

Abstract
Molecular inversion probe (MIP) technology has been demonstrated to be a robust platform for large-scale dual genotyping and copy number analysis. Applications in human genomic and genetic studies include the possibility of running dual germline genotyping and combined copy number variation ascertainment. MIPs analyze large numbers of specific genetic target sequences in parallel, relying on interrogation of a barcode tag, rather than direct hybridization of genomic DNA to an array. The MIP approach does not replace, but is complementary to many of the copy number technologies being performed today. Some specific advantages of MIP technology include: Less DNA required (37 ng vs. 250 ng), DNA quality less important, more dynamic range (amplifications detected up to copy number 60), allele specific information “cleaner” (less SNP crosstalk/contamination), and quality of markers better (fewer individual MIPs versus SNPs needed to identify copy number changes). MIPs can be considered a candidate gene (targeted whole genome) approach and can find specific areas of interest that otherwise may be missed with other methods.
Keywords: Molecular inversion probe, genotyping, gene copy number, microarrays, single nucleotide polymorphisms, alleles
1. Introduction
A molecular inversion probe (MIP) is a single oligonucleotide that recognizes and hybridizes to a specific genomic target sequence with two inverted recognition complementary flanks ranging from 20 to 30 nucleotides (1). The total length of the MIP is 120 nucleotides. After the probe hybridizes to the target DNA, a single base pair gap exists in the middle of the two recognition sequences (Figure 1). The gap can represent either a single nucleotide polymorphism (SNP) or a non-polymorphic nucleotide. With the addition of the specific nucleotide to fill the gap and subsequent ligation after the probe specifically anneals to its complementary genomic sequence, the probe undergoes an intramolecular rearrangement (i.e. circularization) that allows the amplification of a barcode sequence unique to each oligonucleotide. The barcode is queried via a microarray and intensity of a specific barcode reflects a specific SNP and is also quantitative in terms of copy number (2–4).
Figure 1.
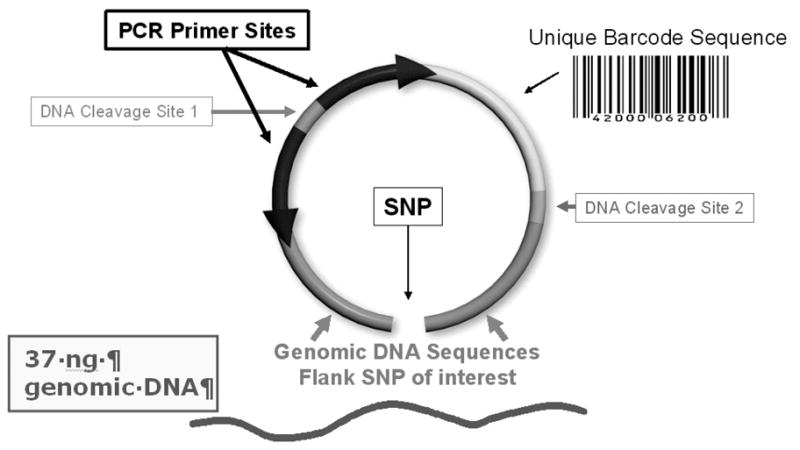
Molecular Inversion Probe (MIP). Each MIP is 120 bp oligonucleotide, with a unique gap fill for SNP of interest. Each probe contains unique tag (barcode) sequence corresponding to interrogated SNP.
The flow process of the reaction is as follow:
In a single sample volume, the MIP probes are mixed with genomic DNA.
Subsequently annealing is carried out. A “gap-fill” ligation reaction is then carried out with a dNTP in the tube.
The samples are then amplified using a single set of common primers and the fragment content of each reaction is hybridized to a standard oligonucleotide chip array.
After the hybridization on the DNA chip, the components are decoded. The relative base incorporation is measured by the fluorescence signal intensity at the corresponding complementary bar code tag site on the DNA array.
The advantages of MIPs technology are derived from the intramolecular nature of the assay (1–4). A single molecule (probe) hybridizes to genomic DNA with two recognition sequences. Even though there are two hybridization events, the second is extremely rapid since the effective local concentration of the second site is in the micromolar range. For this reason, much less probe is required as compared to other molecule assays. We typically use between 5 and 20 attomoles (10-8 moles) of each probe in a 1,000 multiplex reaction for 5 to 20 fentomoles (10–15 moles) of total probe. The benefits of this are that more probes can be added into a single reaction (higher multiplexing) and lower concentration of each probe reduces the likelihood of two probes interacting with each other (lower backgrounds). In the initial report, this technology was used for standard SNP genotyping at high levels of multiplexing (1). We found that this approach was highly quantitative and highly accurate through multiple hybridization and enzymatic processing events (2–4).
