

Abstract
Background
High density lipoprotein (HDL) protects the artery wall by removing cholesterol from lipid-laden macrophages. However, recent evidence suggests that HDL might also inhibit atherogenesis by combating inflammation.
Methods and Results
To identify potential anti-inflammatory mechanisms, we challenged macrophages with lipopolysaccharide (LPS), an inflammatory microbial ligand for Toll-like receptor 4 (TLR4). HDL inhibited the expression of 30% (277 of 911) of the genes normally induced by LPS, microarray analysis revealed. One of its major targets was the type I interferon response pathway, a family of potent viral immunoregulators controlled by TLR4 and the TRAM/TRIF signaling pathway. Unexpectedly, HDL’s ability to inhibit gene expression was independent of macrophage cholesterol stores. Immunofluorescent studies suggested that HDL promoted TRAM translocation to intracellular compartments, which impaired subsequent signaling by TLR4 and TRIF. To examine the potential in vivo relevance of the pathway, we used mice deficient in apolipoprotein (apo) A-I, HDL’s major protein. After infection with Salmonella typhimurium, a Gram-negative bacterium that expresses LPS, apoA-I–deficient mice had 6-fold higher plasma levels of interferon-β –a key regulator of the type I interferon response– than did wild-type mice.
Conclusions
HDL inhibits a subset of LPS-stimulated macrophage genes that regulate the type I interferon response, and its action is independent of sterol metabolism. These findings raise the possibility that regulation of macrophage genes by HDL might link innate immunity and cardioprotection.
Keywords: Lipid-raft, MyD88, chemokine, cytokine, interferon regulatory factor 7
INTRODUCTION
HDL protects against artery disease by removing cholesterol from artery wall macrophages through reverse cholesterol transport1-4. There is mounting evidence, however, that it has additional anti-atherosclerotic effects2, 5-8. One such activity may be modulation of the innate immune system’s inflammatory response9-14.
Two cholesterol-transporting proteins, ABCA1 and ABCG1, might link HDL to both cholesterol removal and regulation of inflammation10-12. Both membrane-associated ATP-binding cassette transporters are found in macrophages. ABCA1 promotes cholesterol efflux to lipid-poor apoA-I, the major HDL protein, while ABCG1 induces cholesterol efflux to intact HDL particles10. Macrophages isolated from mice with genetically engineered deficiencies in ABCA1 and/or ABCG1 overexpress well-known inflammatory genes, such as TNF-α, IL-1β, and IL- 8 when the cells are stimulated with bacterial lipopolysaccharide (LPS)11. In both macrophages and endothelial cells, HDL’s anti-inflammatory effects have been proposed to reflect changes in membrane cholesterol and lipid rafts, with downstream effects on signal transduction and the activities of membrane-associated proteins11, 12, 15.
The innate immune system’s response to infection involves membrane-associated Toll-like receptors (TLRs), which can sense a diverse range of conserved microbial structures16. TLR signal transduction begins when one or more adaptor proteins are recruited to the receptor’s intracellular TIR (Toll/interleukin-1 receptor) domain. These adaptors include MyD88 (myeloid differentiation primary response protein 88), TRAM (TRIF-related adapter molecule), and TRIF (TIR domain-containing adaptor protein inducing interferon).
The classic TLR ligand is LPS, which rapidly upregulates thousands of macrophage genes17-19. LPS binds to TLR4, which in turn activates signaling pathways dependent on MyD88 or TRAM/TRIF20. The MyD88 pathway activates NF-κB and other transcription factors, which in turn induce type II interferon response genes, such as TNF-α and IL-1β. In contrast, the TRAM/TRIF pathway, which is independent of MyD88, induces expression of interferon-β (IFN-β) by activating interferon regulatory factor 3 (Irf3), a transcription factor21, 22. IFN-β then activates other type I interferon-inducible genes. The type II interferon response enhances anti-bacterial responses by macrophages, whereas the type I interferons are potent antiviral immunoregulators23, 24.
We analyzed macrophages with microarrays to search for novel anti-inflammatory actions of HDL and possible roles for ABCA1 and/or ABCG1. We found that HDL exerts potent effects on the type I interferon pathway in LPS-stimulated macrophages without assistance from cholesterol transporters. These observations raise the possibility that therapies that raise HDL levels might act in novel ways to retard atherosclerosis and perhaps other immune disorders.
METHODS
For additional details, see the online-only Data Supplement.
Cell Culture
Macrophages were isolated by lavage from the peritoneum of C57BL/6 mice 5 days after intraperitoneal (i.p.) injection of thioglycolate, adhesion purified, and cultured in Dulbecco’s minimum essential medium (DMEM) supplemented with 2% lipoprotein-deficient bovine serum (LPDS, Sigma). After incubation for 48 h with or without 50 μg/mL of acetylated-LDL in 2% LPDS in DMEM, macrophages were treated for 4 h with serum-free medium supplemented with or without 50 μg/mL of HDL, and then exposed for 4 h to serum-free medium supplemented with or without 100 ng/mL of LPS.
