

Abstract
Nitric oxide (NO) induces mitochondrial biogenesis in skeletal muscle cells via upregulation of the peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α). Further, we have shown that nitric oxide interacts with the metabolic sensor enzyme, AMPK. Therefore, we tested the hypothesis that nitric oxide and AMPK act synergistically to upregulate PGC-1α mRNA expression and stimulate mitochondrial biogenesis in culture. L6 myotubes treated with nitric oxide donors, S-nitroso-N-penicillamine (SNAP, 25 μm) or diethylenetriamine-NONO (DETA-NO, 50 μm), exhibited elevated AMPK phosphorylation, PGC-1α mRNA and protein, and basal and uncoupled mitochondrial respiration (P < 0.05). Pre-treatment of cultures with the AMPK inhibitor, Compound C, prevented these effects. Knockdown of AMPKα1 in L6 myotubes using siRNA reduced AMPKα protein content and prevented upregulation of PGC-1α mRNA by DETA-NO. Meanwhile, siRNA knockdown of AMPKα2 had no effect on total AMPKα protein content or PGC-1α mRNA. These results suggest that NO effects on PGC-1α expression are mediated by AMPKα1. Paradoxically, we found that the AMPK-activating compound, AICAR, induced NO release from L6 myotubes, and that AICAR-induced upregulation of PGC-1α mRNA was prevented by inhibition of NOS with NG-nitro-l-arginine methyl ester (l-NAME, 1 mm). Additionally, incubation of isolated mouse extensor digitorum longus (EDL) muscles with 2 mm AICAR for 20 min or electrical stimulation (10 Hz, 13 V) for 10 min induced phosphorylation of AMPKα (P < 0.05), which was completely prevented by pre-treatment with the NOS inhibitor, l-NG-monomethyl arginine (l-NMMA, 1 mm). These data identify the AMPKα1 isoform as the mediator of NO-induced effects in skeletal muscle cells. Further, this study supports a proposed model of synergistic interaction between AMPK and NOS that is critical for maintenance of metabolic function in skeletal muscle cells.
Introduction
Skeletal muscle accounts for approximately 45% of body mass, and is the main site for glucose disposal under insulin stimulated conditions (Elia, 1992). Metabolic efficiency in muscle is therefore of utmost importance for whole-body metabolic balance. Currently, several reports suggest that nitric oxide (NO) at physiological amounts positively impacts mitochondrial function and is required for mitochondrial biogenesis induced by cold exposure and calorie restriction in different cell types (Nisoli et al. 2003, 2004, 2005). NO has also been linked to upregulation of the glucose transporter GLUT4 in skeletal muscle (Lira et al. 2007). A likely explanation is that NO mediates these adaptations by controlling the expression of the peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α). However, the mechanism involved in the NO-induced improvement in skeletal muscle metabolic efficiency along with increased expression of PGC-1α remains unknown.
NO is a reactive nitrogen molecule that is formed enzymatically by nitric oxide synthases (NOS), via the conversion of l-arginine to l-citrulline. Skeletal muscle expresses three isoforms of NOS, namely the neuronal (nNOS), the endothelial (eNOS), and the inducible (iNOS) (Stamler & Meissner, 2001). Under normal physiological conditions both eNOS and nNOS synthesize NO at low levels, whereas iNOS expression is upregulated during inflammation and results in high levels of NO production (Stamler & Meissner, 2001; Pilon et al. 2004; Ji et al. 2006). Insulin resistant states often correlate with downregulation of both eNOS and nNOS, and upregulation of iNOS in skeletal muscle (Young & Leighton, 1998; Shankar et al. 2000; Pilon et al. 2004; Valerio et al. 2006).
PGC-1α stimulates transcription of nuclear- and mitochondrial-encoded metabolic genes, as well as mitochondrial DNA replication (Wu et al. 1999; Scarpulla, 2008). PGC-1α is upregulated by contraction and is involved in most of the metabolic adaptations that occur in muscle thereafter (Geng et al. 2010; Lira et al. 2010). In addition, PGC-1α levels in muscle have been associated with insulin sensitivity both in animal and human models (Patti et al. 2003; Mootha et al. 2003; Handschin et al. 2007).
The 5′-AMP-activated protein kinase (AMPK) is downregulated in diseases characterized by an unbalanced metabolic state, such as type 2 diabetes and obesity (Sriwijitkamol et al. 2007; Bonnard et al. 2008). AMPK is a heterotrimeric enzyme (α1, α2, β1, β2, γ1, γ2, γ3), that is activated both by direct AMP allosteric regulation and by phosphorylation of its α catalytic subunit (Fryer & Carling, 2005; Hardie et al. 2006). AMPK regulates both acute and delayed responses that improve the cell capacity to resynthesize ATP. Acutely, AMPK stimulates glucose transport and fatty acid oxidation in skeletal muscle (Balon & Jasman, 2001; Smith et al. 2005). The associated delayed responses, or adaptations to AMPK activation include the upregulation of proteins involved either directly or indirectly in substrate availability (e.g. GLUT4) and oxidation capacity (e.g. PGC-1α and mitochondrial genes) (Winder et al. 2000; Zheng et al. 2001). These changes represent important adaptive responses triggered by metabolic challenges such as muscle contraction, energy deprivation and hypoxia (Zhou et al. 2000; Mu et al. 2001; Zong et al. 2002).
We have previously shown that NO regulates GLUT4 expression via AMPK activation (Lira et al. 2007). Of note is the observation that specific pharmacological activation of AMPK with AICAR, and the subsequent induction of the glucose transporter GLUT4, is blunted by inhibition of endogenous NO production. Recently, nitric oxide synthase (NOS) activity has been shown to be involved in AMPK and CaMK-induced mitochondrial biogenesis and PGC-1α expression in L6 myotubes (McConell et al. 2010). Considering these findings together with reports showing that AMPK phosphorylates and activates both eNOS and nNOS (Chen et al. 1999, 2000), we proposed the existence of a positive feedback loop between NO production and AMPK activity in skeletal muscle. However, several questions remain to be addressed: (a) which isoform of the α catalytic subunit of AMPK is primarily involved in NO-dependent improvement in mitochondrial function and regulation of PGC-1α, (b) whether the MEF-2 and CRE cis elements in the proximal 2 kb PGC-1α promoter are required for the NO-dependent induction of the gene, (c) whether endogenous NO production is required for AMPK-induced upregulation of PGC-1α and mitochondrial genes, and (d) whether NOS inhibition affects AMPK activation under pharmacological and physiological stimuli (i.e. AICAR and contraction). Our findings provide further evidence for a positive feedback loop between NO and AMPK that impacts PGC-1α expression and mitochondrial function in skeletal muscle.
Methods
Ethical approval
Animals were housed in the University of Florida Animal Care Services Center according to the guidelines set forth by the Institutional Animal Care and Use Committee. The ethical guidelines established by The Journal of Physiology were followed (Drummond, 2009).
Chemicals and reagents
NG-Nitro-l-arginine methyl ester (l-NAME), l-NG-monomethyl arginine (l-NMMA), S-nitroso-N-penicillamine (SNAP), diethylenetriamine-NONO (DETA-NO), methylamine hexamethylene methylamine-NONO (MAHMA-NO) and 1H-[1,2,4]oxadiazolo [4,3,-a]quinoxalin-1-one (ODQ) were purchased from Cayman Chemical (Ann Arbor, MI, USA). 5-Aminoimidazole-4-carboxamide-1-β-d-ribonucleoside (AICAR) was obtained from Toronto Research Chemicals (North York, ON, Canada). 8-Bromo-cyclicGMP (8-Br-cGMP) was obtained from Tocris Bioscience (Ellisville, MO, USA), and Compound C was from Calbiochem (San Diego, CA, USA). 4-Amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) diacetate was obtained from Invitrogen (Carlsbad, CA, USA) and Krebs–Henseleit buffer was from Sigma-Aldrich (St Louis, MO, USA).
Muscle bath experiments
A total of 28 female adult ICR mice (8–9 weeks old) were purchased from Harlan Sprague Dawley, Inc. (Indianapolis, IN, USA). Animals were maintained on a 12:12 h light–dark cycle and provided with food and water ad libitum. EDL and soleus muscles from both hindlimbs were carefully removed while mice were maintained in a surgical plane of anesthesia with 3–4% isoflurane using oxygen as the carrier gas. Muscles were rapidly transferred to oxygenated (95% O2, 5% CO2) Krebs–Henseleit (KH) buffer with 2 g l−1 glucose, 25 mm sodium bicarbonate, 5 mm Hepes, 2.54 mm calcium chloride and 100 μm l-arginine (pH 7.4) and kept at 30°C (Bonen et al. 1994). Experiments were conducted with muscles at optimal length (L0), which was determined after an initial 30 min equilibration period. Once L0 was established, the buffer was washed from baths, reloaded and equilibrated for an additional 10 min. At this point, muscles were either electrically stimulated at 10 Hz, 13 V, 0.1 ms pulse duration for 10 min, or treated with AICAR (2 mm) for 20 min. The contralateral EDL and soleus muscles served as non-contracting or non-AICAR treated controls. Muscles from 14 animals were exposed to 1 mmNG-monomethyl-l-arginine (l-NMMA) 10 min prior to and during either electrical stimulation or AICAR treatment. Muscles were immediately frozen in liquid N2 following the experimental treatment, and kept at −80°C thereafter.
