

Abstract
Background & Aims
c-Jun N-terminal kinase (JNK) is activated by multiple profibrogenic mediators; JNK activation occurs during toxic, metabolic, and autoimmune liver injury. However, its role in hepatic fibrogenesis is unknown.
Methods
JNK phosphorylation was detected by immunoblot analysis and confocal immunofluorescent microscopy in fibrotic livers from mice after bile duct ligation (BDL) or CCl4 administration and in liver samples from patients with chronic hepatitis C and non-alcoholic steatohepatitis. Fibrogenesis was investigated in mice given the JNK inhibitor SP600125 and in JNK1- and JNK2-deficient mice following BDL or CCl4 administration. Hepatic stellate cell (HSC) activation was determined in primary mouse HSCs incubated with pan-JNK inhibitors SP600125 and VIII.
Results
JNK phosphorylation was strongly increased in livers of mice following BDL or CCl4 administration as well as in human fibrotic livers, occurring predominantly in myofibroblasts. In vitro, pan-JNK inhibitors prevented transforming growth factor (TGF)β-, platelet-derived growth factor (PDGF)-, and angiotensin II-induced murine HSC activation and decreased PDGF and TGFβ signaling in human HSCs. In vivo, pan-JNK inhibition did not affect liver injury but significantly reduced fibrosis after BDL or CCl4. JNK1-deficient mice had decreased fibrosis after BDL or CCl4 whereas JNK2-deficient mice displayed increased fibrosis after BDL but fibrosis was not changed after CCl4. Moreover, patients with chronic hepatitis C who displayed decreased fibrosis in response to the angiotensin receptor type 1 blocker losartan showed decreased JNK phosphorylation.
Conclusion
JNK is involved in HSC activation and fibrogenesis and represents a potential target for antifibrotic treatment approaches.
Introduction
Hepatic fibrosis is the result of chronic hepatocellular injury and the first step towards the development of clinically relevant complications such as cirrhosis and hepatocellular carcinoma1,2. While acute hepatocellular injury leads to the activation of a transient wound healing response and restoration of normal liver architecture, chronic liver injury leads to a persistent activation of wound healing responses and increasing deposition of extracellular matrix (ECM) proteins. In advanced stages, increased ECM leads to changes in the hepatic blood-flow and portal hypertension, as well as the replacement of healthy liver parenchyma by scar tissue1,2. However, the signals that promote the activation of myofibroblasts and deposition of ECM are only incompletely understood.
c-Jun N-terminal kinase (JNK) is a member of the MAP kinase family and involved in the regulation of proliferation, cell death, inflammation and metabolism3,4. JNK1 and JNK2 are ubiquitously expressed whereas JNK3 expression is largely restricted to the central nervous system, testis and heart. JNKs are activated in response to stimuli such as inflammatory cytokines, bacterial products, oxidative stress and irradiation3,4. Many effects of JNKs are mediated through phosphorylation of their target c-Jun that hetero- and homodimerizes with other Jun and Fos family members to form AP-1 transcription factors5. In addition to their archetypical target c-Jun, JNKs also phosphorylate a number of other targets which is likely the basis for their involvement in a wide range of biological processes such as cell death, proliferation and inflammation6. JNKs play a crucial role in liver regeneration7,8 and hepatocyte death9,10. Although JNKs are involved in the regulation of liver injury in response to TNFα, Concanavalin A, ischemia-reperfusion, acetaminophen intoxication, high-fat diet and hepatic carcinogens9,11–16, their role in the subsequent wound healing response is not known. In hepatic stellate cells (HSCs), a cell type that is crucially involved in the promotion of hepatic fibrosis17, profibrogenic mediators such as platelet-derived growth factor (PDGF), transforming growth factor (TGF)β and angiotensin II (AngII) promote JNK activation18,19. Moreover, JNK contributes to α-smooth muscle actin (αSMA) expression in early stages of HSC activation and to HSC migration19,20 supporting a potential role for JNK in the development of liver fibrosis. On the other hand, JNK is also involved in potential anti-fibrogenic effects such as the promotion of TNFα- and sulfasalazine-induced cell death in HSCs and fibroblasts21,22.
Here we investigate the role of JNK in hepatic fibrogenesis using a combination of pharmacologic and genetic in vivo approaches as well as experiments in primary HSCs. We present evidence that JNK activation in the fibrotic liver occurs in myofibroblasts in both mice and humans, and that JNK plays a crucial role in the activation of HSCs and development of liver fibrosis.
Methods
Mice and fibrosis induction
Balb/c mice, JNK1-deficient mice (backcrossed 5 times to C57B1/6, http://jaxmice.jax.org/strain/004319.html) and JNK2-deficient mice (in pure C57Bl/6 background, http://jaxmice.jax.org/strain/004321.html) and C57Bl/6 were obtained from Jackson laboratories. JNK1-deficient mice were crossed with C57Bl/6 mice, and the resulting heterozygous mice were used to establish JNK1-deficient and wild-type JNK1 control strains. Mice were kept in a specific pathogen-free environment at Columbia University. Transgenic mice expressing collagen α1(I)-driven green fluorescent protein (GFP) and αSMA-driven red fluorescent protein (RFP) have been described23,24. Hepatic fibrosis was induced in 8–12 week-old age-matched male mice by ligating the common bile duct (BDL) as described24, by 4–6 gavages or 8 intraperitoneal injections of CCl4 (0.5 µl/g body weight, diluted 1:3 in corn oil, given every 3 days). For pharmacologic JNK inhibition, mice were injected twice daily intraperitoneally with 0.5 or 1 mg SP600125 dissolved in 15 µl DMSO (LC Laboratories), or DMSO vehicle.
Mouse HSC isolation and culture
HSCs were isolated by pronase-collagenase perfusion followed by Nycodenz-based density centrifugation and cultured as described24. Purity as determined by vitamin A autofluorescence routinely exceeded 95%. Experiments in murine HSCs were performed between 2 and 7 days after isolation. All animal procedures were approved by the Columbia University Institutional Animal Care and Use Committee.
Human liver samples and HSCs
Human liver samples were from liver biopsies performed in patients with chronic hepatitis C and non-alcoholic steatohepatitis (NASH). Fragments of normal livers were obtained from resections of liver metastasis of colon cancer. Human HSCs were isolated by density gradient-based centrifugation from normal liver as described25 and used between passage 3 and 5. Procedures involving human materials were approved by the Investigational Review Board of the Hospital Clínic of Barcelona. Paraffin-embedded samples were also obtained from patients with chronic hepatitis C that were enrolled in an open-label study with the angiotensin type 1 receptor blocker losartan (50 mg/day for 18 months)26,27. The protocol was approved by the Ethics Committee of the Hospital Clínic of Barcelona and the Agencia Española del Medicamento as a phase IV trial (ARAHEPC 02-0491), and registered into the protocol registration system (NCT00298714).
Fibrosis evaluation by Sirius red staining and hydroxyproline assay
Sirius red staining of paraffin-embedded liver sections was performed as described24. The Sirius red-positive area was quantified in at least 10 low-power fields using Adobe Photoshop and ImageJ in a blinded fashion. Hydroxyproline assays were performed as described24.
Immunoblot analysis
Electrophoresis and immunblotting were performed as described24. Blots were incubated with anti-αSMA (Sigma) at 1:5000, anti-phospho c-Jun (Santa Cruz), total c-Jun, total JNK or anti-phospho JNK (all Cell Signaling) at 1:1000, and reprobed with anti-actin (MP Biomedicals) or anti-tubulin (Santa Cruz) mouse antibodies.