Microarray Analysis
All microarray data are available from the Gene Expression Omnibus Database (accession number GSE19340).
TRAM translocation
RAW264.7 macrophage cells were stably transfected with retrovirus expressing TRAM-GFP25. Cells were treated with serum-free medium supplemented with HDL or LDL (50 μg/mL) for 4 h. The subcellular location of TRAM-GFP was determined by fluorescence microscopy.
Murine Infection Model
Mice were housed under specific pathogen-free conditions26. C57BL/6 mice and apoA-I deficient C57BL/6 mice27 were injected i.p. with 500 CFU log-phase Salmonella typhimurium in 0.2 mL of PBS. Animals were matched for gender, weight and age.
Statistical analysis
Data are means and SEMs. Comparison of mean values between two groups was evaluated by the two-tailed Student’s t-test. With more than two groups, we used the Kruskal-Wallis (nonparametric ANOVA) with the Wilcoxon signed rank test for comparisons post-test. Differences were considered significant at P< 0.05. Unless otherwise indicated, N=3 per group.
RESULTS
To test the hypothesis that HDL can suppress inflammatory pathways triggered by Toll-like receptors, we first exposed cholesterol-laden macrophages to medium alone or medium supplemented with HDL (50 μg/mL protein), extensively washed the cells, and then challenged them with the LPS (100 ng/mL), a TLR4 ligand (Fig 1A). To determine how HDL affected gene expression, we used transcriptional profiling.
Figure 1. Effect of HDL pretreatment on TRAM/TRIF-dependent and MyD88-dependent genes in cholesterol-laden mouse macrophages challenged with LPS.
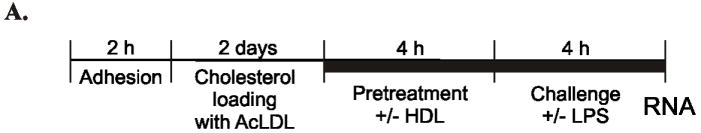



Panel A: Experimental Approach. Thioglycolate-elicited peritoneal macrophages were isolated from mice, adhesion purified, and incubated for 48 h with acetylated-LDL in 2% LPDS. Macrophages were treated for 4 h with serum-free medium or serum-free medium supplemented with 50 μg/mL of mouse HDL, washed, and then exposed for 4 h to serum-free medium or serum-free medium supplemented with 100 ng/mL of LPS.
Panel B: Array analysis of LPS-stimulated macrophages. RNA harvested from LPS-stimulated macrophages, with or without HDL pretreatment, was analyzed on microarrays. Results are presented as a heat map (Red, increased expression; Green, decreased expression). Results represent one of two independent experiments with 4 independent biological replicates per condition.
Panel C: qRT-PCR analysis of LPS-stimulated macrophage genes. mRNA levels are expressed as fold change relative to no HDL pretreatment and no LPS stimulation. per group. * P<0.05 relative to no HDL and no LPS stimulation, # P<0.05 relative to no HDL and LPS stimulation by the Kruskal-Wallis. Results are representative of ≥3 independent experiments.
Panel D: ELISA of macrophage-conditioned medium. per group. * P<0.05 relative to no HDL and no LPS stimulation, # P<0.05 relative to no HDL and LPS stimulation by the Kruskal-Wallis. Results are representative of 2 independent experiments.
HDL selectively inhibits LPS-induced genes that are turned on by the TRAM/TRIF-dependent signaling pathway
In two independent experiments with 4 biological replicates per group, 2,749 of 28,289 genes showed statistically significant differential expression in macrophages challenged with LPS (false discovery rate controlled at level 0.05). We observed particularly striking increases (> 10-fold; Supplemental Table 1) in levels of mRNA for cytokines (Il-1β, Tnf-α, Il-6, and Il-12β) and chemokines (Ccl4, Ccl5, Cxcl2, Cxcl3, and Cxcl10). Using the criterion of a significant change in gene expression that was at least 1.5-fold, we observed 911 genes that were regulated by LPS. There was extensive overlap between the genes regulated by LPS (100 ng/mL) in our study and those reported by investigators using a lower LPS concentration (10 ng/mL)19.
We next explored HDL’s effect on LPS-induced gene expression. Pretreatment of macrophages with HDL prevented (by ≥25%) LPS from inducing 277 genes (Supplemental Table 2 and 3). Strikingly, the most strongly affected genes (Fig. 1B) were viral response genes known to be induced by the TRAM/TRIF-dependent pathway19, 28. Examples were Irf7, Cxcl10 (IP-10), Il-12β, Il-15, Tlr3, Tlr8, Nos2 (iNOS), Ifit1, and Ifit3 (Fig. 1B). In contrast, HDL pretreatment failed to significantly suppress MyD88-dependent pathway genes, such as Tnf-α, Il-6, and Il-1β (Fig. 1B).