Cell culture
Both L6 and C2C12 were obtained from American Type Culture Collection (Manassas, VA, USA) and cultured at 37°C in 5% CO2 and 95% atmospheric air. Cells were maintained in 5 mm glucose Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin until reaching 80% confluence. Differentiation into myotubes was induced with DMEM supplemented with 2% HoS. Treatments were performed in differentiation medium after 5 days of differentiation, unless specified otherwise.
Gene reporter assay experiments
C2C12 cells seeded in 24-well plates once reaching 90% confluence were transfected with 0.5 μg per well of only one of the following: the 2 kb intact PGC-1α promoter (Addgene plasmid 8887), the mutated promoters (PGC-1αΔCRE or PGC-1αΔMEF2, Addgene plasmids 8888 and 8889, respectively), or the pGL3 Control plasmid (Promega, Madison, WI, USA; cat E1741) as previously described (Handschin et al. 2003). In addition, cells were always cotransfected with 0.01 μg per well of the pRL-CMV (Promega, Madison, WI, USA). Lipofectamine 2000 reagent was used for transfections (1 μl per well, Invitrogen; cat. 11668-027), and DNA–Lipofectamine complexes were formed according to the manufacturer's instructions. Briefly, cells were washed in serum-free Opti-MEM (Invitrogen; cat. 31985) before transfection, and then exposed to DNA–Lipofectamine complexes also in serum-free Opti-MEM. Plates were gently swirled every 2 h, and after the initial 6 h of transfection, 12% HoS Opti-MEM was added in a volume sufficient to bring the final concentration of serum in wells to 6% HoS. Cells were then kept in 6% HoS Opti-MEM for additional 18 h. At this point, cells were switched to 2% HoS DMEM. Treatments started 48 h after the beginning of transfection and lasted for 9 h. After treatments, C2C12 myotube cultures were washed once in phosphate buffered saline (PBS) at room temperature and then lysed by addition of 120 μl passive lysis buffer (PLB; Promega Dual-Luciferase Reporter Assay System). Plates were rocked at room temperature for 15 min. Lysates were then transferred to microcentrifuge tubes and centrifuged for 30 s (2000 g) to sediment cellular debris. Supernatants were transferred to new tubes for subsequent use in the assay. The level of transcriptional activity of the intact PGC-1α promoter, the mutated promoters (PGC-1αΔCRE and PGC-1αΔMEF2) as well as of pGL3, represents the ratio between the raw firefly luciferase activity (RLU) and the renilla luciferase activity (RLU) assessed with a manual luminometer (model FB12, Berthold). Values were always reported as relative to the average obtained for the control group.
siRNA experiments
L6 cells were transfected with siRNAs for the α1 and α2 subunits of AMPK at 2 days of differentiation when myotubes were 60–70% confluent. The siRNA sequences used were previously reported (Pilon et al. 2004; Konrad et al. 2005; Wijasekara et al. 2006) and consisted of GCA UAU GCU GCA GGU AGA UdTdT, nucleotides 738 to 756, acc. no. NM019142 – AMPKα1, CGU CAU UGA UGA UGA GGC UdTdT, nucleotides 865–883, acc. no. NM023991 – AMPKα2, and AUU CUA UCA CUA GCG UGA CUU for the unrelated (control) siRNA. All siRNA oligonucleotides were purchased from Dharmacon (Lafayette, CO, USA), and transfection efficiency was tested with fluorophore-conjugated siRNA (siGLO Green, cat. D-001630-01-05). Knockdown efficiencies at mRNA and protein levels were tested at 48 h post-transfection. All treatments were performed after 48 h of transfection. For assessment of the phosphorylation status of AMPK and other proteins, cells were kept in serum-free DMEM (0.5% BSA) for 4 h before and during the 1 h treatments. For mRNA experiments, cells were treated for 3 h in 2% HoS DMEM.
Western blot
Muscle samples were homogenized in 20 mm Tris (pH7.5), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Nonidet P-40, 2.5 mm sodium pyrophosphate, 1 mmβ-glycerol phosphate, 1 mm sodium orthovanadate, 1 μg ml−1 leupeptin, 1 mm PMSF, and 10 μg ml−1 aprotinin containing 1% v/v phosphatase inhibitor (p-5726) from Sigma. L6 and C2C12 cells were washed twice in ice-cold PBS and harvested in non-denaturing lysis (NDL) buffer containing 1% v/v Triton X-100, 0.3 m NaCl, 0.05 m Tris base, 5 mm EDTA, 3.1 μm NaN3, 95 mm NaF, 22 μm Na3VO4, 0.1% v/v protease inhibitors (p-8340), and 1% v/v phosphatase inhibitors (p-5726) from Sigma. Protein concentrations were assessed using the DC Protein Assay Kit (Bio-Rad Laboratories, Richmond, CA, USA). Aliquots of muscle homogenates (50–70 μg), or cell lysates (15–30 μg) were then run in SDS-PAGE gels (either 8% or 4–20% Thermo Scientific Pierce Precise Protein Gels). For PGC-1α immunoblots, however, 150 μg of protein from L6 lysates was used. Primary antibodies were used as follows: rabbit anti-PGC-1α (1:1000, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), rabbit anti-AMPKα and anti-phospho(Thr172)AMPKα (1:1500, Cell Signaling Technology, Inc., Danvers, MA, USA), rabbit anti-ACC (1:1500, Cell Signaling), rabbit anti-phospho(Ser79)ACC (1:500; Upstate Biotechnology, Inc., Lake Placid, NY, USA). Protein expression values were normalized to either β-actin (mouse anti-β-actin 1:5000, Abcam Inc., Cambridge, MA, USA) or α-tubulin (mouse anti-α-tubulin 1:10 000, Santa Cruz). Reactions were developed by using enhanced chemiluminescence detection reagents (ECL Plus; GE Healthcare (Amersham Biosciences), Little Chalfont, UK), and protein levels were determined by densitometry (Kodak 1D Image Analysis Software v. 3.6).
Quantitative real-time PCR
Quantification of mRNA from both L6 and C2C12 myotubes was performed with specific primer and probes using TaqMan technology designed by Applied Biosystems for Quantitative real-time PCR as previously described (Lira et al. 2007). RT reaction was performed with 1–2.5 μg of total RNA using either the Verso cDNA Kit (Thermo Scientific, UK) or SuperScript III First-Strand Synthesis System for RT-PCR (Life Technologies, Carlsbad, CA, USA). Primers and probes used were: AMPKα1 (RefSeq NM_019142.1, assay no. Rn00569558_m1), AMPKα2 (RefSeq NM_023991.1, assay no. Rn00576935_m1), PGC-1α (RefSeq NM_031347.1, assay no. Rn00580241_m1 (rat) and RefSeq NM_008904.1, assay no. Mm00447183_m1 (mouse)), F1ATP synthase (RefSeq NM_134364.1, assay no. Rn01756310_g1), citrate synthase (RefSeq NM_130755.1, assay no. Rn00756225_m1), HPRT (RefSeq NM_013556.2, assay no. Mm01545399_m1). These primers and probes were obtained from Applied Biosystems (Foster City, CA, USA) and their sequences are proprietary and therefore are not reported. The primer and probe sequences for the rat HPRT used are: forward, 5′-GTTGGATACAGGCCAGACTTTGT-3′; reverse, 5′-AGTCAAGGGCATATCCAACAACAA-3′; probe, 5′-ACTTGTCTGGAATTTCA-3′. Quantitative real-time PCR for the target genes were performed in the ABI Prism 7700 Sequence Detection System (Applied Biosystems), using the 2-ΔΔCT method, where CT is threshold cycle.
Fluorescence-based real-time measurement of NO production
Intracellular NO was monitored with DAF-FM, a pH-insensitive fluorescent dye that emits increased fluorescence after reaction with an active intermediate of NO formed during the spontaneous oxidation of NO to NO2− (Pye et al. 2007). L6 were seeded in 24-well plates, grown and differentiated as previously mentioned. After 4 days of differentiation, cells were washed once with phenol-red free and serum-free DMEM, and incubated with 10 μm DAF-FM for 30 min at 5% CO2 and 37°C while protected from light. After loading was completed, cells were rinsed three times with phenol red-free, serum-free DMEM and then exposed to AICAR or MAHMA-NO. Cells were then immediately placed in a SpectraMax M5 multi-detection reader (Molecular Devices, Sunnyvale, CA, USA) for background fluorometric analysis. At this point cells were transferred back to the incubator for 40 min before final NO fluorescence was once more assessed using excitation and emission wavelengths of 488 and 520 nm, respectively. Results represent the difference between the fluorescence seen at 40 min of incubation and the background fluorescence.