There are a number of technologies that assess copy number on the whole genome scale such as array comparative genomic hybridization (CGH). Formats include using bacterial artificial chromosome (BAC) CGH, spotted cDNAs and more recently several types of oligonucleotides arrays including ones which rely on SNP sequences. Some of the newer CGH methodologies allow for allelic information to be obtained. The utility of measurement of allele copy number include the identification of loss of heterozygosity (LOH) events and the allelic genotype at amplified loci. Likewise, molecular inversion probe (MIP) technology has been demonstrated to be a robust platform for human genomics applications such as germline genotyping and copy number variation (CNV) in cancer (2). This system has a number of strengths including minimal DNA requirements, more flexible sequence design constraints because of its reliance on a barcode intermediate as opposed to direct array hybridization, and ability to use a ubiquitous barcode array that is inexpensive (<$90 per array). Because a barcode intermediate is used instead of direct genomic DNA hybridization to an array, one can query any unique, nonrepetitive sequence for simultaneous genotyping and copy number changes (e.g. allelic quantitation) without having to optimize for specific hybridization parameters of array comparative genomic hybridization (CGH) or oligonucleotide arrays. Expanding the number of loci is simply a matter of producing more probes. Gene copy determination is sequence specific, meaning that the resolution of determining gene copy number alterations is oftentimes at an individual sequence level for high performing probes. This enables extremely high resolution delineation of copy number changes which are specific to a given gene. As already mentioned, less DNA is required (37 ng), and the source of the DNA and its quality is less important to accurate allelic quantitation with sources such as paraffin embedded tissues (3,4). The current number of loci which can be analyzed ranges in the tens of thousands but there are efforts to expand probe sets into the hundreds of thousands.
2. Materials
2.1 Reagents and arrays
Molecular inversion probe pool (Affymetrix, Santa Clara, CA)
ExoMix - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
10x Buffer A - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
Enzymer A - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
GapFill Mix - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
AmpMix - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
2nd Stage PCR Mix - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
Cleavage Enzyme - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
Cleavage Tube - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
HY Digest Mix - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
Stain Cocktail - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
Hyb Cocktail - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
dNTPs - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
Arrays, Universal Tag 70K - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
Wash A - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
Wash B - Affymetrix GeneChip® Custom SNP Kit (Affymetrix, Santa Clara, CA)
PicoGreen dsDNA Quantitation Reagent (Molecular Probes, Inc., Eugene, OR)
TE (pH 8.0) buffer
Stratagene Taq DNA Polymerase (Stratagene, San Diego, CA)
TITANIUM™ Taq Polymerase (Clontech, Mountain View CA)
3% Precast Gel, Ready Agarose 96 Plus, TBE (BioRad, Hercules CA)
1kb Molecular Ruler (BioRad, Hercules CA)
BSA (10mg/ml)
10x TBE Buffer (0.89M Tris base, 0.02M EDTA, 0.89M boric acid)
Streptavidin Phycoerythrin (SAPE) (1mg/ml)
2.2 Disposables
200 μl PCR tubes in strips of 12
BD Falcon microplate
Filtered pipette tips
Kim Wipes
Disposable Gloves
50 mL Reagent Reservoir
1.5 mL Eppendorf tube
96 well plates
384 well plates
Thermo-Fast® 96 Rigid Skirted PCR Plate, opaque black (Thermo Fisher Scientific, Rockford, IL)
Clear Adhesive Films (Applied Biosystems, Foster City CA)
2.3 Large Equipment
Perkins Elmer Victor2 1420 Multilabel Counter (Perkin Elmer, Waltham MA)
Two Refrigerators, 4°C refrigerator; 6 cu ft
Two Freezers, −20°C; deep freeze; manual defrost; 17 cu ft 2
Thermal cycler e.g. GeneAmp® PCR System 9700 Thermal Cycler (Applied Biosciences, Foster City, CA)
Two table top centrifuges with swinging bucket rotor holding 96 well plates (e.g. Sorvall Legend RT tabletop centrifuge - Thermo Fisher Scientific Inc., Waltham MA)
Eppendorf® Multipurpose Centrifuge 5804 2 VWR Intl. 53513-800 1 Pre-Amp Lab
GeneChip® Hybridization Oven 640 with carriers (Affymetrix, Santa Clara, CA)
GeneChip® Fluidics Station 450 (Affymetrix, Santa Clara, CA)
Affymetrix GeneChip® Scanner (Affymetrix, Santa Clara, CA)
Standard UV Gel Imager
2.4 Small Equipment
Two sets single channel Pipetters (e.g. Ranin P-2, P-10, P-20, P-200, P-1000 - Ranin, Oakland, CA)
Two sets 12-channel Pipetters (Ranin P-10, P-20, P-200 - Ranin, Oakland, CA)
Two sets 24-channel Pipetters (Ranin P-10, P-20, P-200 - Ranin, Oakland, CA)
Eppendorf tube rack
Two Galaxy Mini Centrifuges or similar model (interchangeable for microtubes and strip tubes)
Two Vortexers
Gel Box, wide Mini-Sub Cell GT cell (BioRad, Hercules CA)
Portable pipette aid
2.5 Computers
7G Instrument Control Workstation: The Instrument Control Workstation is installed in the Post-Amp Lab. It controls the scanner and the fluidics station. This workstation will use Microsoft® Windows XP operating system with Service Pack 1 or 2. This workstation should be operating the Affymetrix GeneChip® Operating Software (GCOS). It should be appropriately networked as per the installation instructions.
-
MIP Post-Amp Lab Workstation: The MIP Post-Amp Lab Workstation includes Microsoft Windows XP operating system with Service Pack 2. It should be appropriately networked as per the installation instructions.