Immunohistochemistry, confocal immunofluorescent microscopy, and image quantification
Paraffin sections were incubated with anti-αSMA (Abcam, 1:100), anti-p-JNK (Cell Signaling, 1:30), anti-keratin (Dako, 1:1500), anti-keratin 8 (DSHB, 1:50), anti-F4/80 (eBioscience, 1:50 to 1:350), anti-GFP (Abcam, 1:100) and either with secondary labeled polymer HRP anti-rabbit envision antibody (Dako) followed by DAB or the fluorescent horseradish-peroxidase substrate TSA solution Cyanin3 (Perkin Elmer), or a secondary AlexaFluor488-conjugated chicken anti-goat or anti-rat antibody (both Molecular Probes). Immunofluorescent staining was visualized on a Zeiss LSM510 confocal microscope using 1 µM plane thickness. αSMA and p-JNK were quantified with Adobe Photoshop and AnalySIS software, respectively.
TGFβ reporter assays
HSCs were co-infected with Ad(CAGA)9-Luc and AdLacZ at a multiplicity of infection of 100 and 10 particles/cell, respectively, for 12 hours as described24. Following serum starvation, cells were treated with recombinant human TGFβ1 (R&D systems) at 10–100 pg/ml for 6h. Luciferase activity was measured in a platereader (Fluostar Optima) and normalized to β-galactosidase as described28.
[3H]-thymidine incorporation
Serum-starved HSCs (0.4 × 105/well) were pretreated with SP600125 (5 µM), JNK inhibitor VIII (Calbiochem, 16 µM) or vehicle (0.1% DMSO) followed by PDGF treatment (20 nM) for 26h. Eighteen hours after PDGF stimulation, cells were pulsed with 1 µCi/mL [3H]-thymidine (Amersham Biosciences) for 8h, followed by TCA-precipitation, lysis, and measurement in a scintillation counter.
Real time PCR
Quantitative real time PCR (qPCR) was performed as previously described using commercial primer-probe pairs (Applied Biosystems), normalization to 18s and relative quantification by standard curve24.
Statistical Analysis
All data are expressed as mean ±SD. Differences between experimental and control groups were assessed by two-tailed unpaired Student t test. p values less than 0.05 were considered significant.
Results
JNK is activated in hepatic myofibroblasts during hepatic fibrogenesis
JNK plays a key role in liver injury after concanavalin A, TNFα, high-fat diet, acetaminophen intoxication, carcinogen exposure or ischemia-reperfusion injury 9,11–16. To test the role of JNK in hepatic wound healing responses to chronic injury, we first analyzed the phosphorylation status of JNK and its target c-Jun in mice following BDL or CCl4 treatment. We observed a strong activation of the JNK pathway as demonstrated by elevated phosphorylated JNK (p-JNK), phosphorylated c-Jun and total c-Jun levels in livers extracts of BDL- and CCl4-treated mice (Fig. 1A–B, Supplementary Fig. 1A). Immunohistochemistry revealed almost no p-JNK in hepatocytes of BDL-treated mice (Fig. 1C–D) but intense p-JNK staining in periportal areas of BDL mice that markedly reproduced the pattern of fibrotic areas (Fig. 1C). In CCl4-induced fibrosis we also detected p-JNK in fibrotic septae, and not within the hepatic parenchyma (Fig. 1D). To assess whether p-JNK was localized in myofibroblasts within the fibrotic areas, mice expressing collagen-driven GFP underwent CCl4 treatment or BDL followed by costaining of liver sections for GFP and p-JNK. Confocal microscopy revealed that myofibroblasts constituted a large proportion of p-JNK-expressing cells both in the BDL and the CCl4 fibrosis model as seen by orange fluorescence (Fig. 1E–F). Following BDL, there was also some p-JNK staining in bile duct epithelial cells as previously reported29 (Supplementary Fig. 1 B,C). There was no p-JNK staining in the great majority of Kupffer cells both in the BDL and the CCl4 model (Supplementary Fig. 1 D,E).
Figure 1. JNK activation during hepatic fibrogenesis occurs in myofibroblasts.
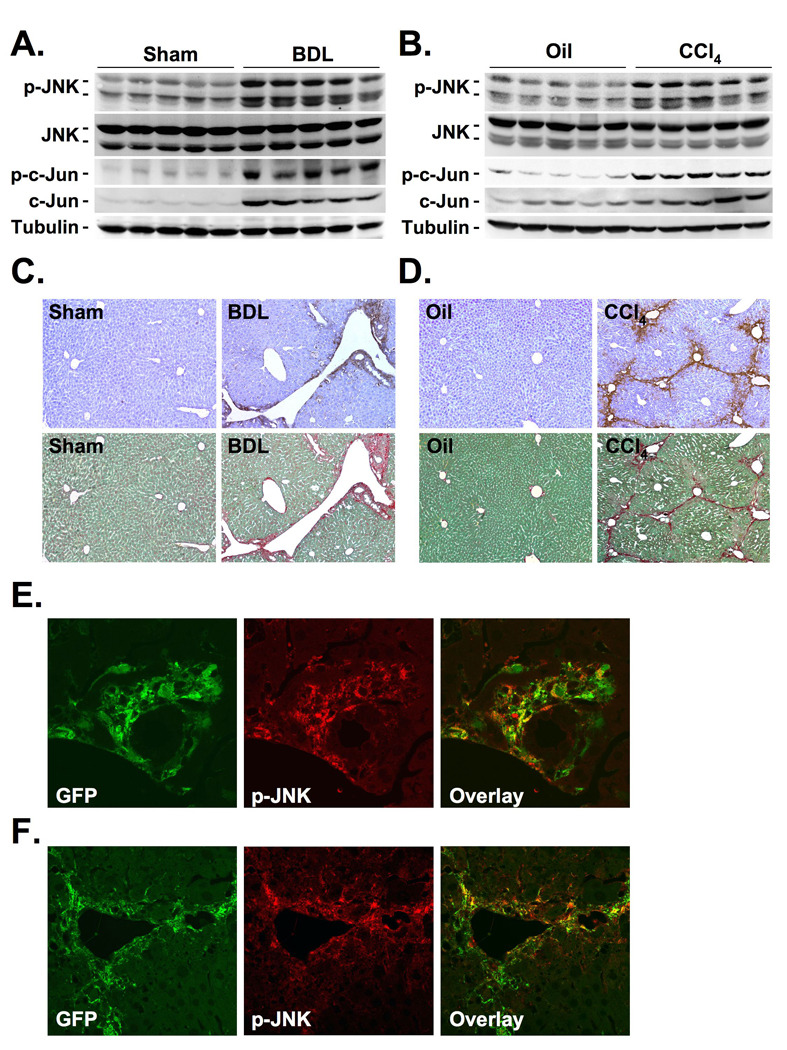
A–D. Balb/c mice underwent BDL or sham operation for 15 days, or 8 gavages of oil or CCl4 (0.5 µl/g). JNK activation was determined by immunoblotting of phospho-c-Jun or p-JNK (A–B.). Serial sections were stained for p-JNK and by picrosirius red (C–D.). E–F. Collagen-GFP mice underwent BDL or sham operation for 15 days (E.), or were treated with 8 injections of CCl4 or oil (F.). Sections were stained for GFP (green staining) and p-JNK (red staining) followed by confocal microscopy.