Citrate synthase assay
Citrate Synthase activity was assayed using a modified protocol from Srere (1969). The assay buffer (230 μl final volume) contained monobasic and dibasic potassium phosphate buffers (36.5 and 63.5 mm, respectively), EDTA (10 mm), DTNB (0.2 mm), acetyl-CoA (0.1 mm) and Triton X-100 (0.05% v/v). The reaction was initiated by the addition of 2 μl of cell lysate and of 10 μl of oxaloacetate. Absorbance at 412 nm (25°C) was measured during 5 min. Values were then normalized to protein content.
Cell respiration
After 2 days of differentiation, L6 myotubes were exposed to treatments of 12 h during either four or five consecutive days. Compound C (10 μm) was added to cells always 30–40 min before the addition of DETA-NO (50 μm). After each treatment, cells were washed once in fresh medium before being kept in fresh 2% HoS medium until the next treatment. Cells were harvested 10 h after the last treatment bout. At that point cells had been in differentiation medium for 7 days and myotubes were fully confluent. Cell respiration assessment was performed as previously described (Koves et al. 2005). Basically, myotubes were washed once in PBS, trypsinized, and centrifuged at 800 g for 3 min. Cell pellets were then washed twice, resuspended in 400 μl of supplemented PBS containing 10 mm glucose, 10 mm Hepes, 0.2% BSA and pH 7.45 and maintained at 37°C thereafter. First, 300 μl of supplemented PBS was added to the oxymeter chamber (Hansatech Instruments, Pentney, UK) and equilibrated for 1 min. Immediately afterwards, 350 μl of the cell suspension solution was added and basal respiration rate was recorded for approximately 4.5 min. At this point, Oligomycin, an inhibitor of F1/F0 ATP synthase, was added to a final concentration of 3 μg ml−1. Subsequently, 6 μm FCCP was added and uncoupled respiration, also referred as maximal respiration, was then monitored for 1.5 min. Basal coupled respiration rates were calculated by subtracting oligomycin respiration rates from basal respiration rates. Results were then normalized to protein concentration on aliquots of cells left in supplemented PBS. For this purpose, cells were lysed by three consecutive freezing-thaw cycles in liquid N2.
Statistical analysis
Data are expressed in means ± standard error of the mean (s.e.m.). One-way or two-way ANOVAs, followed by Newnam–Keuls post hoc tests, were used for all experiments with L6. The effect of NO treatments on AMPK phosphorylation and PGC-1α mRNA in C2C12 was analysed with Student's t test. The effect of electrical stimulation on AMPK phosphorylation in the EDL controls, and EDL treated with l-NMMA, were analysed separately using paired t tests. The same analysis was conducted for the EDL stimulated with AICAR instead of electrical stimulation. Statistical significance was established a priori at P < 0.05.
Results
AMPK inhibition prevents NO-induced upregulation of PGC-1α mRNA
One-hour treatments of L6 myotube cultures with either one of two chemically distinct NO donors, SNAP (25 μm) and DETA-NO (50 μm), were sufficient to significantly increase AMPKα phosphorylation by 2- to 2.2-fold (Fig. 1A and B). Both NO donors upregulated PGC-1α mRNA by approximately 2-fold (P < 0.05) following treatments of 3 h. PGC-1α mRNA remained elevated for 8.5 h only when myotubes were treated with DETA-NO (Fig. 2A). In a separate experiment, 3 h treatment of myotubes with the cell permeant cGMP analogue 8-Br-cGMP increased PGC-1α mRNA similarly to DETA-NO treatment. Co-treatment with the guanylate cyclase inhibitor ODQ ablated the NO-dependent effect on PGC-1α mRNA. Pharmacological inhibition of AMPK with Compound C also prevented the upregulation of PGC-1α mRNA seen with NO treatment alone (Fig. 2B).
Figure 1. NO donors SNAP (25 μm) and DETA-NO (50 μm) increase AMPKα phosphorylation in myotubes.
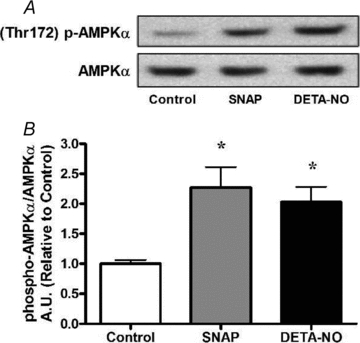
L6 cells were treated for 1 h and immediately harvested. A, representative immunoblots. B, quantification of immunoblots for AMPKα phosphorylation normalized to total AMPKα expression (n = 4/group). *P < 0.05 in comparison to untreated cells (Control).
Figure 2. NO donors SNAP (25 μm) and DETA-NO (50 μm) induce PGC-1α mRNA expression in myotubes which is prevented by pharmacological inhibition of sGC and AMPK.
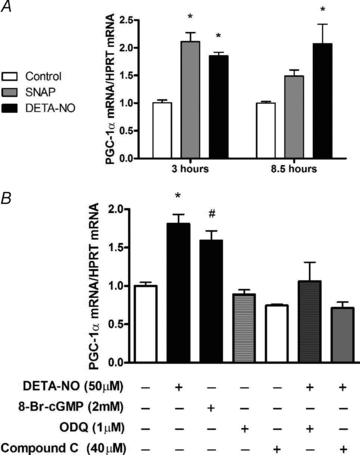
A, L6 cells were treated for 3 h and 8.5 h and immediately harvested. Values represent fold changes (means ±s.e.m.) versus untreated cells (Control) at the same time point (n = 4–6/group). *P < 0.05 in comparison to Control. B, L6 cells were exposed to inhibitors 1 h prior and during the 3 h treatments with DETA-NO. Cells were harvested immediately after treaments. Values represent fold changes (means ±s.e.m.) versus untreated (Control) cells (n = 4–5/group). *P < 0.05 in comparison to all groups except 8-Br-cGMP, #P < 0.05 in comparison to all groups except DETA-NO and ODQ+DETA-NO.
NO-induced AMPK phosphorylation is guanylate cyclase dependent and not dependent upon superoxide or peroxynitrite
To test whether activation of AMPK by NO is mediated by oxidant production or formation of peroxynitrite, a convergent product of NO and superoxide, L6 myotubes were treated for 1 h with DETA-NO or the peroxynitrite generating compound SIN-1, in the presence of uric acid or Tempol to scavenge peroxynitrite and superoxide, respectively. Separate cultures were treated with the guanylate cyclase inhibitor ODQ. DETA-NO or SIN-1 induced phosphorylation of AMPK, and this was unaffected by co-treatment with Tempol. Uric acid treatment alone (without DETA-NO or SIN-1) tended to increase AMPK phosphorylation, but did not reach statistical significance (P = 0.16). Uric acid treatment did not prevent phosphorylation of AMPK by SIN-1, but did prevent significant phosphorylation by DETA-NO (although there was a trend for greater p-AMPK/AMPK, P = 0.12). Finally, ODQ treatment completely prevented the phosphorylation of AMPK by DETA-NO and SIN-1 (Fig. 3).
Figure 3. AMPKα phosphorylation induced by NO donors (24 μm SIN-1 and 50 μm DETA-NO) in myotubes is prevented by pharmacological inhibition of sGC.
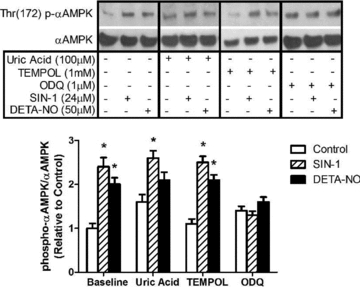
L6 myotubes were treated for 1 h with DETA-NO or the peroxynitrite generating compound, SIN-1, in the presence of uric acid to scavenge peroxynitrite, TEMPOL to scavenge superoxide, or ODQ to inhibit soluble guanylate cyclase (sGC) (n = 5–6/group). *P < 0.05 in comparison to untreated cells (Control).
NO-induced upregulation of mitochondrial function is blocked by AMPK inhibition
Treatment of L6 myotubes for 48 h with either SNAP or 8-Br-cGMP induced an approximate 40% increase in maximal citrate synthase activity (Fig. 4). Chronic treatments of myotubes for either 4 or 5 days with DETA-NO induced ∼30% increase in PGC-1α protein expression and ∼40% increase in basal respiration, respectively. A 15–20% increase in maximal (uncoupled) respiration was also observed after 5 days of treatment. Compound C treatment alone did not reduce PGC-1α protein or basal and uncoupled respiration significantly. However, treatment with Compound C completely ablated the positive effects of NO supplementation on PGC-1α protein and mitochondrial function of these cells (Fig. 5).
Figure 4. Chronic NO and cGMP treatments increase maximal citrate synthase activity in myotubes.
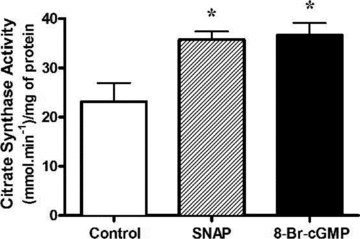
L6 cells were treated for 48 h with either SNAP (25 μm) or 8-Br-cGMP (2 mm) and were immediately harvested. Values represent enzyme activity normalized to protein concentration (means ±s.e.m., n = 6/group). *P < 0.05 compared to Control.