Microsoft® SQL Server 2000, personal edition
Java 2 Platform, Standard Edition, release 1.4.2
Apache Tomcat Web Server 4.9.1
GeneChip® Targeted Genotyping Analysis Software (GTGS) (Java-based)
Microsoft Office Professional
GCOS Runtime Libraries
3. Methods
This method describes the analysis of 48 samples. On previous experience, we have found that data quality is better using larger sample batches (also, see Note 1). Therefore, we typically accrue enough samples to run a minimum of 48 per any given run. Each sample will be tagged with two dinucleotide mixes, dATP/dTTP and dCTP/dGTP, and ultimately each sample will be analyzed by two microarrays totally 96 microarrays. The two microarrays per sample will correspond to the two dinucleotide mixes each samples will be tagged with.
We recommend running 5 to 10 normal diploid genome samples simultaneously that were processed for genomic DNA in a similar fashion to tumor samples. This is necessary for downstream data processing.
Each work area should be separated by a physical boundary such as a wall – it is ideal to have two rooms. The two work areas are named the pre-amplification lab (pre-amp) and post-amplification lab (post-amp). Work flow should always proceed forward from pre-amp to post-amp (see Note 2).
3.1 Measuring the Concentration of Genomic DNA. (pre-amp)
Prepare a working stock of 1x TE Buffer (pH 8.0) by diluting a concentrated stock using sterile DNAse-free water.
Prepare lambda DNA standards by making a dilution series of the 100 ng/μl stock solution with the following concentrations: 0 ng/μl, 2 ng/μl, 8 ng/μl, 14 ng/μl, and 20 ng/μl. Dilute the stock using 1x TE (pH 8.0) buffer and bring the final volume to 1 ml (see Table 1).
Add 10 μl of each lambda DNA standard to a well of a Thermo-Fast® 96 Rigid Skirted PCR Plate. Once the standard DNA solutions have aliquoted, store at 4°C for future use.
To each well containing 10 μl of lambda DNA standard, add 90 μl 1x TE (pH 8.0). Record the location of each sample. Set the plate aside on the bench.
Thaw all DNA samples which have an unknown concentration. Vortex all samples repeatedly to ensure the samples are homogeneous throughout. Pulse centrifuge to remove excess liquid from the side of the tube.
Dilute the DNA samples by adding 2μl DNA to 198μl 1x TE (pH 8.0) in a fresh 1.5 ml eppendorf tube. Vortex to mix.
Add 10μl of this dilute DNA and 90μl 1x TE (pH 8.0) to an assigned well of the same 96 well plate which the lambda DNA standards were previously aliquoted onto. The total dilution of the DNA samples is now 1,000-fold from the original stock. Complete this for each sample.
In a 1.5 ml Eppendorf tube prepare a working stock of PicoGreen reagent by diluting the PicoGreen 200-fold in 1x TE (pH 8.0). For example, pipette 5μl PicoGreen in 995μl TE which is enough for exactly 10 samples (see Note 3).
Add 100μl dilute PicoGreen reagent to the wells containing DNA and lambda DNA standards - the dilution of the sample DNA is now 2,000-fold. Use a 12-channel pipette when processing large numbers of samples to avoid time lapse between incubations.
Allow the Picogreen reaction to incubate for 2–5 minutes at room temperature in the dark.
Measure fluorescence using a fluorescent scanner with a microplate reader such as the Perkin Elmer fluorimeter.
Plot the standard curve intensity data against the known DNA concentration of the standards and derive a linear regression. Use this equation to determine the concentration of each individual sample.
Table 1.
Concentration of lambda DNA standards in both the first dilution series and in the final concentration in the assay.
| Volume (μl) of 100μg/ml DNA Stock | Volume (μl) 1x TE Buffer | Final DNA Concentration | DNA Concentration in Assay |
|---|---|---|---|
| 200 | 800 | 20μg/ml | 1μg/ml |
| 140 | 986 | 14μg/ml | 0.7μg/ml |
| 80 | 982 | 8μg/ml | 0.4μg/ml |
| 20 | 998 | 2μg/ml | 0.1μg/ml |
| 0 | 1000 | Blank | Blank |
3.2 Molecular Inversion Probe Annealing – preparation of the DNA samples and 384-well annealing plate. (pre-amp)
Based on the quantitation of the DNA from the Picogreen assay, normalize each genomic DNA sample to 8 ng/μl. Pipette 10–20 μl of each sample into rows A, B, C and D of a 96 well plate and record the well location for each sample. Label this plate Sample Plate and place the plate on ice (see Note 4). All remaining steps will refer to this plate as the Sample Plate.
Label 384-well plate with the title Anneal Plate. This 384-well plate will be referenced as the Anneal Plate in all subsequent steps. Place the plate on ice.
In a 1.5 mL eppendorff tube, prepare the Probe Master Mix. This master mix contains 10x Buffer A, Probe, and Enzyme A. To the 1.5 mL eppendorff tube add 45 μl 10x Buffer A, 66.6 μl Probe and 2.7 μl Enzyme A (see Note 5). The master mix is designed to create enough volume for 48 samples plus an additional 10% leaving room for pipette error.
Aliquot out 9.2 μl of the Probe Master Mix described above (10x Buffer A + Probe + Enzyme A) into every other well in row A up to well A23 on the Anneal Plate. More explicitly wells A1, A3, A5, A7, A9, A11, A13, A15, A17, A19, A21 and A23 on the Anneal Plate should contain Probe Master Mix (see Note 6).
