. [DOI] [PubMed] [Google Scholar]
- Satin LS, Cook DL. Voltage-gated Ca2+ current in pancreatic B-cells. Pflugers Arch. 1985;404:385–387. doi: 10.1007/BF00585354. [DOI] [PubMed] [Google Scholar]
- Suga S, Kanno T, Nakano K, Takeo T, Dobashi Y, Wakui M. GLP-I(7–36) amide augments Ba2+ current through L-type Ca2+ channel of rat pancreatic β-cell in a cAMP-dependent manner. Diabetes. 1997;46:1755–1760. doi: 10.2337/diab.46.11.1755. [DOI] [PubMed] [Google Scholar]
- Tabcharani JA, Misler S. Ca2+-activated K+ channel in rat pancreatic islet B cells: permeation, gating and blockade by cations. Biochim Biophys Acta. 1989;982:62–72. doi: 10.1016/0005-2736(89)90174-0. [DOI] [PubMed] [Google Scholar]
- Tamarina NA, Wang Y, Mariotto L, Kuznetsov A, Bond C, Adelman J, Philipson LH. Small-conductance calcium-activated K+ channels are expressed in pancreatic islets and regulate glucose responses. Diabetes. 2003;52:2000–2006. doi: 10.2337/diabetes.52.8.2000. [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Sudhof TC, Neher E, Verhage M. Munc18–1 promotes large dense-core vesicle docking. Neuron. 2001;31:581–591. doi: 10.1016/s0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- Yada T, Itoh K, Nakata M. Glucagon-like peptide-1-(7–36)amide and a rise in cyclic adenosine 3′,5′-monophosphate increase cytosolic free Ca2+ in rat pancreatic β-cells by enhancing Ca2+ channel activity. Endocrinology. 1993;133:1685–1692. doi: 10.1210/endo.133.4.8404610. [DOI] [PubMed] [Google Scholar]
- Yan L, Figueroa DJ, Austin CP, Liu Y, Bugianesi RM, Slaughter RS, Kaczorowski GJ, Kohler MG. Expression of voltage-gated potassium channels in human and rhesus pancreatic islets. Diabetes. 2004;53:597–607. doi: 10.2337/diabetes.53.3.597. [DOI] [PubMed] [Google Scholar]
- Zhang M, Houamed K, Kupershmidt S, Roden D, Satin LS. Pharmacological properties and functional role of Kslow current in mouse pancreatic β-cells: SK channels contribute to Kslow tail current and modulate insulin secretion. J Gen Physiol. 2005;126:353–363. doi: 10.1085/jgp.200509312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.