Using Gene Ontology (GO), we characterized cellular processes affected by HDL pretreatment. GO analysis with the DAVID algorithm29 demonstrated a significant enrichment (relative to the entire mouse genome) in “response to virus” genes (p=9×10-14, Supplemental Table 3), while “I-kB/NF-kB cascade” genes activated by LPS were not significantly different (Supplemental Table 3). These results suggest that exposing macrophage foam cells to HDL suppresses the subsequent expression of a subset of genes that is regulated by LPS through the TRAM/TRIF-pathway that does not involve MyD88 –the type I interferon pathway23, 30.
We used qPCR to quantify mRNA levels for 6 important genes in this pathway: Irf7, Ip-10, Il-12b, Tlr3, Tlr8, and Il-15. All are activated by the TRAM/TRIF pathway when cells are challenged with TLR4 agonists23, 24. Five of the 6 genes (Irf7, Ip-10, Il-12b, Tlr3, and Il-15) were significantly induced by LPS (Fig. 1C, p <0.05). However, increased gene expression was almost completely abrogated by pretreating macrophages with HDL (Fig. 1C). HDL’s inhibitory effect depended on its concentration in the medium (Supplemental Fig. 1). Furthermore, both mouse and human HDL were inhibitory. We obtained similar results with macrophages derived from mouse bone marrow (Supplemental Fig. 2), demonstrating that HDL exerts similar anti-inflammatory effects on the type 1 interferon response in both elicited macrophages and bone marrow-derived macrophages.
HDL inhibits LPS-stimulated secretion of interferon-β, IP-10, and IL-12 from macrophages
To determine whether our mRNA data were paralleled at the protein level, we monitored levels of IFN-β, IP-10, and IL-12β in macrophage-conditioned medium. LPS stimulated the expression of IFN-β (7-fold), IP-10 (>1,000-fold), and IL-12β (1,000-fold) protein. IFN-β was readily detected in media harvested from LPS-stimulated cells, but was undetectable by our assay in 2 of the 3 control cell media and in 1 of 3 media from HDL-pretreated cells (Fig. 1D). HDL significantly lowered levels of IP-10 and IL-12β in the medium of macrophages challenged with LPS (Fig. 1D). These data indicate that at both the mRNA and protein levels, HDL suppresses TRIM/TRAM pathway genes in macrophages challenged with LPS.
HDL inhibits TLR4 agonist-induced genes that are turned on by the TRAM/TRIF pathway
To explore how HDL affects the TRIM/TRAM pathway, we determined whether pretreating macrophages with HDL prevented a TLR4 agonist antibody from regulating gene expression. Irf7, Ip-10, and Tnf-α were all induced when the macrophages were incubated with the antibody (Fig. 2). Pretreating the macrophages with HDL significantly inhibited the expression of the TRAM/TRIF-dependent pathway genes Irf7 and Ip-10. Thus, HDL suppresses activation of the TRIM/TRAM pathway when TLR4 is activated by a lipid (LPS) or protein (antibody) agonist.
Figure 2. Effect of HDL on cholesterol-laden macrophages challenged with a TLR4 agonistic antibody.

Macrophages were treated for 4 h with serum-free medium or serum-free medium supplemented with 50 μg/mL of HDL, washed, and then exposed for 4 h to serum-free medium supplemented with 16 ng/mL of a TLR4 activating antibody. mRNA levels were quantified by qRT-PCR. per group. ** P<0.05 relative to no HDL and no LPS stimulation, ## P<0.05 relative to no HDL and LPS stimulation by the Kruskal-Wallis. Results are representative of 2 independent experiments.
HDL’s suppressive effects do not require LPS to bind lipoproteins
It is well established that HDL, LDL, and other lipoproteins bind to LPS to prevent it from activating TLR431, 32. To determine whether this mechanism is responsible for HDL’s ability to inhibit gene expression, we exposed macrophages to HDL or LDL and quantified the LPS-induced expression of Irf7 and Ip-10 (two TRAM/TRIF-dependent genes) and Tnf-α (a MyD88- dependent pathway gene). LPS significantly increased the expression of all 3 genes (Fig. 3; p<0.001): Irf7 (20-fold), Ip-10 (500-fold), and Tnf-α (100-fold). HDL suppressed this induction of Irf7 and Ip-10, but did not significantly affect Tnf-α expression (Fig. 3). In contrast, the same concentration of human LDL (50 μg/mL protein) had little affect on gene expression (Fig. 3).