Using a 12-channel pipette dispense 1.9 μl/well of the Probe Master Mix into rows B, C, D and E of the Anneal Plate. The 12-channel pipette will dispense the Probe Master Mix into every other column on the Anneal Plate similar to the way the Probe Master Mix was dispensed into row A. The 1.9 μl will be taken from the 9.2 μl which was aliquoted into the 12 wells in row A (step 4).
From the 96-well Sample Plate, using a 12-channel pipette, add 4.7 ul of the genomic DNA into the wells containing the Probe Master Mix, mix up and down 10 times to mix while avoiding bubbles. More explicitly; 4.7 μl of sample from row A, row B, row C and row D on the Sample Plate should be transferred to row B, row C, row D, and row E on the Anneal Plate respectively. Because the DNA was transferred using a 12-channel pipette, the sample will be distributed in every other well just as the Probe Master Mix was dispensed. Ensure that the well location of each DNA sample is recorded.
Tightly seal the Anneal Plate with a clear adhesive film.
Spin the Anneal Plate in a balanced table top centrifuge at 1,000 rpm (110 rcf) for 15 sec. All subsequent spins should be done in this table top centrifuge.
Transfer the Anneal Plate to the thermal cycler and start the Meg Anneal program (see Figure 2a) (see Note 7).
One minute into the 58°C incubation pause the thermal cycling program, take the Anneal Plate out and place it on ice for 2 min.
Spin the Anneal Plate at 1,000 rpm for 15 seconds (see Note 8). Put the Anneal Plate back onto the thermal cycler and resume the thermal cycling program, 58°C anneal overnight.
Annealing time is 16 hours (+/− 1hour), e.g. start annealing at five pm on day one. The next stage can start at 9am on day two (see Note 9).
Figure 2.


Thermal cycling programs. Each program should be programmed into the appropriate thermal cycler, pre-amplification lab or post-amplification lab, prior to commencement of the molecular inversion probe assay.
Figure 2a. Meg Anneal thermal cycling program to be programmed in the pre-amplification lab.
Figure 2b. Meg 3-5-7 K thermal cycling program to be programmed in the pre-amplification lab.
Figure 2c. Hy-Titanium-10cycles thermal cycling program to be programmed in the post-amplification lab.
Figure 2d. Meg Hydigest- a thermal cycling program to be programmed in the post-amplification lab
3.3 Molecular inversion probe 1st Stage PCR – Creating the Assay Plate, aliquoting Gap Fill and dNTPs. (pre-amp)
On day two, take a new 96-well plate and label it Assay Plate. Place the plate on ice. The remaining steps will refer to this 96-well plate as the Assay Plate.
Into 12-strip PCR tubes, pipette 11 μl of the gap fill mix into all 12 wells of the strip. The gap fill mix is a pre-made reagent. Label the strip tube Gap Fill and set the 12-strip PCR tubes aside on ice.
Prepare 1x Buffer A by adding 100μl of the 10x Buffer A solution into 900 μl of H20. The 10x Buffer A solution is a pre-made reagent.
Into 12-strip PCR tubes, pipette 80 μl of 1x Buffer A into all 12 wells of the strip. Label the strip tube as 1x Buffer A and set aside on ice.
Remove the Anneal Plate from the thermal cycler where it has been held at 58°C overnight and place the plate on ice for two minutes. Spin the plate at 1,000 rpm for 15 seconds.
Using a 12-channel pipette and the PCR strip tubes containing the 11 μl of gap fill mix previously aliquoted as your stock, add 1.25 μl of the gap fill mix to each well containing sample on the Anneal Plate. The wells containing sample are every other column in rows B, C, D and E up to column number 23. Mix well by pipetting up and down three times avoiding bubbles.
Using a 12-channel pipette and the PCR strip tubes containing the 80 μl of 1x Buffer A previously aliquoted as your stock, add 12 μl of the 1x Buffer A to each well containing sample on the Anneal Plate. These are the same wells the gap fill mix was just added to. Mix well by pipetting up and down twenty times avoiding bubbles.
All samples from the Anneal Plate will now be transferred to the Assay Plate. Using a 12-channel pipette transfer 9 μl from row B of the 384-well annealing plate to rows A and B of the Assay Plate. Repeat this step transferring this time 9 μl from row C of the Anneal Plate to rows C and D of the Assay Plate. Now transfer 9 μl from row D of the Anneal Plate to rows E and F of the Assay Plate. Finally transfer 9 μl from row E of the Anneal Plate to rows G and H of the Assay Plate. Seal the wells of the Assay Plate with a clear adhesive film. Spin the Assay Plate at 1,000 rpm for 15 seconds. At this point there should be very little or nothing left in the Anneal Plate.
Start the thermal cycler program Meg 3-5-7 K (see Figure 2b). Once the thermal cycler has reached 58°C, load the Assay Plate and close the lid. The reagents which will be prepared in the following steps will be added to the Assay Plate while it remains on the thermal cycler. Be aware of the thermal cycler and which stage it is at, as dNTPs addition will occur when the timer reads 1 minute remaining in the second 58°C period.
The nucleotides (dNTPs) come in a 96-well plate. Spin the dNTP plate at 1,000 rmp for 15 seconds. Place the plate on ice.
Carefully remove the adhesive film covering the wells. The rows on the dNTP plate will be clearly labeled indicating which nucleotide is in each row.