Pharmacologic JNK inhibition reduces experimental fibrogenesis in mice
As our data showed p-JNK activation predominantly in myofibroblasts of the fibrotic liver, we next evaluated whether JNK is functionally involved in fibrogenesis. For this purpose, we inhibited JNK activation in mice that underwent BDL or CCl4 treatment using SP600125, a well-characterized JNK inhibitor that has shown significant effects in acetaminophen-induced liver injury13,14. SP600125 significantly reduced hepatic fibrosis after BDL as assessed by Sirius Red staining, hydroxyproline quantification, αSMA immunohistochemistry and immunoblot analysis in two independent cohorts (Fig. 2A–D, Supplementary Fig. 2A). Five days after BDL, a time point at which injury is most prominent, we found no significant reduction of hepatocellular injury as determined by serum ALT levels, and quantification of bile infarcts (Fig. 2E–F). A similar reduction of the Sirius Red positive area, hydroxyproline content and αSMA expression but no reduction in injury was found in SP600125-treated mice that received CCl4 (Fig. 3A–E). Quantification of keratin-positive cholangiocytes revealed no significant difference in bile duct proliferation by SP600125 (Supplementary Fig. 2 B). There was a reduction of some inflammatory parameters in mice treated with SP600125 with significant reductions of MCP-1 in the BDL model, and TNFα in the CCl4 model (Supplementary Fig. 3A,B). There was no significant difference in the infiltration of F4/80-positive macrophages between SP600125- and vehicle-treated mice in both fibrosis models (Supplementary Fig. 2 C, D).
Figure 2. JNK inhibition ameliorates bile duct ligation-induced hepatic fibrosis.
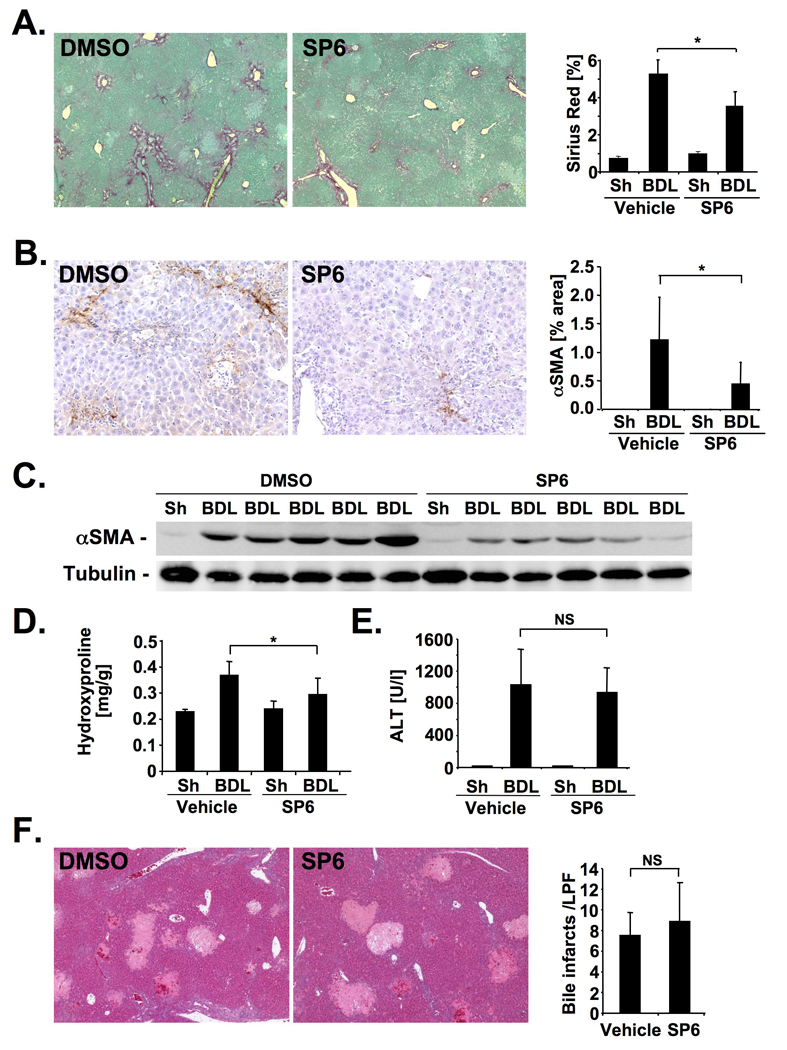
A–D. Male Balb/c mice underwent BDL for 15 days, and were treated with 1 mg SP600125 twice daily (n=7) or DMSO vehicle (n=10). Hepatic fibrosis was evaluated by Sirius Red staining (A), αSMA immunohistochemistry (B), αSMA immunoblotting (C), and hydroxyproline content (D). E.–F. Hepatic injury in SP600125- (n=7) or vehicle-treated (n=8) mice was determined by ALT levels (E) and bile infarcts 5 days after BDL (F). * p<0.05
Figure 3. JNK inhibition ameliorates CCl4-induced hepatic fibrosis.
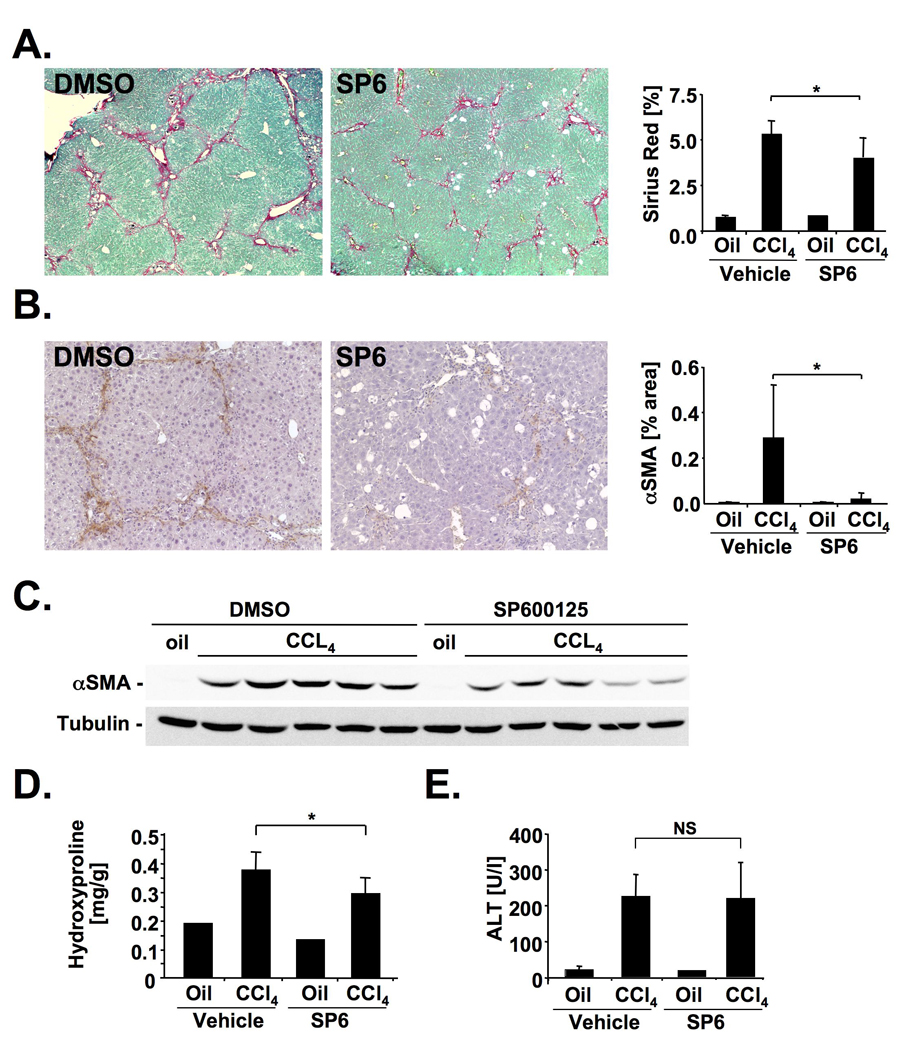
A–E. SP600125- treated (n=9) and vehicle-treated (n=9) Balb/c mice received 4 gavages of CCl4. Hepatic fibrosis and injury were evaluated by Sirius Red staining (A), αSMA immunohistochemistry (B), αSMA immunoblotting (C), hydroxyproline content (D) and ALT levels 48h after the last CCl4 injection (E). * p<0.05
JNK activity is required for TGFβ-, AngII- and PDGF-induced HSC activation and proliferation
As myofibroblasts were the only cell population in which pJNK was found consistently at high levels in both models of fibrogenesis, we next determined the role of JNK in the activation of HSCs, an important source of hepatic myofibroblasts. TGFβ strongly promoted JNK activation in HSCs, and JNK inhibitors SP600125 and inhibitor VIII efficiently reduced TGFβ-induced JNK activation (Fig. 4A). Neither inhibitor had toxic effects in HSCs (Supplementary Fig. 4). We found a significant reduction of TGFβ-driven CAGA activity by SP600125 and inhibitor VIII (data not shown) confirming previous findings that JNK inhibition blocks the activity of a Smad-dependent PAI-I reporter19. Notably, incubation of HSCs with SP600125 or JNK inhibitor VIII blunted αSMA induction by TGFβ (Fig. 4B). Moreover, JNK inhibitors drastically reduced TGFβ-induced α-SMA reporter activity in HSCs isolated from transgenic mice expressing RFP under the control of the αSMA promoter (Fig. 4C). AngII, another well-established profibrogenic mediator, also strongly induced c-Jun phosphorylation which was efficiently inhibited by both SP600125 and inhibitor VIII (Fig. 4D). Moreover, SP600125 and inhibitor VIII blunted the strong induction of Ang-induced αSMA expression and αSMA-dependent RFP reporter activity (Fig. 4E–F). Moreover, SP600125 and inhibitor VIII markedly reduced PDGF-mediated [3H]-thymidine uptake (Supplementary Fig. 5).