Figure 3. Effects of HDL and LDL on LPS-stimulated macrophage genes.

Macrophages were challenged with LPS as described in the legend to Fig. 1. Cells were pretreated with HDL or LDL (50 μg protein/mL). mRNA levels were quantified by qRT-PCR. per group. * P<0.05 relative to no HDL and no LPS stimulation, # P<0.05 relative to no HDL and LPS stimulation by the Kruskal-Wallis. Results are representative of 2 independent experiments.
When macrophages were challenged with a 10-fold lower concentration of LPS (10 ng/mL), pretreatment with HDL inhibited the induction of Irf7, but not of TNF-α (Supplemental Fig. 3). These results do not support the hypothesis that HDL selectively inhibits type 1 interferon response genes because the TRAM/TRIF pathway is more sensitive than the MyD88 pathway to lower concentrations of LPS.
Next, we considered the possibility that HDL might inhibit LPS activity by binding to and sequestering CD14 or TLR4 on the cell surface. However, pretreating the cells for 1 min with HDL—or even adding HDL at the same time as LPS—did not significantly prevent LPS from upregulating those genes (Supplemental Figs. 4 and 5A). Under these conditions, HDL pretreatment still suppressed the expression of TRAM/TRIF-dependent genes (Supplemental Fig. 5B). Taken together with the TLR4 agonist antibody results (Fig. 2), these data indicate that HDL does not need to bind to LPS to suppress TRAM/TRIF-dependent genes.
Inhibition of TRAM/TRIF-dependent genes by HDL is independent of macrophage cholesterol content
Previous work strongly supports the proposal that HDL decreases macrophages’ inflammatory state by enabling ABCA1 or ABCG1 to remove excess cellular sterol10, 11. To determine whether these transporters are involved when HDL suppresses genes in the TRAM/TRIF-dependent pathway, we downregulated ABCA1 and ABCG1, using siRNA (Fig. 4). Immunoblot analysis confirmed decreased protein expression (Fig. 4A). However, HDL pretreatment suppressed LPS-induced expression of Irf7 or Ip-10 to the same extent (Fig. 4B). Thus, HDL’s suppressive effect is not mediated through ABCA1 or ABCG1, and its anti-inflammatory properties appear to be independent of cholesterol stores.
Figure 4. Suppressive effects of HDL in LPS-stimulated cholesterol-laden macrophages exposed to siRNAs for ABCA1 or ABCG1.
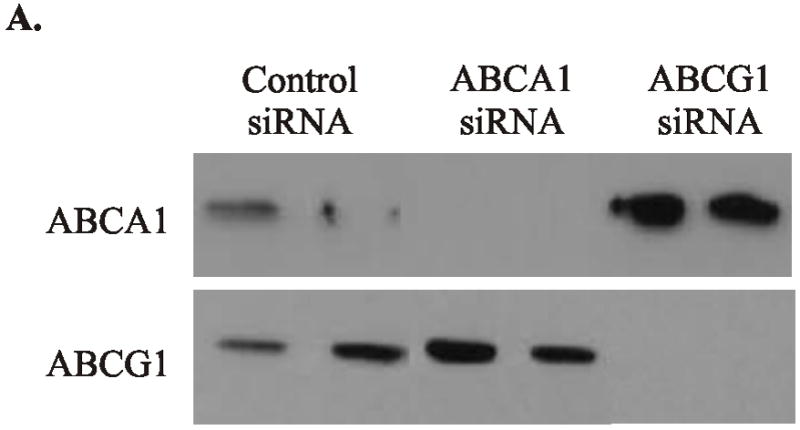

Isolated macrophages were incubated with 2% LPDS and transfected with specific siRNA for 24 h to knockdown ABCA1 and ABCG1. Macrophages were then loaded with cholesterol by changing the medium to 2% LPDS supplemented 50 μg protein/mL of acetylated LDL.
Panel A: Immunoblot analysis. Cellular lysates were fractionated on a 5%–15% gradient SDS-PAGE and then subjected to immunoblot analysis with antibodies to ABCA1 or ABCG1.
Panel B: qRT-PCR analysis. per group. * P<0.05 relative to no HDL and no LPS stimulation, # P<0.05 relative to no HDL and LPS stimulation by the Kruskal-Wallis. Results are representative of 2 independent experiments.
We next depleted macrophages of cholesterol by treating them for 4 h with β-methylcyclodextrin (β-MCD)33. β-MCD pretreatment did not alter HDL’s ability to suppress LPS-induced Irf7 or Il-12b induction (Fig. 5A), and β-MCD itself did not inhibit LPS response (Fig. 5B). We also determined whether lipid-free apoA-I, HDL’s major protein, would suppress LPS-stimulated gene expression. We first showed that a 4-h incubation with 10 μg/mL of apoA-I promoted cholesterol efflux from acetyl-LDL-loaded macrophages to about the same extent as 50 μg/mL of HDL. Then we demonstrated that 10 μg/mL of apoA-I did not significantly effect LPS’s ability to induce the expression of Irf7 (Fig. 5C).