The dNTP plate contains approximately 20 μl of nucleotide per well. Using a multichannel pipette prepare a dinucleotide mix by combining 20 μl dATP with 20 μl dTTP to a labeled 12-strip PCR tubes. Repeat this creating a dinucleotide mix of dCTP with dGTP and combining 20 μl of each of these two nucleotides to a new labeled 12-strip PCR tubes. Each 12-strip PCR tubes, dATP/dTTP and dCTP/dGTP, will now contain approximately 40 μl per well.
When the thermal cycler timer reads 1 minute remaining for the second 58°C period, pause the thermal cycler, remove the Assay Plate and place it on ice for 1 minute.
Carefully remove the clear adhesive film from the Assay Plate.
Using a 12-channel pipette, add 4μl of the dATP/dTTP dinucleotide mix to rows A, C, E and G of the Assay Plate – this is every other row. Mix well by pipetting up and down 20 times avoiding bubbles.
Using a 12-channel pipette, add 4μl of the dCTP/dGTP dinucleotide mix to rows B, D, F and H of the Assay Plate – again this is every other row. Mix well by pipetting up and down 20 times avoiding bubbles.
Seal the Assay Plate with a clear adhesive film. Vortex the plate to mix. Spin the plate at 1,000 rpm for 15 seconds.
Return the Assay Plate to the thermal cycler, close the lid and resume the Meg 3-5-7 K program. Be conscious of the thermal cycling program. New reagents will be added to the plate with 14 minutes remaining in the first 37°C period. These reagents will be prepared in Molecular inversion probe 1st Stage PCR – aliquoting ExoMix, AmpMix and Cleavage Reagent Mix.
3.4 Molecular inversion probe 1st Stage PCR – aliquoting ExoMix, AmpMix and Cleavage Reagent Mix. (pre-amp)
Prepare two 12-strip PCR tubes. Pipette 45 μl of the exonuclease mix into each tube of one of the 12-strip PCR tubes. The exonuclease mix is a pre-made reagent. Using a 12-channel pipette transfer 22.5 μl of the exonuclease mix from the first 12-strip PCR tubes into the second 12-strip PCR tubes. Label the two 12-strip PCR tubes Exo Mix and set aside on ice.
When the thermal cycler reads 14:00 minutes remaining in the first 37°C period, pause the Meg 3-5-7 K program.
Remove the Assay Plate from the thermal cycler and place on ice for 2 minutes.
Spin the Assay Plate at 1,000 rpm for 15 seconds and return the plate to ice.
Carefully remove the clear adhesive film from the Assay Plate.
Using a 24-channel pipette, transfer 4 μl of Exo Mix from the two sets of 12-strip PCR tubes into each well on the Assay Plate, mixing 20 times with each addition. Avoid bubbles.
Reseal the Assay Plate with clear adhesive film and spin the plate at 1,000 rpm for 15 seconds.
Place the Assay Plate back on the thermal cycler, close the lid and resume the Meg 3-5-7 K thermal cycling program.
Prepare the Cleavage Reagent by adding 24 μl of the pre-made cleavage enzyme to each 3 mL cleavage tube, also pre-made. This mixture of cleavage reagent and cleavage enzyme will now be referred to as the Cleavage Reagent Mix. Place the mix on ice.
Prepare two 12-strip PCR tubes. Pipette 240 μl of the Cleavage Reagent Mix into each tube of one of the two 12-strip PCR tubes. Using a 12-channel pipette transfer 120 μl of the Cleavage Reagent Mix from the first 12-strip PCR tubes into the second 12-strip PCR tubes. Label the 12-strip PCR tubes Cleavage Mix and place on ice.
Prepare the Amp Mix by adding 67 μl Stratagene Taq Polymerase to the pre-made amp mix tube. This mixture of Stratagene Taq Polymerase and amp mix will now be referred to as the Amp Mix. Place the mix on ice.
Prepare two 12-strip PCR tubes. Pipette 240 μl of the Amp Mix into each tube of one of the 12-strip PCR tubes. Using a 12-channel pipette transfer 120 μl of the Amp Mix from the first 12-strip PCR tubes into the second 12-strip PCR tubes. Label the 12-strip PCR tubes Amp Mix and set aside on ice.
When the thermal cycler reads 9:00 minutes remaining in the second 37°C period, pause the Meg 3-5-7 K program.
Leaving the Assay Plate on the thermal cycler, carefully remove the clear adhesive film.
Using a 24-channel pipette, transfer 25 μl of Cleavage Mix from the two sets of 12-strip PCR tubes previously made (step 10) into each well on the Assay Plate. Mix 10 times by pipetting up and down with each addition of Cleavage Mix. Avoid bubbles.
Reseal the Assay Plate with clear adhesive film, close the lid and resume the Meg 3-5-7 K thermal cycling program.
When the thermal cycler reaches the first 60°C period pause the Meg 3-5-7 K program.
Leaving the Assay Plate on the thermal cycler, carefully remove the clear adhesive film.
Using a 24-channel pipette, transfer 25 μl of Amp Mix from the two sets of 12-strip PCR tubes previously made (steps 11 and 12) into each well on the Assay Plate. Mix 10 times by pipetting up and down with each addition of Amp Mix. Avoid bubbles.
Reseal the Assay Plate with clear adhesive film, close the lid and resume the Meg 3-5-7 K thermal cycling program.