Figure 4. JNK promotes hepatic stellate activation and proliferation.
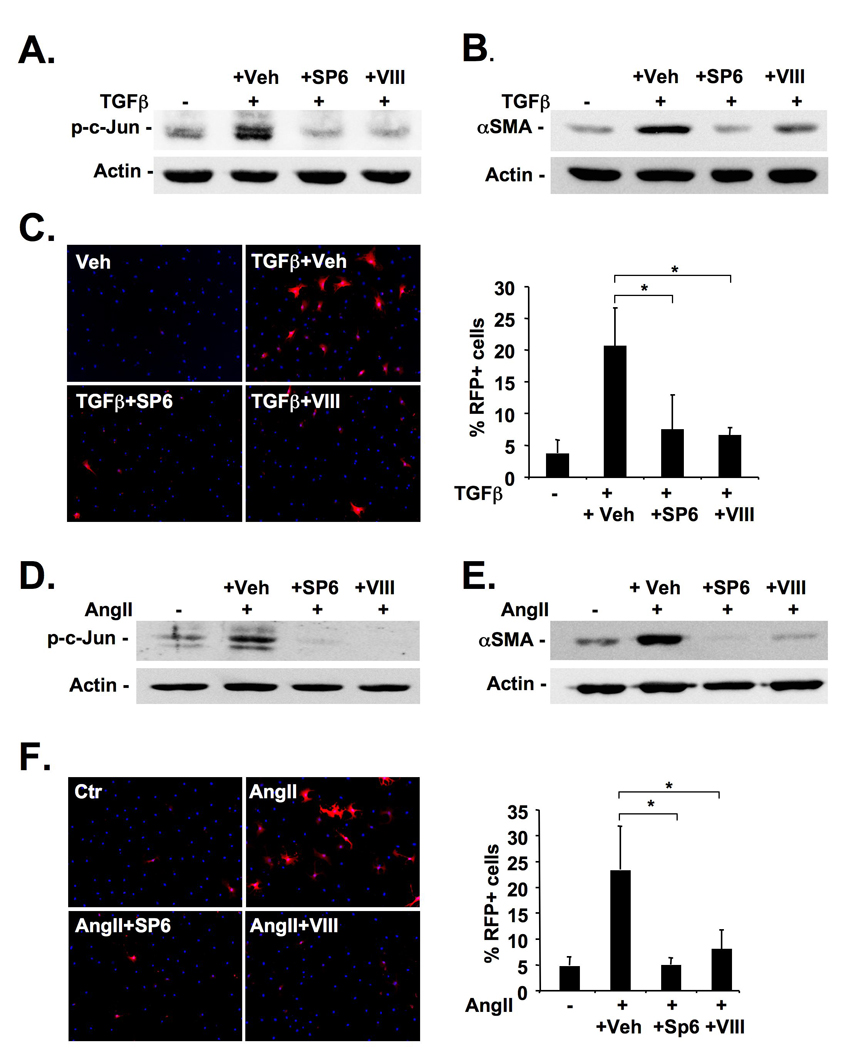
A–B. Primary mouse HSCs were pretreated with SP600125 (5 µM), inhibitor VIII (16 µM) or vehicle (0.1% DMSO) followed by TGFβ (2 ng/ml) treatment. c-Jun phosphorylation (A) and αSMA expression (B) were determined by immunoblot 60 minutes and 48h after TGFβ treatment, respectively. C. HSCs were isolated from αSMA-RFP mice, pretreated with JNK inhibitors or vehicle for 24h and treated with TGFβ (2 ng/ml) for 48h followed by Hoechst 33342 staining and quantification of RFP expression. D–E. One day after isolation, primary mouse HSCs were pretreated with JNK inhibitors or vehicle (see above) followed by treatment with AngII (10−7 M). c-Jun phosphorylation (D) and αSMA expression (E) were determined by immunoblot 20 minutes and 48h after AngII treatment, respectively. F. HSCs from αSMA-RFP mice were pretreated as described above followed by treatment with AngII for 48h and evaluation of RFP expression.
JNK1 but not JNK2 promotes fibrogenesis
To confirm the profibrogenic role of JNK by a genetic approach and to assess the contribution of different JNK isoforms, we next performed BDL in JNK1- and JNK2- deficient mice. We found a significant reduction of Sirius red staining in JNK1-deficient mice (Fig. 5A) whereas the Sirius red positive area was increased in two cohorts of JNK2-deficient mice (Fig. 6A, Supplementary Fig. 6A). Moreover, JNK1-deficient mice displayed a decrease in hepatic hydroxyproline content and αSMA protein (Fig. 5 B–D) whereas JNK2-deficient mice displayed increased αSMA expression and hydroxyproline content (Fig. 6B–D). JNK1-deficiency did not reduce hepatocellular injury 5 days after BDL (Fig. 5 E–F), and showed no changes in hepatic MCP-1 and TNFα expression (Supplementary Fig. 7A). There was a trend towards less bile duct proliferation in JNK1-deficient mice but this did not reach statistical significance (Supplementary Fig. 7B) In contrast, JNK2-deficient mice displayed significant decreased ALT levels and bile infarcts (Fig. 6 E–F) but no significant changes in MCP-1 and TNFα expression (Supplementary Fig. 6). In a second model of fibrogenesis, CCl4 treatment, JNK1-deficient mice again showed a significant reduction of fibrosis (Supplementary Fig. 8) but no significant changes in hepatic inflammation (data not shown) whereas fibrosis remained unchanged in JNK2-deficient mice.
Figure 5. JNK1 promotes hepatic fibrosis.