Figure 5. Suppressive effects of HDL on macrophages loaded or not loaded with cholesterol and then challenged with LPS.




Panel A: Pretreatment with cyclodextrin and/or HDL. Macrophages incubated with acetyl-LDL were treated (or not treated) with 200 μg/mL of cyclodextrin for 2 h. And then cells were treated with 50 μg protein/mL of HDL and challenged with LPS. mRNA levels were quantified by qRT-PCR. per group. * P<0.05 relative to no HDL and no LPS stimulation, # P<0.05 relative to no HDL and LPS stimulation by the Kruskal-Wallis. Results are representative of 2 independent experiments.
Panel B and C: Pretreatment with cyclodextrin or lipid-free apoA-I. Macrophages incubated with acetyl-LDL were treated (or not treated) with 200 μg/mL of cyclodextrin, 10 μg protein/ml of lipid-free apoA-I or 50 μg protein/ml of HDL, and then challenged with LPS. mRNA levels were quantified by qRT-PCR. per group. * P<0.05 relative to no HDL and no LPS stimulation, # P<0.05 relative to no HDL and LPS stimulation by the Kruskal-Wallis. Results are representative of 2 independent experiments.
Panel D: Array analysis of macrophages exposed or not exposed to acetyl-LDL. RNA harvested from LPS-stimulated macrophages, with or without cholesterol-loading and with or without HDL pretreatment, was analyzed on microarrays. Results represent 4 independent biological replicates and are presented as a heat map (Red, increased expression; Green, decreased expression).
To further investigate the role of cholesterol loading, we performed microarray analysis on macrophages that had been incubated for 48 h with either 2% lipoprotein-deficient serum or serum alone supplemented with 50 μg/mL of acetyl-LDL. HDL inhibited the expression of LPS-induced genes in the TRAM/TRIF-dependent pathway to a similar degree under both conditions (Fig. 5D). Thus, cholesterol loading or depletion has little effect on HDL’s ability to suppress genes in the TRAM/TRIF-dependent pathway when macrophages are challenged with LPS.
HDL suppresses interferon pathway genes through TLR4 but not through TLR2, TLR3, TLR7/8 or CD14
We used poly I:C (a TLR3 ligand) and R848 (a TLR 7/8 ligand) to determine whether HDL specifically suppresses the TLR4 pathway. Each ligand induced macrophages to express representative type I and type II interferon response genes: Irf7, iNOS, Ip-10, Il-15, and Il-12β (Fig. 6). However, HDL pretreatment suppressed those genes significantly only when the macrophages were challenged with LPS (p<0.05 for all conditions). Thus, HDL’s ability to inhibit gene expression in macrophages appears to be specific for LPS and a TLR4 agonist antibody, strongly suggesting that it selectively affects TLR4 signaling.
Figure 6. HDL’s effects on cholesterol-laden macrophages challenged with various TLR agonists.

Macrophages were loaded with cholesterol as described in the legend to Fig. 1. The cells were treated for 4 h with HDL, and then challenged for 4 h with 100 ng of LPS, 1 μg/mL of polyI:C (a TLR3 agonist), or 1 μg/mL of R848 (a TLR7/8 agonist). mRNA was harvested and quantified by qRT-PCR. per group. * P<0.05 relative to no HDL pretreatment by the Kruskal-Wallis. Results are representative of 3 independent experiments.
Because HDL acted on the TLR4 signaling pathway, we wondered if CD14, a key regulator of both the MyD88-independent and -dependent pathways, was its target34. We found, however, that HDL had no effect on TLR2 signaling (Supplemental Fig. 6), which also involves CD14. Moreover, HDL also failed to affect CD14 expression in macrophages (as measured by flow cytometry), and soluble CD14 was unable to overcome HDL’s suppressive action (data not shown). Thus, CD14 does not appear to be involved in HDL’s action on innate immunity genes.
HDL induces TRAM translocation to intracellular compartments
Recent studies suggest that TLR4 activates the TRAM/TRIF pathway by signaling from early endosomes25, 35. The proposed mechanism involves LPS-induced endocytosis of TLR and then TRAM/TRIF-dependent signaling from an endosomal compartment. To determine whether this pathway might be involved when HDL regulates type 1 interferon response genes, we used a macrophage cell line stably transfected with a TRAM-GFP fusion protein25.