Once the Meg 3-5-7 K thermal cycler program has reached completion all subsequent steps should be run in the post amplification lab. No products from this point should be moved into the pre amplification lab (see Note 2).
3.5 Molecular inversion probe quality control – Checking the 1st Stage PCR. (post-amp)
Once the Meg 3-5-7 K program has reached completion remove the Anneal Plate from the thermal cycler, transfer the Anneal Plate to the post-amp lab and place the plate on ice. Carefully remove the clear adhesive film.
Label a new 96-well plate 1st QC Plate and place the plate on ice. Using a 24-channel pipette transfer 15 μl from each well on the Anneal Plate into the corresponding wells on the 1st QC Plate.
To each well on the 1st QC Plate add 2 μl loading dye to the sample.
Prepare a working stock of 1x TBE Buffer. For instance, add 100 mL 10x TBE Buffer to 900 mL H2O
Load the BioRad precast 3% gel into a gel box containing 1x TBE Buffer. Ensure the empty wells of the gel are completely submerged with buffer.
In the first and last well of the agarose gel, load 2.5 μl BioRad 1kb Ladder.
Using a 12-channel pipette load each row of the 1st QC Plate into the wells on the gel.
Run the gel for 13 minutes at 150 volts.
Visualize the gel using a standard UV gel imager. A clear distinct band should be present at 120 bp for each sample. No bands should appear below or above the prominent band.
3.6 Molecular inversion probes 2nd stage PCR – aliquoting Allele Tube Mix and 1st stage PCR to the Label Plate. (post-amp)
Prepare the Allele Tube Mix by adding 22 μl of Titanium Taq Polymerase to each 2nd Stage PCR Mix tube, these are both pre-made reagents. Mix by pipetting up and down at least 10 times.
Pour the Allele Tube Mix into a 50 ml reagent reservoir and place on ice.
Take a new 96-well plate and label it Label Plate. This plate will be referred to as the Label Plate in all subsequent steps. Place the plate on ice.
Using a 12-channel pipette, aliquot 15.5 μl of the Allele Tube Mix from the 50 ml reagent reservoir into all wells on the Label Plate.
Prepare the Assay Plate by removing the clear adhesive film and placing the plate on ice.
Using a 12-channel pipette, transfer 2 μl from each row of the Assay Plate to the corresponding row on the Label Plate. The Label Plate will also contain 15.5 μl of the Allele Tube Mix which was aliquoted in step 4.
Set a 12-channel pipette to dispense 10 μl. Using this setting, mix the contents of the Label Plate in each row by pipetting up and down 5 times, avoiding bubbles.
Seal the Label Plate with clear adhesive film and spin the plate at 1,000 rpm for 15 seconds.
Place the Label Plate on a thermal cycler and close the lid.
Start the HY-Titanium-10cycle program (see Figure 2c).
When the HY-Titanium-10cycle program is complete, remove the Label Plate from the thermal cycler and spin at 1,000 rpm for 15 seconds. Return the plate to ice.
3.7 Molecular inversion probe target digest – Creating the Hyb Plate and aliquoting the Digest Mix. (post-amp)
Prepare a new 96-well plate by labeling it Hyb Plate. All subsequent steps will refer to this plate as the Hyb Plate.
Carefully remove the clear adhesive film from the Label Plate.
Using a 12-channel pipette, transfer 17 μl from each well of the Label Plate to the corresponding well on the Hyb Plate.
Prepare two 12-strip PCR tubes. Pipette 6 μl of the HY Digest Mix, a pre-made reagent, into each tube of one of the 12-strip PCR tubes. Using a 12-channel pipette transfer 3 μl of the HY Digest Mix from the first 12-strip PCR tubes into the second 12-strip PCR tubes. Label the 12-strip PCR tubes HY Digest Mix.
Using a 24-channel pipette, transfer 1.6 μl of HY Digest Mix from the two sets of 12-strip PCR tubes previously made, into each well on the Hyb Plate. The Hyb Plate will also contain 17 μl of sample transferred in step 3. Set a 24-channel pipette to 15 μl and mix each well by pipetting up and down 20 times, avoiding bubbles.
Seal the Hyb Plate with clear adhesive film and spin the plate at 1,000 rpm for 15 seconds.
Plate the Hyb Plate on the thermal cycler and start the Meg Hydigest-a program (see Figure 2d).
3.8 Molecular inversion probe quality control – Checking the 2nd Stage PCR. (post-amp)
Prepare a new 96-well plate and label it 2nd QC Plate.
When the thermal cycler reads 5 minutes remaining in the second 37°C period of the Meg Hydigest-a program, pause the thermal cycler and remove the Hyb Plate. Place the plate on the bench.
Remove the clear adhesive film from the Hyb Plate and using a 24-channel pipette remove 1 μl from each well and transfer it to the corresponding well of the 2nd QC Plate.
Re-seal the Hyb Plate with clear adhesive film, return the plate to the thermal cycler and resume the Meg Hydigest-a program.
To the 2nd QC Plate add 7 μl H20 to each well. This can be down by adding H20 to a 50 ml reagent reservoir and using a 24-channel pipette to aliquot the H20 to the wells on the 2nd QC Plate.
Add 2 μl loading dye to each well of the 2nd QC Plate.