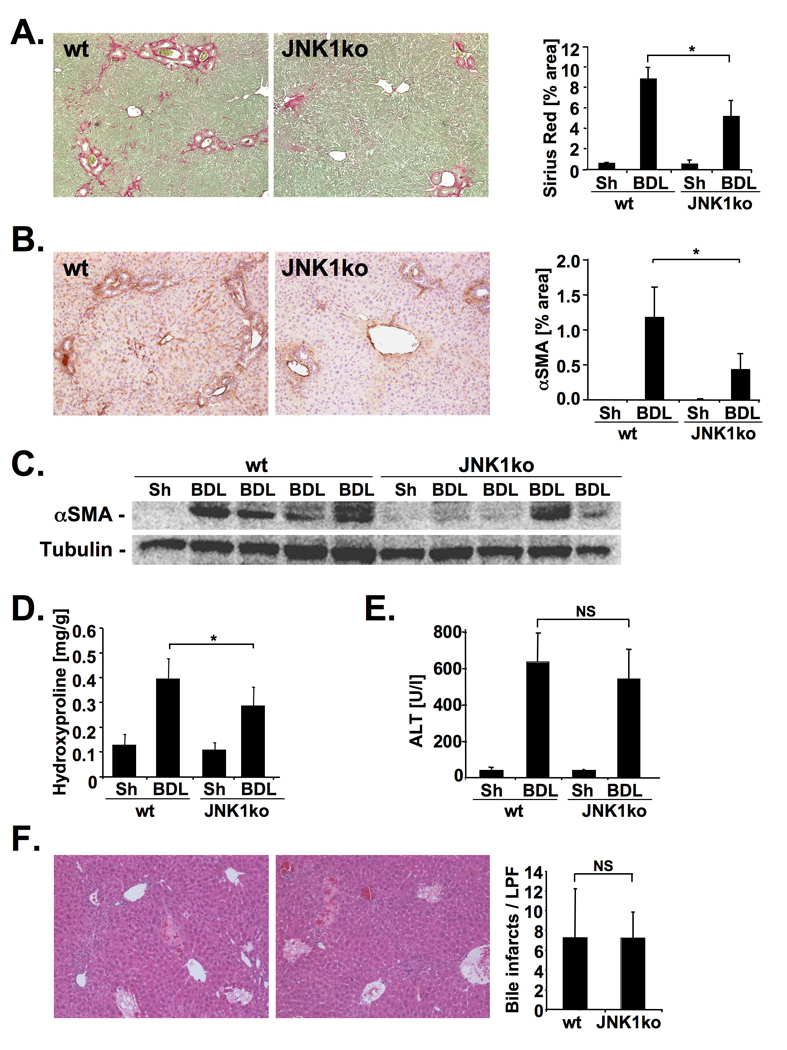
A.–D. JNK1-deficient (n=10) or control mice (n=7) underwent BDL for 21 days. Hepatic fibrosis was evaluated by Sirius Red staining (A), αSMA immunohistochemistry (B), αSMA immunoblotting (C), and hydroxyproline content (D). E–F. Hepatic injury was quantified by ALT levels (E) and bile infarcts (f) in JNK1-deficient (n=9) and control mice (n=9) five days after BDL. * p<0.05
Figure 6. JNK2 decreases hepatic fibrosis.
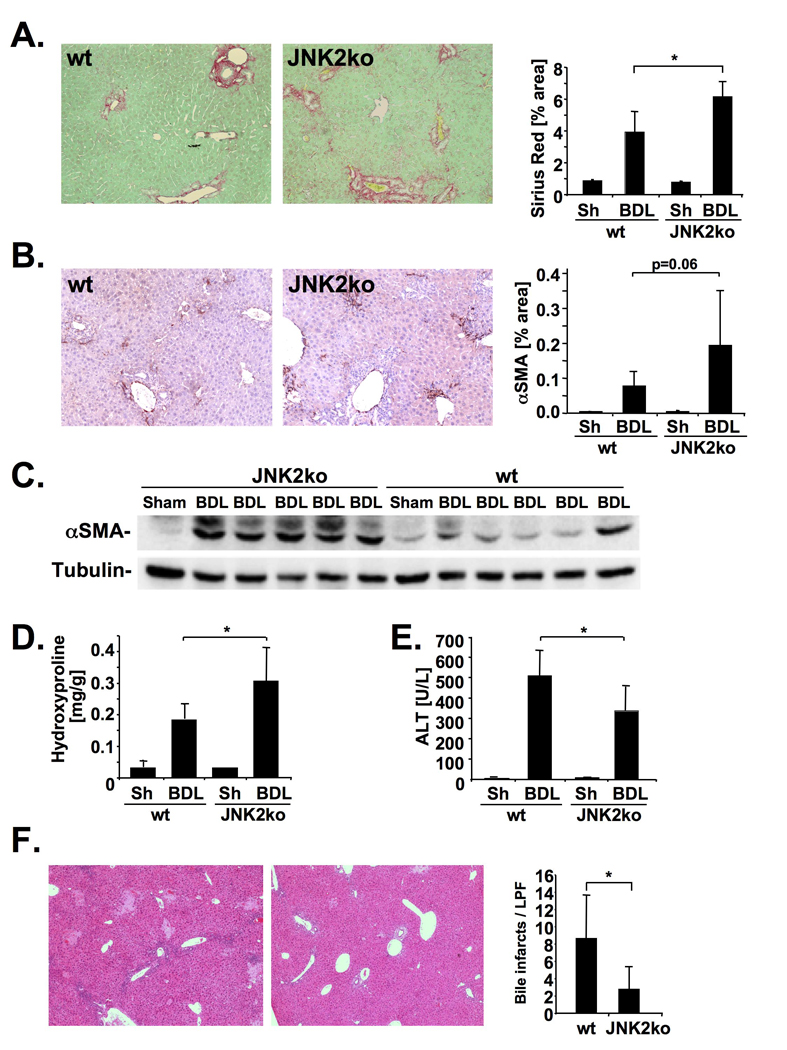
A–D. JNK2-deficient mice (n=9) or C57Bl/6 control mice (n=10) underwent BDL for 21 days. Hepatic fibrosis was evaluated by Sirius Red staining (A), αSMA immunohistochemistry (B), αSMA immunoblotting (C), and hydroxyproline content (D). E–F. Hepatic injury was quantified by ALT levels (E.) and bile infarcts (F.) in JNK2-deficient (n=8) and C57Bl/6 control mice (n=6) five days after BDL. * p<0.05
JNK is activated in human fibrogenesis
Immunohistochemistry of fibrotic livers from patients with chronic hepatitis C and NASH displayed a significant increase in the expression of p-JNK (Suppl. Fig. 9). Although previous studies had described p-JNK in apoptotic hepatocytes in cirrhotic human liver30, we found p-JNK predominantly in fibrotic areas and in areas with high αSMA expression similar to the results observed in mice (Fig. 7A). Notably, confocal immunofluorescent microscopy demonstrated co-localization of p-JNK and αSMA in patients with hepatitis C (n=3) or NASH (n=3) as seen by the orange fluorescence (Fig. 7B). In primary human HSCs, both PDGF and TGFβ strongly increased c-Jun phosphorylation, which was efficiently inhibited by SP600125 and inhibitor VIII (Fig. 7 C,D). Pharmacological inhibition of JNK strongly reduced TGFβ-induced CAGA reporter activity (Fig. 7C), and significantly decreased PDGF-induced [3H]-thymidine uptake (Fig. 7D). Notably, SP600125 and inhibitor VIII had no toxic effects on human HSCs (Supplementary. Fig. 4). To provide additional evidence for the involvement of JNK in human fibrogenesis, p-JNK expression was investigated in patients with chronic hepatitis C who were enrolled in an interventional study to study anti-fibrotic effects of the angiotensin type 1 receptor blocker losartan26,27. We detected a significant reduction of p-JNK expression in patients who responded to losartan with a reduction of fibrosis whereas p-JNK expression remained unchanged in patients who did not respond to losartan (Suppl. Fig. 10).