When macrophages were incubated for 4 h in serum-free medium (Fig. 7) or medium supplemented with 2%FBS (data not shown), fluorescence microscopy demonstrated that virtually all of the TRAM-GFP fusion protein was associated with the macrophages’ plasma membrane. However, cells incubated with 50 μg/mL of HDL translocated TRAM to intracellular compartments (Fig. 7) but did not induce Irf7 (data not shown). LDL failed to affect TRAM’s cellular location (Fig. 7). These observations suggest that HDL induces TRAM translocation to endosomes or other intracellular compartments, which might account for the impairment in TLR4 signaling.
Figure 7. Subcellular visualization of TRAM in macrophages.

RAW264.7 macrophages expressing TRAM-GFP were treated for 4 h with serum-free medium or serum-free medium supplemented with 50 μg/mL of LDL or HDL. The subcellular location of TRAM-GFP was determined by fluorescence microscopy. Arrows highlight fluorescent material. Results are scores for plasma membrane vs. intracellular localization and are representative of 3 independent experiments.
Plasma interferon-β levels increase markedly in apoA-I deficient mice challenged with Salmonella typhimurium
To determine whether HDL prevents LPS from inducing TRAM/TRIF genes in vivo, we infected wild-type and apoA-I-deficient mice (both in the C57BL/6 background) with Salmonella typhimurium, a Gram-negative bacterium that expresses LPS. HDL-C levels in the apoA-I-deficient mice were ~20% those in the wild-type mice (Fig. 8A). By 10 days after the bacterial challenge, the same number of animals in each group had died (8 of 10; Fig. 8B), suggesting that apoA-I deficiency did not significantly affect survival. In striking contrast, plasma levels of IFN-β were 6-fold greater in the apoA-I–deficient mice, even though the two strains had similar plasma levels of TNF-α (Fig. 8C). Thus, HDL appeared to selectively modulate the expression of IFN-β –a key mediator of the type I interferon response– in this model of Gram-negative sepsis.
Figure 8. Plasma levels of interferon-β in wild-type and apoA-I-deficient mice infected with Salmonella typhimurium.
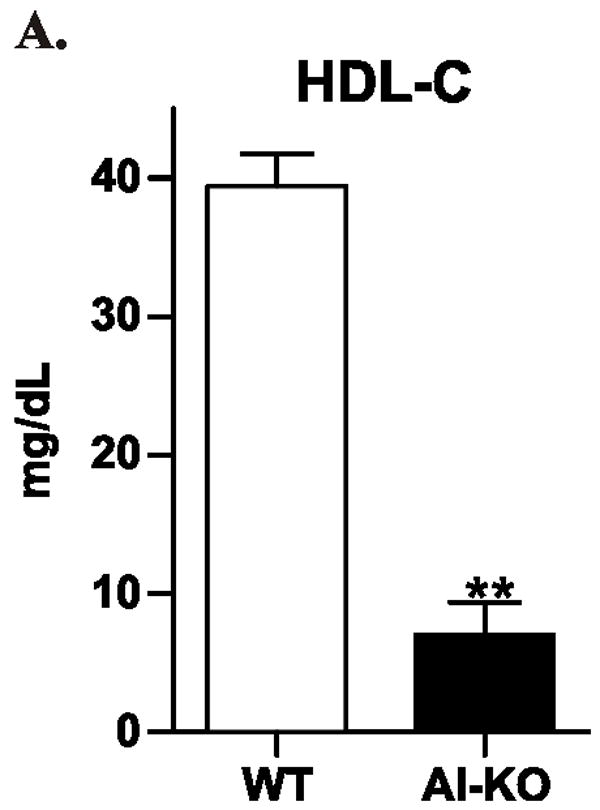
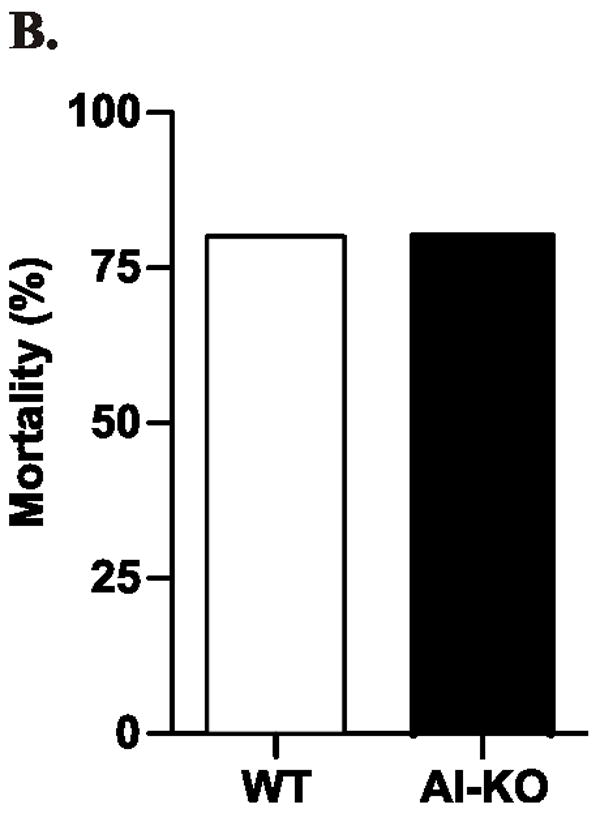

C57BL/6 mice and apoA-I-deficient C57BL/6 mice were injected intraperitoneally with Salmonella typhimurium. Animals (N=6 per group) were matched for gender, weight, and age.