Figure 5. AMPK inhibition prevents chronic NO-dependent upregulation of PGC-1α protein and basal and maximal mitochondrial respiration in myotubes.
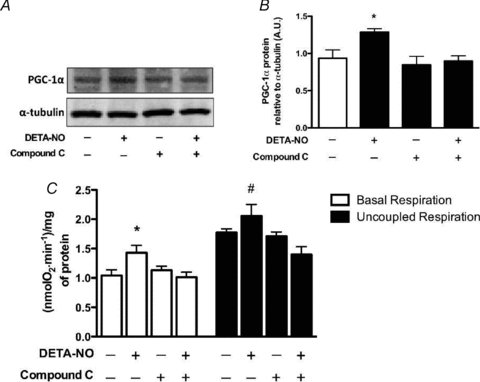
L6 cells were treated for 4 days (PGC-1α protein) or 5 days (mitochondrial respiration), 12 h day−1 with DETA-NO (50 μm) and Compound C (10 μm) and harvested 10 h after the last treatment (refer to Methods for details). A, representative immunoblots for PGC-1α protein. B, quantification of immunoblots for PGC-1α protein expression. C, quantification of mitochondrial respiration rates normalized to protein concentration in L6 myotubes (means ±s.e.m., n = 5/group). *P < 0.05 in comparison to all groups (except for Compound C in mitochondrial respiration), #P < 0.05 in comparison to DETA-NO + Compound C (P = 0.054 in comparison to Control).
NO-dependent upregulation of PGC-1α requires normal AMPKα1 expression
We used siRNA to knock down AMPKα1 and AMPKα2 catalytic subunits to test their role on the NO-dependent regulation of PGC-1α. Experiments were performed to optimize siRNA concentrations for maximal efficacy and specificity for knockdown of α1 and α2 subunits of AMPK (Supplemental Fig. 1). Uptake efficiency of siRNAs assessed 24 h after transfection was ∼90% in myotubes (Supplemental Fig. 1A). Transfection with 120 nmα1 siRNA knocked down AMPKα1 mRNA by 60%, while 200 nmα2 siRNA knocked down AMPKα2 mRNA by 95%. These knockdown effects were analysed separately and were achieved without compensatory upregulation of the other α isoform (Supplemental Fig. 1B and C). The combination of 120 nmα1 siRNA and 200 nmα2 siRNA was the most effective in knocking down both AMPKα1 and AMPKα2 mRNA levels, causing a 60–70% reduction in total AMPKα protein expression at 48 h, 56 h and 64 h after transfection (Supplemental Fig. 1D and E). All siRNA experiments thereafter were performed at 48 h after transfection. The siRNAs either together or independently did not cause any significant effect on PGC-1α mRNA (Supplemental Fig. 1F).
As shown in Fig. 6A and B, a significant reduction in total AMPKα expression was only observed in myotubes treated with either α1 siRNA, or both α1 and α2 siRNAs. Regardless of the degree of total AMPKα expression, DETA-NO treatment consistently caused a 2- to 2.5-fold increase in AMPKα phosphorylation (Fig. 6C). However, NO supplementation was able to induce a significant increase in AMPKα activity, indirectly assessed by ACC phosphorylation (Fig. 6D), only when total AMPKα and AMPKα1 expression were unaffected (i.e. unrelated siRNA and α2 siRNA conditions). Consistent with the effects of NO on AMPK activation and ACC phosphorylation, we observed a 2-fold increase in PGC-1α gene expression only in cells possessing normal AMPKα1 levels (Fig. 6E).
Figure 6. Knockdown of AMPKα1 prevents NO-dependent increase in PGC-1α mRNA.
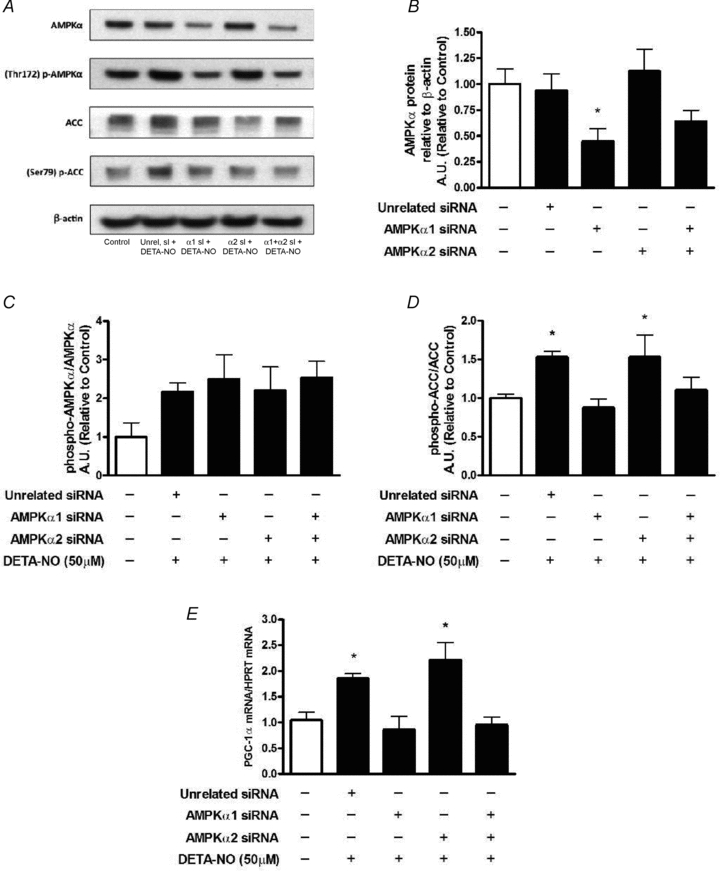
L6 cells were exposed to DETA-NO treatments after 48 h of transfection (n = 6–9/group, refer to Methods for details). Protein expression was assessed after treatments of 1 h. mRNA expression was analysed in cells treated for 3 h. A, representative immunoblots of proteins assessed and their phosphorylation status. B, AMPKα expression (*P < 0.05 in comparison to Control, Unrelated siRNA and AMPKα2 siRNA, P = 0.09 between Control and AMPKα1 siRNA +AMPKα2 siRNA groups). C, phospho-to-total AMPKα ratio. D, phospho-to-total ACC ratio (*P < 0.05 in comparison to Control and AMPKα1, P = 0.07 between either Unrelated siRNA or AMPKα2 siRNA and AMPKα1 siRNA +AMPKα2 siRNA groups). E, PGC-1α mRNA (*P < 0.05 in comparison to Control, AMPKα1 siRNA and AMPKα1 siRNA +AMPKα2 siRNA groups).
The CRE and MEF2 cis elements are not required for NO-dependent upregulation of PGC-1α promoter activity
NO supplementation induced AMPKα phosphorylation and PGC-1α mRNA induction in C2C12 myotubes to levels seen in L6 myotubes (Fig. 7A–C). Treatment of myotubes with DETA-NO increased activity of the intact PGC-1α promoter by 80%. Mutation of the CRE element (ΔCRE promoter construct) did not affect the ability of DETA-NO treatments to upregulate the PGC-1α promoter. The mutation of the MEF2 binding site was also shown not to be critical for the NO-mediated effect in the PGC-1α promoter, since DETA-NO treatment upregulated the ΔMEF2 promoter activity by ∼80% as well (Fig. 7).
Figure 7. NO-dependent activation of the 2 kb PGC-1α promoter in myotubes.
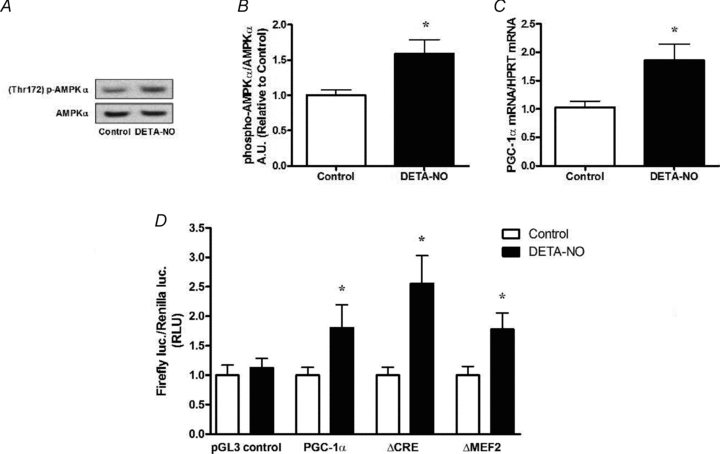
A, representative immunoblots of non-transfected C2C12 cells treated for 1 h with DETA-NO (50 μm). B, quantification of immunoblots for AMPKα phosphorylation normalized to total AMPKα expression (n = 4/group). C, PGC-1α mRNA induction in non-transfected C2C12 cells treated with DETA-NO (50 μm) for 3 h (n = 4–6/group). D, activity of the intact 2 kb PGC-1α promoter and mutated promoters in C2C12 cells treated 48 h after transfection with DETA-NO (50 μm) for 9 h (n = 12–15/group in pGL3 control & PGC-1α experiments; n = 8–12 in ΔCRE and ΔMEF2 experiments). Values represent fold changes (means ±s.e.m.) versus untreated (control) cells (refer to Methods for details). *P < 0.05 compared to Control.