Figure 7. JNK is activated in human liver fibrosis and promotes TGFβ and PDGF signaling in human hepatic stellate cells.
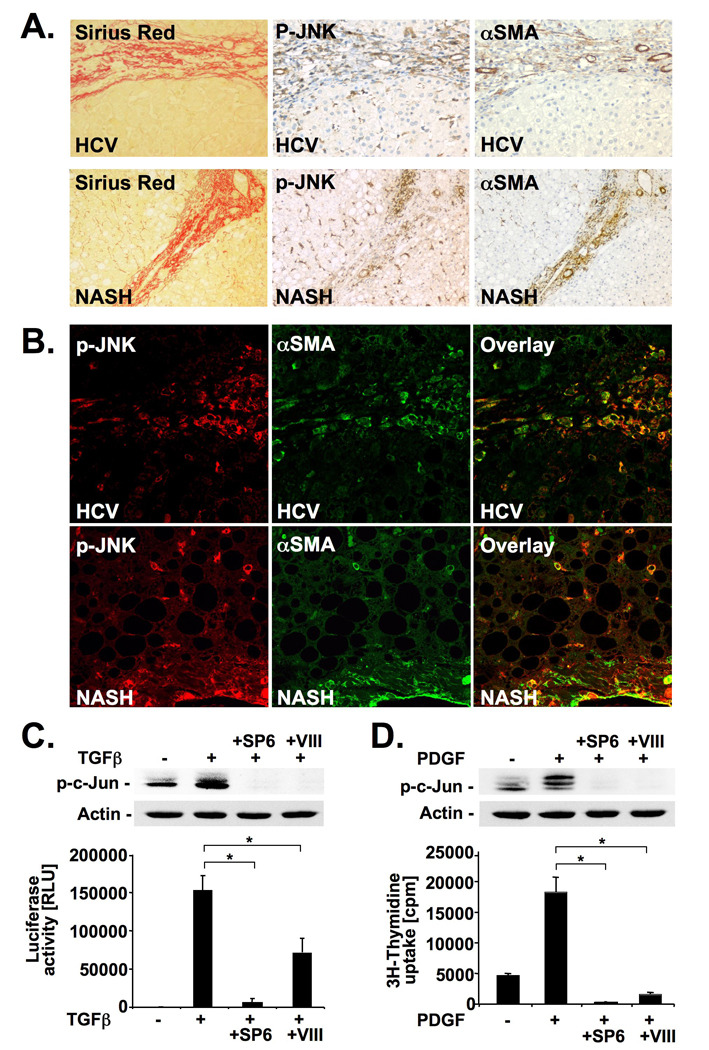
A–B. Serial sections of fibrotic livers from patients with HCV (n=3) or NASH (n=3) were stained for p-JNK, αSMA or by picrosirius red. Single sections were stained for αSMA (green) and p-JNK (red) followed by confocal microscopy. Shown are representative images from one patient each (B). C. Primary human HSCs were pretreated with SP600125 or JNK inhibitor VIII. c-Jun phosphorylation was determined by immunoblot 60 minutes after TGFβ (2 ng/ml) treatment (upper panel). (CAGA)9-driven luciferase was determined 6 hours after TGFβ (10 pg/ml) (lower panel). D. Primary human HSCs were pretreated with JNK inhibitors followed by PDGF (20 ng/ml) treatment. Phosphorylation of c-Jun (upper panel) and [3H]-thymidine incorporation (lower panel) were determined 15 minutes and 26h after PDGF stimulation, respectively. * p<0.05
Discussion
The activation of JNK occurs in response to a wide array of cellular stress including injury and wound healing responses31–33. Moreover, JNK plays an important role in fibroblasts31,34 and HSCs19,20, and has been implicated in the regulation of injury in toxic, metabolic, autoimmune and neoplastic liver disease9,11,13–16. Data from our study demonstrate for the first time a significant decrease in fibrosis by JNK inhibition in models of toxic and biliary liver fibrosis. Moreover, these results were further expanded by genetic studies which revealed a significant reduction of fibrosis in JNK1-deficient mice in two models of fibrogenesis. Interestingly, we did not find any significant effects of pharmacologic JNK inhibition or genetic JNK1 deficiency on hepatocellular injury suggesting that the reduced fibrogenesis was not secondary to reduced injury. These results are in agreement with previous studies in which CCl4-induced liver injury was not reduced by the JNK inhibitor SP60012513,14. Thus, it is likely that JNK inhibition exerts cytoprotective effects in the liver only under specific conditions, e.g. in TNF-mediated acute liver injury. The absence of p-JNK in parenchymal cells in both the BDL and CCl4 models of fibrogenesis further corroborates our finding that JNK inhibition does not protect from hepatocellular injury in these two models. Although our study found a reduction of some inflammatory mediators in fibrotic mice treated with SP600125, these parameters were not consistently reduced in both models of fibrosis, and there was no significant reduction in JNK1-deficient mice. Moreover, we found no activation of JNK in the majority of Kupffer cells, and there was no reduction in macrophage infiltration in SP600125-treated mice suggesting that modulation of inflammation is not the main mechanism by which JNK signaling promotes toxic and biliary fibrosis.
A common finding in both models of liver fibrosis in our study was the strong staining of p-JNK in collagen-producing cells supporting the hypothesis that JNK is directly involved in fibrogenic signaling. This notion is supported by our finding that inhibition of JNK by two different pharmacologic inhibitors not only attenuated TGFβ-induced HSC activation, but also decreased AngII-induced HSC activation as well as PDGF-induced HSC proliferation of HSCs, a crucial fibrogenic cell population of the liver17. These data extend previous studies that demonstrated a role for JNK in TGFβ- and PDGF-mediated HSC migration as well as serum-induced HSC proliferation and α-SMA expression19,20. Thus, inhibition of several key profibrogenic signaling pathways appears to be the key mechanism by which JNK inhibition reduces myofibroblast activation and fibrosis in vivo.