Panel A: HDL-C was measured in uninfected mice. Panel B: Mortality was documented 10 days after infection. Panel C: Plasma cytokine levels were determined by ELISA 4 days after infection. ** P<0.01 by Student’s t-test. N.S., not significant.
DISCUSSION
Our observations indicate that HDL selectively prevents the activation of type I interferon response genes when macrophages are challenged with LPS, the classic TLR4 ligand, or a TLR4 agonist antibody. In contrast, HDL has little effect on gene expression triggered by ligands for TLR2, TLR3, or TLR 7/8. Although previous studies have demonstrated that cholesterol accumulation in the plasma membrane of macrophages deficient in ABCA1 and/or ABCG1 promotes TLR4 signaling and the expression of inflammatory genes (e.g., TNF-α, IL-6)11, 12, we found that HDL suppressed the type I interferon response regardless of whether macrophages were loaded with cholesterol or ABCA1 or ABCG1 expression was inhibited. Our observations suggest that regulation of the type I interferon response in macrophages might contribute to HDL’s cardioprotective and anti-inflammatory effects by mechanisms that do not involve sterol metabolism.
The type I interferon response is triggered when IFN-β is upregulated by TLR4-dependent activation of the transcription factor Irf721, 22. We found that plasma levels of IFN-β were greater in apoA-I–deficient mice than in wild-type mice when the animals were challenged with Salmonella typhimurium, a Gram-negative bacterium that expresses LPS. In contrast, the two mouse strains had similar plasma levels of TNF-α, suggesting that HDL regulates the type I interferon response but not the type II response in this model of infection. Regulation of the innate immune system might also contribute to HDL’s cardioprotective effects.
Using ligands specific for the various TLRs, we showed that HDL selectively inhibits the expression of type I interferon response genes regulated by TLR4. Moreover, those genes appear to require the adaptor proteins TRAM and TRIF (but not MyD88) for signaling36. Thus, we found that HDL failed to inhibit cytokine and chemokine expression by poly I:C, a TLR3 ligand for a pathway requiring TRIF but not TRAM. In contrast, signaling by TLR4 requires both TRAM and TRIF. The simplest interpretation is that HDL inhibits TLR4 signaling at a step before TRIF, possibly by a mechanism involving TRAM. Because HDL’s ability to inhibit TLR4 signaling appeared little affected by macrophage sterol balance, the underlying mechanism is unlikely to be regulated directly by cholesterol.
Signaling by TRAM requires endocytosis of TLR425, 35. We found that TRAM translocates to intracellular compartments when macrophages are treated with HDL and that translocated TRAM cannot induce Irf7 expression. In contrast, LDL failed to change TRAM’s cellular location. These observations suggest that HDL induces TRAM translocation to an intracellular compartment, thereby impairing subsequent signaling by TLR4. We therefore propose that HDL depletes the plasma membrane of TRAM, a key adaptor molecule that activates TRIF in endosomal compartments.
Recent observations support our suggestion that the TLR4/TRAM/TRIF signaling cascade in macrophages is regulated at a step proximal to TRIF. In mice, Wang et al found that a deficiency in the hemochromatosis gene, Hfe, also suppresses the expression of TRAM/TRIF-dependent genes by a pathway that requires TLR4 but not TLR337.
It is possible that activation of the type I interferon response in macrophages contributes to atherogenesis. For example, TLR4 deficiency reduces atherosclerosis in hypercholesterolemic mice38, suggesting that ligands for this receptor activate macrophages in atherosclerotic lesions. Moreover, TLR4 signaling via TRAM/TRIF induces the type I interferon response gene Cxcl10 (IP-10), a T cell chemokine39. IP-10 is highly expressed in murine and human atherosclerotic tissue40, and lesion formation is retarded in hypercholesterolemic mice deficient in IP-10 or its receptor41.
Type I inteferons play a critical role in the antiviral response of cells42. A key component of the system is a feed-forward loop, which amplifies the initial response to the invading virus. However, positive feedback is deleterious when the response is excessive43. HDL may be a physiologically relevant regulator of amplification because an apoA-I mimetic peptide, D-4F, reduced viral lung titers and tissue inflammation in mice infected with influenza virus44. In cultured cells infected with virus, D-4F suppressed secretion of IFN-α and IFN-β, suggesting inhibition of the type I interferon response45. Moreover, type I interferons promote disease amplification in autoimmune rheumatological disorders43, and apoA-I regulates T cell activation in hypercholesterolemic mice46. These observations raise the possibility that HDL controls inflammation during infection and the immune response against host tissue.