Load a BioRad precast 3% gel into a gel box containing 1x TBE Buffer. Ensure the empty wells of the gel are completely covered with buffer.
In the first and last well of the agarose gel load 2.5 μl BioRad 1kb Ladder.
Using a 12-channel pipette load each row of the 2nd QC Plate into the wells on the gel.
Run the gel for 13 minutes at 150 volts.
Visualize the gel using a standard UV gel imager. A clear distinct band should be present at 80bp. No bands should appear below or above the prominent band.
3.9 Molecular inversion probe hybridization – Creating the Digest Plate and aliquoting the Hyb Cocktail. (post-amp)
When the Meg Hydigest-a program has reached completion remove the Hyb Plate and place it on ice.
Prepare a new 96-well plate and label the plate Digest Plate. All subsequent steps will refer to this plate as the Digest Plate. Place this plate on ice.
Using a 12-channel pipette, transfer 1 μl of material from all wells on the Hyb Plate to the corresponding wells on the Digest Plate.
Prepare a 12-strip PCR tubes. Pipette 294 μl of the Hyb Cocktail, a pre-made reagent, into each tube of the 12-strip PCR tubes. Label the 12-strip PCR tubes Hyb Cocktail. Place the 12-strip PCR tubes on ice.
Using a 12-channel pipette, add 33.3 μl Hyb Cocktail from the 12-strip PCR tubes to each well on the Digest Plate. Pipette up and down 10 times to mix.
Pour approximately 1 mL of H2O into a 50 ml reagent reservoir.
Using a 12-channel pipette, add 65.7 μl H2O from the reagent reservoir to each well on the Digest Plate. Pipette up and down 10 times to mix.
Seal the Digest Plate with clear adhesive film and spin the plate at 1,000 rpm for 15 seconds.
Place the Digest Plate on the thermal cycler and start the program Meg Denature.
Immediately following the completion of the Meg Denature program, place the Digest Plate on ice and cover with aluminum foil to prevent light exposure.
Let the Digest Plate cool for 2 minutes.
Spin the Digest Plate at 1,000 rpm for 15 seconds. Set the plate aside covered with aluminum foil.
3.10 Molecular inversion probe microarrays– Preparing the microarrays and loading the microarrays samples. (post-amp)
Set the GeneChip® Hybridization Oven 640 to 39°C. It is crucial that this type of oven is used due to the reagent’s sensitivity to light. If an alternative oven is used, ensure it is enclosed and the inside of the oven is not exposed to light.
Remove the 96 arrays form the 4°C refrigerator. Remove the arrays from their packaging and label two arrays per sample (see Note 10).
Allow the arrays to warm to room temperature with the face of the array facing down. Place the arrays on a soft surface to ensure the front window is not scratched.
While the arrays are warming to room temperature, insert a 200 μl pipette tip into the upper right septum of the array.
Set a single channel pipette to 90 μl.
Pipette 90 μl from each well of the Digest Plate and load the sample onto the appropriately labeled array through the lower left septum. It is helpful to hold the array while loading the sample and watch the front window fill with the sample. For more information regarding which sample is in each well of the Digest Plate refer to steps 8–16 in Method section 3.3.
Once all 96 samples have been loaded onto the appropriate array, remove the 200 μl pipette tip from the upper right septum.
Load the arrays onto the rotators in the GeneChip® Hybridization Oven which was previously set to 39°C. Set the rotator to 25 rpms.
Close the GeneChip® Hybridization Oven and leave the arrays in the oven for 12-16 hours. This time should be kept consistent between experiments. Consistency is imperative (see Note 1).
3.11 Molecular inversion probe microarrays– Staining, washing and scanning the microarrays. (post-amp)
Prepare the Stain Cocktail mixing the following reagents; 104.54 mL Wash A, 528 μl BSA (10mg/ml) and 528 μl SAPE (1mg/ml). This volume of Stain Cocktail is enough for 96 arrays plus an additional 10% material to account for pipette error.
For each array, 96 total, aliquot 360 μl Stain Cocktail to a 1.5 ml eppendorf tube. This tube will be placed in position one on the GeneChip® Fluidics Station 450.
To prepare the GeneChip® Fluidics Station 450, load Wash A and Wash B onto the fluidics station. There is a position to hold each bottle of reagents on the right hand side of the machine. Each tube is labeled and should be submerged into the corresponded reagent. The waste bottle should be monitored and emptied when full.
Open the GCOS software and click on the “Fluidics” button. Open the “Protocol” menu and select “Prime_450.” The computer will prompt you to complete actions on the fluidics station. Follow the prompts.
When “Prime_450” has reached completion select the protocol in GCOS indicated by your Affymetrix representative to begin the staining and washing of the arrays.
Each GeneChip® Fluidics Station 450 will process only four arrays at a time. When prompted by the program load the first four arrays onto the fluidics station, the window of the array facing outwards. Do not push the lever down which will lock the array into place until prompted to do so by the program.
When prompted load the Stain Cocktail into position one on the GeneChip® Fluidics Station 450 and two empty 1.5 ml eppendorf tubes into the other two positions. Again, do not push the lever down which will bring the needles down into the eppendorf tubes, wait until prompted to do so by the program.
Once the GeneChip® Fluidics Station 450 has loaded the first four arrays and lowered the needles into the eppendorf tubes, the washing and staining of the arrays will begin.