Our studies in JNK-deficient mice not only support a role for JNK in fibrogenesis but also suggest distinct functions for JNK1 and JNK2 in the fibrogenic response with JNK1 acting as the major profibrogenic isoform. Whereas JNK1-deficiency protected from BDL and CCl4-induced fibrosis, JNK2-deficiency promoted BDL-induced fibrosis and had no effect on fibrosis in the CCl4 model. There was a moderate (54%) upregulation of JNK1 mRNA levels in the JNK2ko mice but this was statistically not significant (p=0.197). Thus, it is unlikely that increased levels of JNK1 contribute to enhanced fibrogenesis in JNK2-deficient mice. The different effect of JNK2-deficiency in the two different fibrosis models is likely due to different activation of profibrogenic pathways in these models such as the TGFβ pathway35. Our data are comparable to a recent study in which JNK1-deficiency reduced liver fibrosis in a dietary model of NASH36. However, in this model the profibrotic effects of JNK1 were indirectly mediated by JNK1-dependent promotion of inflammation. Thus, it is possible that JNK1 promotes fibrosis by different mechanisms depending on the underlying liver disease. Interestingly, preliminary data show an association of single nucleotide JNK1 polymorphisms with development of HCV-related liver fibrosis but no association of JNK2 polymorphisms (personal communication from Javier Chaves, Valencia, Spain) further emphasizing the potential role for JNK1 in liver fibrogenesis. Distinct and often opposing functions of JNK1 and JNK2 in the liver have been reported for a number of other hepatic diseases such as MCD- and high-fat diet-induced hepatic steatosis and steatohepatitis15,36,37, TNFα-induced injury11, and DEN-induced hepatocellular carcinoma8. However, the molecular basis for these differences is not well understood, and likely to be based on yet-uncharacterized differences in substrate specificities between JNK1 and JNK2. For example, it has been suggested that JNK1 is the major c-Jun phosphorylating kinase whereas JNK2 regulates c-Jun stability34. In the liver, the Jun family member and JNK target JunD exerts profibrogenic effects38. However, this profibrogenic effect was independent of JNK-mediated phosphorylation38 suggesting that the JNK and JunD promote fibrogenesis through 2 distinct mechanisms. In our study, differences in liver injury could not explain the different fibrosis-promoting effects of JNK1 and JNK2 as liver injury in JNK1-deficient mice was unchanged, and liver injury in JNK2-deficient mice was decreased (which would explain a decrease but not the observed increase in fibrosis). Data from our study display similarities to the role of JNK1 in kidney and lung fibrosis. In the lung, JNK1 mediates bleomcyin- and TGFβ-induced pulmonary fibrosis as well as the expression of TGFβ-regulated genes and genes associated with epithelial-mesenchymal transition (EMT)39,40. Since the role of EMT in the liver remains controversial, and a recent study using an advanced cell tracking system found no evidence for EMT of hepatocytes41, we consider it unlikely that JNK1 predominantly contributes to hepatic fibrosis through this mechanism. In the kidney, JNK1-deficient but not JNK2-deficient mice display a significant reduction of αSMA-expressing cells after unilateral ureter obstruction42. Morever, unilateral ureter obstruction activates JNK1 in an AngII-dependent manner as demonstrated by the ability of the angiotensin type 1 receptor blocker candesartan to diminish JNK1 activation and to decrease renal fibrosis43. Thus, JNK1 appears to be a common mediator of the fibrogenic response, and a key component of the signaling pathway of the profibrogenic TGFβ and AngII pathways in several organs.
Our data in murine fibrogenesis was confirmed in human HSCs and in patients with NASH and HCV: (i) There was a strong upregulation of p-JNK expression in fibrotic HCV and NASH livers. (ii) p-JNK was localized in myofibroblasts not only in chronic hepatitis C but surprisingly also in NASH livers. (iii) JNK was involved in TGFβ and PDGF signaling in human HSCs. (iv) We detected a significant decrease in p-JNK expression in livers from patients who had responded to the angiotensin receptor 1 blocker losartan with reduced fibrosis but found no decrease in non-responders. The latter results suggest a relationship between the degree of fibrosis and the amount of p-JNK in the liver. Considering that JNK activity was required for AngII-mediated HSC activation, one could also speculate that AngII represents an important mediator of JNK activation in the fibrotic liver. In view of the unchanged p-JNK expression in non-responders, it is most likely that the decreased expression of p-JNK in responders is a consequence of decreased fibrosis. Together with our results in primary HSCs and murine models of liver fibrosis, our human data strongly suggest a role for JNK in the fibrogenic process and render JNK a potential target for anti-fibrotic treatment. Despite our functional data in human HSCs and the co-localization of p-JNK and α-SMA in human fibrosis, we cannot exclude that JNK contributes to fibrosis through additional mechanisms in patients with chronic liver disease: (i) Human and experimental murine fibrosis display considerable differences: Mice undergo massive injury with a large percentage of necrosis and rapidly progress to fibrosis in the BDL and CCl4 models whereas most patients display only minor injury with predominantly apoptotic cell death, and slow disease progression over decades. (ii) Although p-JNK was expressed in fibrotic areas and co-localized with α-SMA in chronic HCV or NASH, there was also some p-JNK staining in other areas including the inflammatory infiltrate, especially in patients with HCV. Thus we cannot exclude that JNK inhibition in patients might exert anti-fibrotic effects through additional mechanisms, e.g. reduced inflammation.
In view of our data in JNK1- and JNK2-deficient mice, a JNK1-specific inhibitor would be most suitable for anti-fibrotic therapies. However, our data clearly show efficacy of a pan-JNK inhibitor in murine fibrogenesis and no role for JNK2 in toxic fibrogenesis, suggesting that even pan-JNK inhibition might represent a potential anti-fibrotic approach. A decrease in liver regeneration might constitute one side effect of JNK inhibition as JNK has been shown to be involved in hepatocellular proliferation and hepatic regeneration after partial hepatectomy7,8. However, under most clinical circumstances, such massive loss of liver mass does not occur and one would not expect defective regeneration to represent a significant clinical challenge in patients treated with JNK inhibitors. On the contrary, blocking proliferative effects of JNK may be beneficial as JNK1 promotes hepatocarcinogenesis in mice, and is activated in a high percentage of human HCCs8,16. In view of the strong association of HCC with advanced liver fibrosis, and preliminary data showing JNK activation in fibroblasts surrounding HCC lesions (RB and RFS, unpublished observation), JNK activation in HSCs may contribute to the microenvironment that promotes tumor formation in the liver. Thus, future investigations need to establish whether JNK inhibition may represent a strategy for the prevention of both hepatic fibrosis and hepatocellular carcinoma, especially in patients with high risk for disease progression.
Supplementary Material
Acknowledgements
We would like to thank Dr. David Brenner (La Jolla, CA) for providing αSMA-RFP and Coll-GFP mice, Dr. Javier Chaves (Valencia, Spain) for information on JNK1 SNPs, and Christina Millan (Barcelona, Spain) for excellent technical support.
Grant support: This study was supported by grants U54CA126513 (PI of Project 2: RFS), DK076920 (to RFS) and the 2008 Charles Trey, MD Memorial Postdoctoral Research Fellowship from the American Liver Foundation (to JK).
Abbreviations
- α-SMA
α smooth muscle actin
- AngII
angiotensin II
- BDL
bile duct ligation
- GFP
green fluorescent protein
- JNK
c-Jun N-terminal kinase
- HSC
hepatic stellate cell
- NASH
non-alcoholic steatohepatitis
- p-JNK
phospho-JNK
- RFP
red fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions: Johannes Kluwe was involved in acquisition, analysis and interpretation of data, statistical analysis, experimental design and drafting of the manuscript. Jean-Philippe Pradere, Geum-Youn Gwak, Ali Mencin, Samuele De Minicis, Christoph H. Osterreicher and Jordi Colmenero were involved in acquisition, analysis and interpretation of data and statistical analysis. Ramon Bataller was involved in experimental design and analysis, and interpretation of data. Robert F. Schwabe was responsible for study concept and design, drafting of the manuscript, obtained funding and supervised the study.
Disclosures: No conflicting interests exist.