NOS activity is required for AICAR-induced expression of PGC-1α and mitochondrial markers
L6 myotubes treated for 16 h with AICAR presented a significant, ∼10-fold increase in PGC-1α and F1ATP synthase mRNA levels, and a 4-fold increase in citrate synthase mRNA. Co-treatment with the NOS inhibitor l-NAME significantly blunted the AICAR effect on both PGC-1α and F1ATP synthase mRNA expression, and completely prevented the AICAR effect on citrate synthase mRNA (Fig. 8). In a separate experiment, addition of the nitric oxide-detecting fluorophore, DAF-FM, to the culture medium revealed that incubation of myotubes for 40 min with AICAR produced a significant dose-dependent release of NO (Fig. 9).
Figure 8. NOS inhibition blunts AMPK-dependent upregulation of mitochondrial biogenesis markers in myotubes.
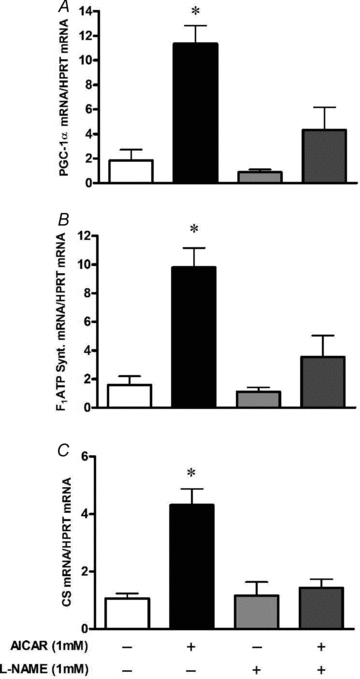
L6 cells were exposed to l-NAME 1 h prior and during the AICAR treatments of 16 h. Cells were harvested immediately after treatments. Values represent fold change versus untreated (Control) cells (means ±s.e.m., n = 5–7/group). Effect of AICAR treatment alone or in combination with l-NAME in PGC-1α mRNA (A), F1ATP synthase mRNA (B) and citrate synthase mRNA (C). *P < 0.05 in comparison to all other groups.
Figure 9. AMPK activation stimulates NO production in myotubes.
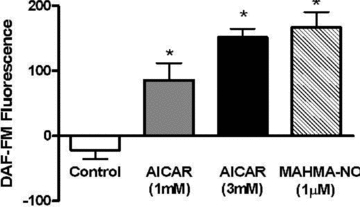
L6 cells were pre-loaded with DAF-FM, after which background fluorescence was assessed. Final fluorescence was determined following treatments of 40 min with either AICAR or MAHMA-NO (refer to Methods for details). Values represent the fluorescence at 40 min of incubation minus the background fluorescence, and are presented as means ±s.e.m. of n = 6/group. *P < 0.05 compared to Control.
NOS activity is required for AICAR and contraction-induced phosphorylation of AMPK in mouse skeletal muscle, in vitro
To extend examination of AMPK/NO interactions into adult skeletal muscle, we investigated whether NOS inhibition could reduce AMPK activation by either electrical stimulation or AICAR. Our protocols involving AICAR incubation or electrical stimulation were very effective in inducing AMPK activation (phosphorylation) in the glycolytic EDL muscle (∼2.3-fold and ∼4-fold increase in phospho-to-total AMPK ratio, respectively; Fig. 10). We observed that NOS inhibition with l-NMMA prevented AMPK activation by AICAR and electrical stimulation in vitro (Fig. 10).
Figure 10. NOS inhibition prevents AMPKα phosphorylation induced by electrical stimulation or AICAR.
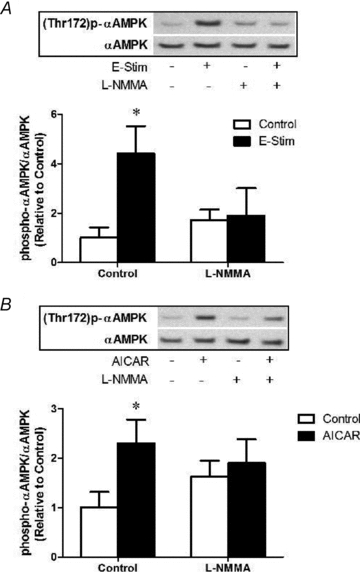
A, muscles were exposed to l-NMMA 10 min prior and during stimulation at 10 Hz, 13 V for 10 min (refer to Methods for details). Quantification of immunoblots for AMPKα phosphorylation normalized to total AMPKα expression. Values represent means ±s.e.m. (n = 6–8/group). *P < 0.05 compared to Control. B, muscles were exposed to l-NMMA 10 min prior to and during subsequent treatment with AICAR (2 mm) for 20 min (refer to Methods for details). Quantification of immunoblots for AMPKα phosphorylation normalized to total AMPKα expression. Values represent means ±s.e.m. (n = 6–8/group). *P < 0.05 compared to Control.
Discussion
Several studies have shown that NO induces mitochondrial biogenesis and upregulates PGC-1α expression in different tissues, including skeletal muscle (Nisoli et al. 2003, 2004, 2005). These reports, however, do not delineate the mechanisms of PGC-1α regulation mediated by NO-dependent signalling. Our lab has recently shown that NO increases GLUT4 expression via AMPK stimulation in skeletal muscle (Lira et al. 2007). Since both GLUT4 expression and mitochondrial biogenesis can be regulated by PGC-1α (Michael et al. 2001; Handschin et al. 2003; Handschin et al. 2007; Wende et al. 2007), and AMPK-dependent signalling induces PGC-1α expression and activation (Zong et al. 2002; Jäger et al. 2007), our central hypothesis was that NO regulates mitochondrial biogenesis via AMPK activation, and subsequent induction of PGC-1α. McConell et al. (2010) recently reported that both AICAR and caffeine (which increases intracellular calcium) induce mitochondrial biogenesis and PGC-1α expression in L6 myotubes. Furthermore, these authors reported evidence that AMPK and calcium–calmodulin-dependent protein kinase (CaMK) regulation of PGC-1α is at least partially mediated by nitric oxide or NOS activity, and that NO donors are sufficient to induce mitochondrial biogenesis in myotubes. These authors conclude that NO plays an important role in the maintenance of skeletal muscle mitochondrial volume in the basal state. However, based on their earlier work (Wadley et al. 2007) it seems that NOS activity is not required for exercise-induced mitochondrial biogenesis. Our findings extend our understanding of the mechanisms linking NO, AMPK-dependent signalling and PGC-1α expression in skeletal muscle, and confirm that NO-induced PGC-1α expression is mediated through different mechanisms from those reported for exercise effects.
The first set of experiments demonstrated that NO induces PGC-1α mRNA in myotubes within 3 h of treatments, and that AMPK activation occurs very rapidly (i.e. within 1 h of exposure of cells to NO donors). The use of two structurally distinct NO donors, namely SNAP (5 h half-life at 37°C and pH 7.4) and DETA-NO (20 h half-life at 37°C and pH 7.4) (Borniquel et al. 2006), reinforces the specificity of NO as the trigger of the signalling cascade leading to AMPK activation and subsequent upregulation of PGC-1α gene expression. We are unaware of other studies looking at short-term regulation of PGC-1α mediated by NO; however, a comparable increase in PGC-1α mRNA (∼80%) in response to a 6-day treatment with 50 μm DETA-NO in L6 cells was previously reported (Nisoli et al. 2004).
Next, we used pharmacological inhibition and molecular knockdown approaches to test whether the NO-dependent regulation of PGC-1α and mitochondrial function requires functional AMPK. Whereas both NO and cGMP increased PGC-1α mRNA, pharmacological inhibition of AMPK prevented the NO-dependent upregulation of the gene. Forty-eight-hour treatment of L6 myotubes with an NO donor or a cGMP analogue caused ∼40% increase in citrate synthase activity. This finding is consistent with the acute upregulation of PGC-1α mRNA in response to 3 h NO donor treatment, the 30% increase in PGC-1α protein expression and the 40% increase in basal coupled respiration rates observed in L6 myotubes following NO treatment for 4–5 days (12 h day−1). Interestingly, L6 cells have been reported to upregulate basal respiration rates by 140% when treated with a cGMP analogue continuously for 6 days (Nisoli et al. 2004). It is possible that a longer and continuous manipulation of a more distal player in the signalling cascade (i.e. cGMP versus NO) may induce more pronounced adaptations. In this context, Koves et al. (2005) reported a 2.5-fold increase in mitochondrial respiration rates of L6 cells transfected with an adenovirus containing the mouse PGC-1α. Consistent with these findings, our results show a lower, but physiologically significant augmentation of mitochondrial volume and respiratory rate following intermittent exposure to NO.