The fluidics station should periodically be monitored to ensure there have been no errors (see Note 11).
Once the first four arrays have been stained, washed and the protocol reaches completion, load the next four arrays onto the fluidics station and again follow the prompts of the protocol. In is imperative a fresh eppendorf tube of Stain Cocktail be loaded into position one for each array.
Staining and washing should be repeated for the first 48 arrays.
For the arrays that have been stained and washed, load each into the carousel of the Affymetrix GeneChip® Scanner. The arrays will only fit one way; do not force the arrays into the slots.
The arrays can be held in the scanner until the first 48 arrays have been stained and washed. The Affymetrix GeneChip® Scanner only holds 48 arrays.
When the first 48 arrays have been loaded onto the Affymetrix GeneChip® Scanner begin the scanning by clicking on the “Start” button.
Repeat steps 6–14 for the second batch of 48 arrays.
3.12 Data analysis
GeneChip® Targeted Genotyping Analysis Software is used for analysis of the MIP data sets read from the arrays. Signal from each chip is background subtracted, color separated, normalized and genotypes called as described previously within the software package (5).
Using normal diploid genome reference samples run at the same time as the tumor sample, the average signal in each of the three clusters (two homozygous clusters and a heterozygous cluster) for each marker as well as the standard deviation of the signal after removing (15%) outliers are calculated. The average signal in a cluster is then set to denote two copies since most reference normal samples will be diploid at any given point in the genome. For homozygous clusters we only consider the signal in the relevant allele and ignore the signal in the other allele for the computation of copy number. For heterozygous clusters we consider both signals and analyze them in two (orthogonal) directions: summing them together (copy sum analysis) and taking their ratio (allele ratio analysis). If a marker in a test sample has an allele imbalance, it may be classified as homozygous and therefore the signal in the other allele ignored.
Acknowledgments
We would like to thank Drs. Yuker Wang and Malek Faham for their contribution in developing molecular inversion probe technology and providing information about the assay. This work was supported by NIH grants K08 CA96879, R21 CA109190 and 2P01HG000205.
Footnotes
It is imperative that the method remain consistent run to run. Consistency includes but is not limited to; number of times pipetted, thermal cyclers used, pipetman, benches, etc. If the method and related procedures are not kept constant, the quality of data will be negatively affected.
Two separated work areas should be created to minimize contamination. Never move any reagents, supplies or equipment from the post-amp lab to the pre-amp lab, it is especially important that no PCR amplified products are ever present in the pre-amp work area or brought into the pre-amp area. Each method will indicate the location the procedure should be completed.
PicoGreen reagent is degraded by light; all original and working stocks should therefore be wrapped in aluminum foil to prevent light exposure.
Be sure to properly label plates prior to starting subsequent protocol steps or any PCR run.
Enzyme A should be added last as it is extremely heat sensitive, minimize warming of this enzyme by keeping it on ice at all times.
The row containing the master mix is used the same way 12-strip PCR tubes would be used; it is simply an easier way to make additional aliquots to subsequent rows in the same plate. The master mix is placed in every other lane to allow the use of a 12-channel pipette in the steps which follow.
Thermal cycling programs should be preprogrammed and properly named prior to begin experiments.
This step is designed to remove liquid from sides of the wells which may have formed from bubbles bursting during the 95°C stage. It is imperative that the entire volume of the reaction take place in the overnight annealing.
Best results are obtained when the annealing time is kept consistent between experiments. For example, if the annealing time is 16 hours and 30 minutes for one experiment try to replicate that time for the experiment which follows.
Keep in mind each sample has been labeled with dATP/dTTP and dCTP/dGTP meaning each sample is now located in two wells of the Digest Plate. It is ideal to label each array with the sample name and which two nucleotides it has been tagged with.
If errors occur it is best to contact Affymetrix technical support for help.
References
- 1.Hardenbol P, Yu F, Belmont J, Mackenzie J, Bruckner C, Brundage T, Boudreau A, et al. Highly multiplexed molecular inversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res. 2005;15:269–275. doi: 10.1101/gr.3185605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ji H, Kumm J, Zhang M, Farnam K, Salari K, Faham M, Ford JM, Davis RW. Molecular inversion probe analysis of gene copy alterations reveals distinct categories of colorectal carcinoma. Cancer Res. 2006;66:7910–7919. doi: 10.1158/0008-5472.CAN-06-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Moorhead M, Karlin-Neumann G, Falkowski M, Chen C, Siddiqui F, Davis RW, Willis TD, Faham M. Allele quantification using molecular inversion probes (MIP) Nucleic Acids Res. 2005;33:e183. doi: 10.1093/nar/gni177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Moorhead M, Karlin-Neumann G, Wang NJ, Ireland J, Lin S, Chen C, Heiser LM, Chin K, Esserman L, Gray JW, Spellman PT, Faham M. Analysis of molecular inversion probe performance for allele copy number determination. Genome Biol. 2007;8:R246. doi: 10.1186/gb-2007-8-11-r246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moorhead M, Hardenbol P, Siddiqui F, Falkowski M, Bruckner C, Ireland J, Jones HB, Jain M, Willis TD, Faham M. Optimal genotype determination in highly multiplexed SNP data. Eur J Hum Genet. 2006;14:207–215. doi: 10.1038/sj.ejhg.5201528. [DOI] [PubMed] [Google Scholar]