References
- 1.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Minden A, Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim Biophys Acta. 1997;1333:F85–F104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- 4.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 6.Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwabe RF, Bradham CA, Uehara T, Hatano E, Bennett BL, Schoonhoven R, Brenner DA. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology. 2003;37:824–832. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- 8.Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest. 2008;118:3943–3953. doi: 10.1172/JCI37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 10.Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for c-Jun NH2-terminal kinase in TNFalpha- and Fas-mediated apoptosis in hepatocytes. FASEB J. 2004;18:720–722. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uehara T, Bennett B, Sakata ST, Satoh Y, Bilter GK, Westwick JK, Brenner DA. JNK mediates hepatic ischemia reperfusion injury. J Hepatol. 2005;42:850–859. doi: 10.1016/j.jhep.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 13.Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–178. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 14.Henderson NC, Pollock KJ, Frew J, Mackinnon AC, Flavell RA, Davis RJ, Sethi T, Simpson KJ. Critical role of c-jun (NH2) terminal kinase in paracetamol- induced acute liver failure. Gut. 2007;56:982–990. doi: 10.1136/gut.2006.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential Effects of JNK1 and JNK2 inhibition on Murine Steatohepatitis and Insulin Resistance. Hepatology. 2009;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci U S A. 2006;103:10544–10551. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters JJ, Brenner DA. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida K, Matsuzaki K, Mori S, Tahashi Y, Yamagata H, Furukawa F, Seki T, Nishizawa M, Fujisawa J, Okazaki K. Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am J Pathol. 2005;166:1029–1039. doi: 10.1016/s0002-9440(10)62324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schnabl B, Bradham CA, Bennett BL, Manning AM, Stefanovic B, Brenner DA. TAK1/JNK and p38 have opposite effects on rat hepatic stellate cells. Hepatology. 2001;34:953–963. doi: 10.1053/jhep.2001.28790. [DOI] [PubMed] [Google Scholar]
- 21.Oakley F, Meso M, Iredale JP, Green K, Marek CJ, Zhou X, May MJ, Millward-Sadler H, Wright MC, Mann DA. Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology. 2005;128:108–120. doi: 10.1053/j.gastro.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Dietrich N, Thastrup J, Holmberg C, Gyrd-Hansen M, Fehrenbacher N, Lademann U, Lerdrup M, Herdegen T, Jaattela M, Kallunki T. JNK2 mediates TNF-induced cell death in mouse embryonic fibroblasts via regulation of both caspase and cathepsin protease pathways. Cell Death Differ. 2004;11:301–313. doi: 10.1038/sj.cdd.4401353. [DOI] [PubMed] [Google Scholar]
- 23.Magness ST, Bataller R, Yang L, Brenner DA. A dual reporter gene transgenic mouse demonstrates heterogeneity in hepatic fibrogenic cell populations. Hepatology. 2004;40:1151–1159. doi: 10.1002/hep.20427. [DOI] [PubMed] [Google Scholar]
- 24.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 25.Schwabe RF, Bataller R, Brenner DA. Human hepatic stellate cells express CCR5 and RANTES to induce proliferation and migration. Am J Physiol Gastrointest Liver Physiol. 2003;285:G949–G958. doi: 10.1152/ajpgi.00215.2003. [DOI] [PubMed] [Google Scholar]
- 26.Colmenero J, Bataller R, Sancho-Bru P, Dominguez M, Moreno M, Forns X, Bruguera M, Arroyo V, Brenner DA, Gines P. Effects of Losartan on Hepatic Expression of Non-phagocytic NADPH Oxidase and Fibrogenic Genes in Patients with Chronic Hepatitis C. Am J Physiol Gastrointest Liver Physiol. 2009 doi: 10.1152/ajpgi.00162.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oakley F, Teoh V, Ching-A-Sue G, Bataller R, Colmenero J, Jonsson JR, Eliopoulos AG, Watson MR, Manas D, Mann DA. Angiotensin II stimulation of RelA Serine 536 phosphorylation (P-Ser536-RelA) promotes myofibroblast survival and liver fibrosis. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.02.081. (Tentatively accepted) [DOI] [PubMed] [Google Scholar]
- 28.Schwabe RF, Sakurai H. IKKbeta phosphorylates p65 at S468 in transactivaton domain 2. FASEB J. 2005;19:1758–1760. doi: 10.1096/fj.05-3736fje. [DOI] [PubMed] [Google Scholar]
- 29.Svegliati-Baroni G, Ridolfi F, Caradonna Z, Alvaro D, Marzioni M, Saccomanno S, Candelaresi C, Trozzi L, Macarri G, Benedetti A, Folli F. Regulation of ERK/JNK/p70S6K in two rat models of liver injury and fibrosis. J Hepatol. 2003;39:528–537. doi: 10.1016/s0168-8278(03)00291-5. [DOI] [PubMed] [Google Scholar]
- 30.Toivola DM, Ku NO, Resurreccion EZ, Nelson DR, Wright TL, Omary MB. Keratin 8 and 18 hyperphosphorylation is a marker of progression of human liver disease. Hepatology. 2004;40:459–466. doi: 10.1002/hep.20277. [DOI] [PubMed] [Google Scholar]
- 31.Javelaud D, Laboureau J, Gabison E, Verrecchia F, Mauviel A. Disruption of basal JNK activity differentially affects key fibroblast functions important for wound healing. J Biol Chem. 2003;278:24624–24628. doi: 10.1074/jbc.M301942200. [DOI] [PubMed] [Google Scholar]
- 32.Bosch M, Serras F, Martin-Blanco E, Baguna J. JNK signaling pathway required for wound healing in regenerating Drosophila wing imaginal discs. Dev Biol. 2005;280:73–86. doi: 10.1016/j.ydbio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 33.Mattila J, Omelyanchuk L, Kyttala S, Turunen H, Nokkala S. Role of Jun N-terminal Kinase (JNK) signaling in the wound healing and regeneration of a Drosophila melanogaster wing imaginal disc. Int J Dev Biol. 2005;49:391–399. doi: 10.1387/ijdb.052006jm. [DOI] [PubMed] [Google Scholar]
- 34.Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol Cell. 2004;15:713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 35.Khimji AK, Shao R, Rockey DC. Divergent transforming growth factor-beta signaling in hepatic stellate cells after liver injury: functional effects on ECE-1 regulation. Am J Pathol. 2008;173:716–727. doi: 10.2353/ajpath.2008.071121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, Osterreicher CH, Schnabl B, Seki E, Brenner DA. JNK1 from Hematopoietic Cells Mediates Progression from Hepatic Steatosis to Steatohepatitis and Fibrosis in Mice. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 38.Smart DE, Green K, Oakley F, Weitzman JB, Yaniv M, Reynolds G, Mann J, Millward-Sadler H, Mann DA. JunD is a profibrogenic transcription factor regulated by Jun N-terminal kinase-independent phosphorylation. Hepatology. 2006;44:1432–1440. doi: 10.1002/hep.21436. [DOI] [PubMed] [Google Scholar]
- 39.Alcorn JF, van der Velden J, Brown AL, McElhinney B, Irvin CG, Janssen-Heininger YM. c-Jun N-terminal Kinase 1 is Required for the Development of Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2008-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alcorn JF, Guala AS, van der Velden J, McElhinney B, Irvin CG, Davis RJ, Janssen-Heininger YM. Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by TGF-beta1. J Cell Sci. 2008;121:1036–1045. doi: 10.1242/jcs.019455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taura K, Miura K, Kodama Y, Oesterreicher CH, Oesterreicher MP, Brenner DA. Hepatocytes do not undergo epithelial mesenchymal transition during CCl4-induced liver fibrosis in mice. Hepatology. 2008;48:381A. doi: 10.1002/hep.23368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma FY, Flanc RS, Tesch GH, Han Y, Atkins RC, Bennett BL, Friedman GC, Fan JH, Nikolic-Paterson DJ. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J Am Soc Nephrol. 2007;18:472–484. doi: 10.1681/ASN.2006060604. [DOI] [PubMed] [Google Scholar]
- 43.Wamsley-Davis A, Padda R, Truong LD, Tsao CC, Zhang P, Sheikh-Hamad D. AT1A-mediated activation of kidney JNK1 and SMAD2 in obstructive uropathy: preservation of kidney tissue mass using candesartan. Am J Physiol Renal Physiol. 2004;287:F474–F480. doi: 10.1152/ajprenal.00452.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.