To confirm the necessity of AMPK for NO-induced upregulation of PGC-1α and to identify the isoform of the AMPKα catalytic subunit responsive to NO, we performed experiments to knock down the expression of both AMPKα1 and AMPKα2 using siRNA. Although both isoforms of AMPKα were detectable at the mRNA level, and both siRNAs were effective in reducing the expression of their target mRNAs, only knockdown of AMPKα1 caused a reduction in total AMPKα protein expression. These findings suggest that AMPKα1 is the main isoform expressed at the protein level in L6 myotubes. Previous studies using the same siRNA sequences reported similar reductions in total AMPKα protein expression (Pilon et al. 2004; Konrad et al. 2005; Wijesekara et al. 2006), but did not attempt to knock down each isoform of AMPKα separately. Our results show that AMPKα1 is required for the NO-dependent regulation of AMPK activity and PGC-1α expression. This is in agreement with observations that the NO donors sodium nitroprusside and spermine NONOate, preferentially activate AMPKα1 in isolated rat EDL muscles (Higaki et al. 2001) and human muscle strips (Deshmukh et al. 2010), respectively. Interestingly, exposure of isolated rat muscle to H2O2 (Toyoda et al. 2004), low-intensity contractions (Toyoda et al. 2006), or caffeine (Jensen et al. 2007) preferentially induces phosphorylation and activity of AMPKα1 without affecting the α2 isoform. Further, the α1 isoform has recently been shown to be critical for twitch-contraction-stimulated glucose uptake in isolated mouse muscle (Jensen et al. 2008). Clearly, the AMPKα1 isoform is sensitive to redox conditions, as shown by Toyoda et al. (2004). Nevertheless, we found that TEMPOL, a powerful scavenger of superoxide, had no effect on NO-induced phosphorylation of AMPK. Scavenging peroxynitrite with uric acid affected baseline AMPK phosphorylation but did not prevent phosphorylation by SIN-1. On the other hand, NO is known to produce many physiological effects via activation of soluble guanylate cyclase and production of cGMP. We found that inhibition of guanylate cyclase with ODQ completely prevented phosphorylation of AMPK by NO donors. Therefore, our data suggest that the primary effects of NO on AMPK are mediated through activation of guanylate cyclase and are generally not dependent on peroxynitrite or superoxide formation.
The distinct effects of NO may be classified as cGMP dependent and cGMP independent (Sugita et al. 2005). NO-mediated mitochondrial biogenesis and vasodilation are examples of cGMP-dependent effects, which are related to eNOS and nNOS activity, involving relatively low NO concentrations. On the other hand, cGMP-independent effects usually result from high NO levels and include nitrogen species-mediated nitrosative modification of proteins, lipids, and/or DNA (Stamler & Meissner 2001). NO-sensitive guanylyl cyclase responds to nanomolar concentrations of NO, with maximal activation reported in the range of 1–100 nm (Friebe & Koesling 2003). Hirota et al. (2001) reported NO concentration in culture medium of 285 nm with 100 μm SNAP treatment. Therefore, our 25 μm SNAP treatment (and presumably the 50 μm DETA-NO treatment which produced similar results) were likely to have formed nanomolar levels of NO, which would be expected to activate cGMP production.
The isoform-specific functions of AMPKα1 and AMPKα2 in skeletal muscle, and the physiological significance of AMPK signalling in muscle remain somewhat confusing. Although AMPKα2 seems to be the primary isoform activated by exercise (Treebak et al. 2007), an AMPKα2 kinase-dead mouse model has been reported to have normal (Maarbjerg et al. 2010) and impaired (Lee-Young et al. 2009) glucose uptake during in vivo exercise. Conversely, the α1 isoform may be more important during rest and periods of low energetic stress (Toyoda et al. 2006). Jorgensen et al. (2005) found that running-induced PGC-1α mRNA was normal in AMPKα1 knockout mice. This is not inconsistent with our finding of NO regulation of AMPKα1 since eNOS and nNOS knockout mice also show normal induction of PGC-1α with exercise (Wadley et al. 2007). It seems that AMPKα1 is required for muscle responses to low-intensity muscle activity and, therefore, may be important for maintenance of muscle phenotype during normal muscle activity. Perhaps NO regulation of AMPKα1 represents a control of basal metabolic phenotype. Since insulin resistance syndrome occurs in sedentary individuals over time via gradual changes in baseline skeletal muscle metabolic properties, aberrant regulation of AMPKα1 may be involved in the development of this disorder and presents an attractive target for intervention.
Another interesting observation was that DETA-NO caused very similar levels of AMPK phosphorylation across siRNA treated groups. However, phospho-to-total ACC, an indirect assessment of AMPK activity, was unchanged after AMPKα1 knockdown and accompanying reduction in total AMPKα expression. This observation suggests that reductions of ∼40–60% of AMPKα expression are sufficient to physiologically compromise the enzyme's function in these cells, even when the remaining enzyme is phosphorylated normally. These data imply that the level of total AMPKα expression should be considered in addition to the degree of phosphorylation of AMPKα when assessing AMPK-dependent signalling in skeletal muscle.
Regulation of transcription of the PGC-1α gene has been examined in recent years. It has been shown that calcium–calmodulin-dependent protein kinase (CaMKIV)- and calcineurin (CaN)-dependent induction of PGC-1α requires functional myocyte enhancer factor-2 (MEF2) and cAMP response element (CRE) DNA sequences in the PGC-1α promoter (Handschin et al. 2003). Further, PGC-1α can regulate its own expression through interaction with MEF2 at its own promoter (Handschin et al. 2003). Therefore, we assessed whether the MEF2 or CRE sites are required for NO-dependent upregulation of the proximal 2 kb PGC-1α promoter activity. We observed that NO is capable of inducing reporter activity in both mutated promoters (i.e. ΔCRE-PGC-1α and ΔMEF2-PGC-1α) to a similar extent to the induction seen with the intact PGC-1α promoter. To our knowledge this is the first study to examine the requirement of these binding sites for the NO-dependent induction of PGC-1α promoter activity. Although our data do not identify the specific promoter elements which are responsive to NO in the PGC-1α gene, we can say that upregulation of PGC-1α mRNA in response to NO appears to differ from exercise-related activation of the PGC-1α promoter, which requires both MEF2 and CRE sites to be intact (Akimoto et al. 2004). This is not overly surprising since Wadley et al. (2007) reported that although eNOS and nNOS-null mice exhibit changes in basal PGC-1α, acute exercise-induced upregulation of PGC-1α mRNA occurred normally. Other mechanisms exist by which NO and AMPK could induce PGC-1α activity and expression. For example, exercise causes a rapid movement of skeletal muscle PGC-1α from the cytosol to the nucleus, followed by an increase in PGC-1α expression (Wright et al. 2007). NO may be involved this early response. It was recently demonstrated that AMPK can directly phosphorylate PGC-1α, increasing its activity (Jäger et al. 2007; Canto et al. 2009). Further, an AMPK-dependent increase in NAD+ increases SIRT1 mediated deacetylation of PGC-1α, which is also necessary for activation (Canto et al. 2009). Activated PGC-1α can then co-activate its own promoter (Handschin et al. 2003). In addition, AMPK in skeletal muscle can phosphorylate and activate cAMP response element binding protein (CREB) (Thomson et al. 2008), a transcription factor known to stimulate PGC-1α expression (Handschin et al. 2003). Therefore, it seems that NO regulates basal PGC-1α activity and expression through a mechanism distinct from exercise-induced activation of the PGC-1α promoter.
Although our data suggest that AMPK is downstream of NO signalling, we have previously reported evidence of feedback interaction between AMPK activity and NOS activity in the regulation of GLUT4 expression in myotubes. Therefore, we examined whether AMPK-dependent regulation of PGC-1α and mitochondrial genes would be affected by inhibition of NO production. Similarly to our observations with GLUT4 (Lira et al. 2007), NOS inhibition blunted AMPK induction of PGC-1α, F1ATP synthase, and citrate synthase mRNA in L6 myotubes. McConell et al. (2010) also report evidence of feedback interaction between AMPK and NOS in the regulation of mitochondrial proteins. These observations are consistent with our finding that AMPK activation, via AICAR, increases NO production in L6. It appears that the AMPK-dependent effects of AICAR require basal endogenous NO production, and that once AMPK activation levels increase, due to AICAR exposure for example, AMPK stimulates NO production even further, most likely due to direct phosphorylation of NOS enzymes (Chen et al. 1999, 2000). This relationship between NOS and AMPK is not limited to myotubes as we observed that AICAR and electrical stimulation-induced phosphorylation of AMPK in isolated mouse EDL muscle was prevented by inhibition of NOS activity.
Significance and conclusions
The results presented here support our central hypothesis that NO regulates PGC-1α expression via AMPK-dependent signalling. Although previous reports have linked eNOS expression and NO production to mitochondrial biogenesis in different tissues (Nisoli et al. 2003, 2004, 2005), the mechanism was still elusive. We demonstrate that functional AMPK is required for NO-dependent increase in the expression of PGC-1α and mitochondrial genes, as well as improvement of mitochondrial respiration in skeletal muscle cells. More specifically, we provide evidence for an important role of AMPKα1 expression in the NO-mediated regulation of PGC-1α in L6 myotubes. Further, our observations suggest that NO does not require intact CRE or MEF2 sites in the proximal 2 kb PGC-1α promoter to induce its activity, raising the possibility that NO and AMPK may influence PGC-1α activity by affecting non-genomic mechanisms such as phosphorylation or nuclear translocation of the PGC-1α protein. Finally, we demonstrate that endogenous NOS activity is required for AMPK-dependent upregulation of PGC-1α, F1ATP synthase and citrate synthase mRNA in L6 myotubes.
Our current and previous findings (Lira et al. 2007) support a model where NO and AMPK, particularly AMPKα1, interact through a positive feedback loop that impacts the metabolic phenotype of skeletal muscle including the regulation of PGC-1α, mitochondrial genes and GLUT4 (Fig. 11). According to this model, basal NO production is required for normal AMPK activity at basal levels and during metabolically challenging conditions (e.g. AICAR and electrical stimulation). Once AMPK is activated, the enzyme induces increases in NO production by direct interaction with NOS. Further, the model suggests that increases in NO production, which can occur independent of AMPK activation status, increases AMPK phosphorylation and activation and impacts metabolic gene expression in skeletal muscle. Our current experiments with siRNA provide the first evidence for a functional requirement for AMPKα1 activation on NO-dependent regulation of muscle metabolic phenotype. This positive feedback loop between NO and AMPK is likely to be important in the maintenance and fine tuning of PGC-1α and associated metabolic genes in skeletal muscle representing potential alternative therapeutic targets for metabolic disorders.
Figure 11. Proposed model for NO and AMPK interaction that impacts PGC-1α expression and metabolic phenotype in skeletal muscle.
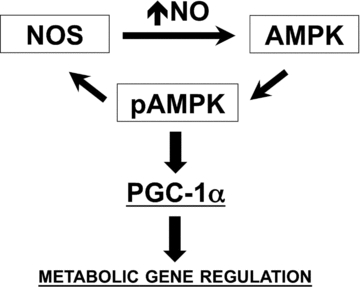
Our cumulative observations provide evidence for a positive feedback loop between NO production and AMPK activation that impacts the expression of PGC-1α, mitochondrial genes and GLUT4. Although both AMPK and NOS enzymes can be regulated by multiple mechanisms, our data with NOS inhibitors suggest that basal NOS activity and basal intracellular NO levels are required for AMPK to respond to AICAR and electrical stimulation in skeletal muscle. The observation of NOS inhibitors also preventing AICAR-mediated upregulation of PGC-1α, mitochondrial genes and GLUT4 in skeletal muscle myotubes supports this notion as well. The mechanisms for such regulation remain to be determined, but possibly involve regulation of AMPK kinases and/or AMPK phosphatases. Our current results further demonstrate that small elevations in intracellular NO levels upregulate PGC-1α and other metabolic genes (e.g. GLUT4) in a process that requires functional AMPKα1. Differently from the exercise-mediated induction of PGC-1α, NO-dependent regulation does not require the MEF2 and CRE cis elements at the 2 kb proximal promoter of PGC-1α (refer to text for details).
Acknowledgments
This work was supported by AHA Award 0530196N. The authors acknowledge and thank Drs Scott Powers and Zhen Yan for their assistance in completing this project.
Glossary
Abbreviations
- AICAR
5-aminoimidazole-4-carboxamide-1-β-d-ribonucleoside
- AMP
adenosine monophosphate
- AMPK
5′-AMP-activated protein kinase
- 8-Br-cGMP
8-bromo-cyclic guanosine monophosphate
- CRE
cyclic AMP response element
- DAF-FM
4-amino-5-methylamino-2′,7′-difluorofluorescein
- DETA-NO
diethylenetriamine-NONO
- eNOS
endothelial NOS
- iNOS
inducible NOS
- MAHMA-NO
methylamine hexamethylene methylamine-NONO
- MEF2
myocyte enhancer factor-2
- l-NAME
NG-nitro-l-arginine methyl ester
- l-NMMA
l-NG-monomethyl arginine
- NO
nitric oxide
- NOS
nitric oxide synthase
- nNOS
neuronal NOS
- ODQ
1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one
- PGC-1α
peroxisome proliferator-activated receptor-γ coactivator 1α
- siRNA
small interfering ribonucleic acids
- SNAP
S-nitroso-N-penicillamine
Author contributions
V.A.L. and D.S.C. conceived and designed the experiments. V.A.L., D.L.B., A.K.L., A.N.K., Q.A.S. and E.H.Z. performed the experiments. V.A.L. and D.S.C. analysed the data and wrote the manuscript. All authors have approved the final version of the manuscript. All work was performed at the University of Florida.
Supplemental material
supplemental Figure S1
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Akimoto T, Sorg BS, Yan Z. Real-time imaging of peroxisome proliferator-activated receptor-γ coactivator-1α promoter activity in skeletal muscles of living mice. Am J Physiol Cell Physiol. 2004;287:C790–C796. doi: 10.1152/ajpcell.00425.2003. [DOI] [PubMed] [Google Scholar]
- Balon TW, Jasman AP. Acute exposure to AICAR increases glucose transport in mouse EDL and soleus muscle. Biochem Biophys Res Commun. 2001;282:1008–1011. doi: 10.1006/bbrc.2001.4677. [DOI] [PubMed] [Google Scholar]
- Bonen A, Clark MG, Henriksen EJ. Experimental approaches in muscle metabolism: hindlimb perfusion and isolated muscle incubations. Am J Physiol Endocrinol Metab. 1994;266:E1–E16. doi: 10.1152/ajpendo.1994.266.1.E1. [DOI] [PubMed] [Google Scholar]
- Bonnard C, Durand A, Vidal H, Rieusset J. Changes in adiponectin, its receptors and AMPK activity in tissues of diet-induced diabetic mice. Diabetes Metab. 2008;34:52–61. doi: 10.1016/j.diabet.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Borniquel S, Valle I, Cadenas S, Lamas S, Monsalve M. Nitric oxide regulates mitochondrial oxidative stress via the transcriptional coactivator PGC1α. FASEB J. 2006;20:1889–1891. doi: 10.1096/fj.05-5189fje. [DOI] [PubMed] [Google Scholar]
- Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZP, McConell GK, Michell BJ, Snow RJ, Canny BJ, Kemp BE. AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. Am J Physiol Endocrinol Metab. 2000;279:E1202–E1206. doi: 10.1152/ajpendo.2000.279.5.E1202. [DOI] [PubMed] [Google Scholar]
- Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- Deshmukh AS, Long YC, Castro Barbosa T, Karlsson HKR, Glund S, Zavadoski WJ, Gibbs EM, Koisinen HA, Wallbrg-Henriksson H, Zierath JR. Nitric oxide increases cyclic GMP levels, AMP-activated protein kinase (AMPK)α1-specific activity and glucose transport in human skeletal muscle. Diabetologia. 2010;53:1142–1150. doi: 10.1007/s00125-010-1716-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia M. Organ and tissue contribution to metabolic rate. In: Kinney JM, Tucker HN, editors. Energy Metabolism: Tissue Determinants and Cellular Corollaries. New York: Raven Press; 1992. pp. 62–68. [Google Scholar]
- Friebe A, Koesling D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ Res. 2003;93:96–105. doi: 10.1161/01.RES.0000082524.34487.31. [DOI] [PubMed] [Google Scholar]
- Fryer LG, Carling D. AMP-activated protein kinase and the metabolic syndrome. Biochem Soc Trans. 2005;33:362–366. doi: 10.1042/BST0330362. [DOI] [PubMed] [Google Scholar]
- Geng T, Li P, Okutsu M, Yin X, Kwek J, Zhang M, Yan Z. PGC-1α plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not in fiber-type transformation in mouse skeletal muscle. Am J Physiol Cell Physiol. 2010;298:C572–C578. doi: 10.1152/ajpcell.00481.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C, Choi CS, Chin S, Kim S, Kawamori D, Kurpad AJ, Neubauer N, Hu J, Mootha VK, Kim Y-B, Kulkarni RN, Shulman GI, Spiegelman BM. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1a knockout mice reveals skeletal muscle-pancraetic β cell crosstalk. J Clin Invest. 2007;117:3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1α expression in muscle. Proc Natl Acad Sci U S A. 2003;100:7111–7116. doi: 10.1073/pnas.1232352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase: development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higaki Y, Hirshman MF, Fujii N, Goodyear LJ. Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes. 2001;50:241–247. doi: 10.2337/diabetes.50.2.241. [DOI] [PubMed] [Google Scholar]
- Hirota Y, Ishida H, Genka C, Obama R, Matsuyama S, Nakazawa H. Physiological concentration of nitric oxide induces positive inotropic effects through cGMP pathway in isolated rat ventricular myocytes. Jpn J Physiol. 2001;51:455–461. doi: 10.2170/jjphysiol.51.455. [DOI] [PubMed] [Google Scholar]
- Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TE, Rose AJ, Hellsten Y, Wojtaszewski JF, Richter EA. Caffeine-induced Ca2+ release increases AMPK-dependent glucose uptake in rodent soleus muscle. Am J Physiol Endocrinol Metab. 2007;293:E286–E292. doi: 10.1152/ajpendo.00693.2006. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Schjerling P, Viollet B, Wojtaszewski JF, Richter EA. AMPK alpha1 activation is required for stimulation of glucose uptake by twitch contraction, but not by H2O2, in mouse skeletal muscle. PLoS One. 2008;3:e2102. doi: 10.1371/journal.pone.0002102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji LL, Gomez-Cabrera MC, Vina J. Exercise and hormesis: activation of cellular antioxidant signaling pathway. Ann NY Acad Sci. 2006;1067:425–435. doi: 10.1196/annals.1354.061. [DOI] [PubMed] [Google Scholar]
- Jørgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA, Pilegaard H. Effects of α-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J. 2005;19:1146–1148. doi: 10.1096/fj.04-3144fje. [DOI] [PubMed] [Google Scholar]
- Konrad D, Rudich A, Bilan PJ, Patel N, Richardson C, Witters LA, Klip A. Troglitazone causes acute mitochondrial membrane depolarisation and an AMPK-mediated increase in glucose phosphorylation in muscle cells. Diabetologia. 2005;48:954–966. doi: 10.1007/s00125-005-1713-7. [DOI] [PubMed] [Google Scholar]
- Koves TR, Li P, An J, Akimoto T, Slentz D, Ilkayeva O, Dohm GL, Yan Z, Newgard CB, Muoio DM. Peroxisome proliferator-activated receptor-γ Co-activator 1α-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem. 2005;280:33588–33598. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- Lee-Young RS, Griffee SR, Lynes SE, Bracy DP, Ayala JE, McGuinness OP, Wasserman DH. Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J Biol Chem. 2009;284:23925–23934. doi: 10.1074/jbc.M109.021048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira VA, Soltow QA, Long JH, Betters JL, Sellman JE, Criswell DS. Nitric oxide increases GLUT4 expression and regulates AMPK signaling in skeletal muscle. Am J Physiol Endocrinol Metab. 2007;293:E1062–E1068. doi: 10.1152/ajpendo.00045.2007. [DOI] [PubMed] [Google Scholar]
- Lira VA, Benton CR, Yan Z, Bonen A. PGC-1α regulation by exercise training and its influences on muscle function and insulin sensitivity. Am J Physiol Endocrinol Metab. 2010;299:E145–161. doi: 10.1152/ajpendo.00755.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarbjerg SJ, Jorgensen SB, Rose AJ, Jeppesen J, Jensen TE, Treebak JT, Birk JB, Schjerling P, Wojtaszewski JF, Richter EA. Genetic impairment of α2-AMPK signaling does not reduce muscle glucose uptake during treadmill exercise in mice. Am J Physiol Endocrinol Metab. 2010 doi: 10.1152/ajpendo.90653.2008. (in press) [DOI] [PubMed] [Google Scholar]
- McConell GK, Ng GPY, Phillips M, Ruan Z, Macaulay SL, Wadley GD. Central role of nitric oxide synthase in AICAR and caffeine-induced mitochondrial biogenesis in L6 myocytes. J Appl Physiol. 2010;108:589–595. doi: 10.1152/japplphysiol.00377.2009. [DOI] [PubMed] [Google Scholar]
- Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, Kelly DP, Spiegelman BM. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci U S A. 2001;98:3820–3825. doi: 10.1073/pnas.061035098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, Cantoni O, Carruba MO, Moncada S, Clementi E. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci U S A. 2004;101:16507–16512. doi: 10.1073/pnas.0405432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisoli E, Tonello C, Cardeile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilon G, Dallaire P, Marette A. Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J Biol Chem. 2004;279:20767–20774. doi: 10.1074/jbc.M401390200. [DOI] [PubMed] [Google Scholar]
- Pye D, Palomero J, Kabayo T, Jackson MJ. Real-time measurement of nitric oxide in single mature mouse skeletal muscle fibres during contractions. J Physiol. 2007;581:309–318. doi: 10.1113/jphysiol.2006.125930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Shankar RR, Wu Y, Shen H, Zhu J-S, Baron AD. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes. 2000;49:684–687. doi: 10.2337/diabetes.49.5.684. [DOI] [PubMed] [Google Scholar]
- Smith AC, Bruce CR, Dyck DJ. AMP-kinase activation with AICAR simultaneously increases fatty acid and glucose oxidation in resting rat soleus muscle. J Physiol. 2005;565:547–553. doi: 10.1113/jphysiol.2004.081679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srere PA. Citrate synthase. Methods Enzymol. 1969;13:3–5. [Google Scholar]
- Sriwijitkamol A, Coletta DK, Wajcberg E, Balbontin GB, Reyna SM, Barrientes J, Eagan PA, Jenkinson CP, Cersosimo E, DeFronzo RA, Sakamoto K, Musi N. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes. 2007;56:836–848. doi: 10.2337/db06-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- Sugita H, Fujimoto M, Yasukawa T, Shimizu N, Sugita M, Yasuhara S, Jeevendra Martyn JA, Kaneki M. Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. J Biol Chem. 2005;280:14203–14211. doi: 10.1074/jbc.M411226200. [DOI] [PubMed] [Google Scholar]
- Thomson DM, Herway ST, Filmore N, Kim H, Brown JD, Barrow JR, Winder WW. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J Appl Physiol. 2008;104:429–438. doi: 10.1152/japplphysiol.00900.2007. [DOI] [PubMed] [Google Scholar]
- Toyoda T, Hayashi T, Miyamoto L, Yonemitsu S, Nakano M, Tanaka S, Ebihara K, Masuzaki H, Hosoda K, Inoue G, Otaka A, Sato K, Fushiki T, Nakao K. Possible involvement of the α1 isoform of 5′AMP-activated protein kinase in oxidative stress-stimulated glucose transport in skeletal muscle. Am J Physiol Endocrinol Metab. 2004;287:E166–E173. doi: 10.1152/ajpendo.00487.2003. [DOI] [PubMed] [Google Scholar]
- Toyoda T, Tanaka S, Ebihara K, Masuzaki H, Hosoda K, Sato K, Fushiki T, Nakao K, Hayashi T. Low-intensity contraction activates the α1-isoform of 5′-AMP-activated protein kinase in rat skeletal muscle. Am J Physiol Endocrinol Metab. 2006;290:E583–E590. doi: 10.1152/ajpendo.00395.2005. [DOI] [PubMed] [Google Scholar]
- Treebak JT, Birk JB, Rose AJ, Kiens B, Richter EA, Wojtaszewski JF. AS160 phosphorylation is associated with activation of α2β2γ1- but not α2β2γ3-AMPK trimeric complex in skeletal muscle during exercise in humans. Am J Physiol Endocrinol Metab. 2007;292:E715–E722. doi: 10.1152/ajpendo.00380.2006. [DOI] [PubMed] [Google Scholar]
- Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L, Cantoni O, Clementi E, Moncada S, Carruba MO, Nisoli E. TNF-α downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest. 2006;116:2791–2798. doi: 10.1172/JCI28570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadley GD, Choate J, McConell GK. NOS isoform-specific regulation of basal but not exercise-induced mitochondrial biogenesis in mouse skeletal muscle. J Physiol. 2007;585:253–262. doi: 10.1113/jphysiol.2007.141309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende AR, Schaeffer PJ, Parker GJ, Zexhner C, Han D-H, Chen M, Hancock CR, Lehman JJ, Huss JM, McClain DA, Holloszy JO, Kelly DP. A role for the transcriptional co-activator PGC-1α in muscle refueling. J Biol Chem. 2007;282:36642–36651. doi: 10.1074/jbc.M707006200. [DOI] [PubMed] [Google Scholar]
- Wijesekara N, Tung A, Thong F, Klip A. Muscle cell depolarization induces a gain in surface GLUT4 via reduced endocytosis independently of AMPK. Am J Physiol Endocrinol Metab. 2006;290:E1276–E1286. doi: 10.1152/ajpendo.00573.2005. [DOI] [PubMed] [Google Scholar]
- Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 2000;88:2219–2226. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- Wright DC, Han DH, Garcia-Roves PM, Geiger PC, Jones TE, Holloszy JO. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1α expression. J Biol Chem. 2007;282:194–199. doi: 10.1074/jbc.M606116200. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Young ME, Leighton B. Evidence for altered sensitivity of the nitric oxide/cGMP signaling cascade in insulin-resistant skeletal muscle. Biochem J. 1998;329:73–79. doi: 10.1042/bj3290073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, MacLean PS, Pohnert SC, Knight JB, Olson AL, Winder WW, Dohm GL. Regulation of muscle GLUT-4 transcription by AMPactivated protein kinase. J Appl Physiol. 2001;91:1073–1083. doi: 10.1152/jappl.2001.91.3.1073. [DOI] [PubMed] [Google Scholar]
- Zhou M, Lin BZ, Coughlin S, Vallega G, Pilch PF. UCP-3 expression in skeletal muscle: effects of exercise, hypoxia, and AMPK-activated protein kinase. Am J Physiol Endocrnol Metab. 2000;279:E622–E629. doi: 10.1152/ajpendo.2000.279.3.E622. [DOI] [PubMed] [Google Scholar]
- Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.