Classically, the type I interferon response produces potent antiviral immunoregulators24 while the type II response promotes anti-bacterial activities of macrophages42. However, emerging evidence also implicates the type I interferon response in host defense during bacterial infection23. Our observations are consistent with previous suggestions that HDL regulates the cellular response to viral infection44, 45, 47. These observations raise the possibility that HDL controls inflammation during infection and the immune response against host tissue.
Supplementary Material
Acknowledgments
Microarray analyses were performed at the Center for Array Technologies at the University of Washington. The authors thank Drs. Ferric Fang and Steven Libby for helpful discussions.
FUNDING SOURCES TV and AH were supported by Pilot and Feasibility Awards from the CNRU and the DERC, University of Washington. This research was supported by grants from the National Institutes of Health (HL086798, HL092969, P30ES07033, P30DK017047, P01HL018645, P01HL030086).
Footnotes
DISCLOSURES We have no conflicts to disclose.
References
- 1.Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. HDL, ABC transporters and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–375. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 2.deGoma EM, deGoma RL, Rader DJ. Beyond high-density lipoprotein cholesterol levels evaluating high-density lipoprotein function as influenced by novel therapeutic approaches. J Am Coll Cardiol. 2008;51:2199–2211. doi: 10.1016/j.jacc.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas MJ, Bhat S, Sorci-Thomas MG. Three-dimensional models of HDL apoA-I: implications for its assembly and function. J Lipid Res. 2008;49:1875–1883. doi: 10.1194/jlr.R800010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davidson WS, Thompson TB. The structure of apolipoprotein A-I in high density lipoproteins. J Biol Chem. 2007;282:22249–22253. doi: 10.1074/jbc.R700014200. [DOI] [PubMed] [Google Scholar]
- 5.Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, Fogelman AM. Mechanisms of disease: proatherogenic HDL--an evolving field. Nat Clin Pract Endocrinol Metab. 2006;2:504–511. doi: 10.1038/ncpendmet0245. [DOI] [PubMed] [Google Scholar]
- 6.Shao B, Heinecke JW. HDL, lipid peroxidation, and atherosclerosis. J Lipid Res. 2009;50:599–601. doi: 10.1194/jlr.E900001-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Getz GS, Wool GD, Reardon CA. Apoprotein A-I mimetic peptides and their potential anti-atherogenic mechanisms of action. Curr Opin Lipidol. 2009;20:171–175. doi: 10.1097/MOL.0b013e32832ac051. [DOI] [PubMed] [Google Scholar]
- 8.Feig JE, Shamir R, Fisher EA. Atheroprotective effects of HDL: beyond reverse cholesterol transport. Curr Drug Targets. 2008;9:196–203. doi: 10.2174/138945008783755557. [DOI] [PubMed] [Google Scholar]
- 9.Vaisar T, Pennathur S, Green PS, Gharib SA, Hoofnagle AN, Cheung MC, Byun J, Vuletic S, Kassim S, Singh P, Chea H, Knopp RH, Brunzell J, Geary R, Chait A, Zhao XQ, Elkon K, Marcovina S, Ridker P, Oram JF, Heinecke JW. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117:746–756. doi: 10.1172/JCI26206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oram JF, Heinecke JW. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- 11.Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, Maeda N, Parks JS. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem. 2008;283:22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curtiss LK, Tobias PS. The toll of Toll-like receptors, especially toll-like receptor 2, on murine atherosclerosis. Curr Drug Targets. 2007;8:1230–1238. doi: 10.2174/138945007783220605. [DOI] [PubMed] [Google Scholar]
- 14.Moore KJ, Freeman MW. Targeting Innate Immunity for CV Benefit. Drug Discov Today Ther Strateg. 2008;5:15–23. doi: 10.1016/j.ddstr.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy AJ, Woollard KJ, Hoang A, Mukhamedova N, Stirzaker RA, McCormick SP, Remaley AT, Sviridov D, Chin-Dusting J. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler Thromb Vasc Biol. 2008;28:2071–2077. doi: 10.1161/ATVBAHA.108.168690. [DOI] [PubMed] [Google Scholar]
- 16.O’Neill LA. ‘Fine tuning’ TLR signaling. Nat Immunol. 2008;9:459–461. doi: 10.1038/ni0508-459. [DOI] [PubMed] [Google Scholar]
- 17.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 18.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bjorkbacka H, Fitzgerald KA, Huet F, Li X, Gregory JA, Lee MA, Ordija CM, Dowley NE, Golenbock DT, Freeman MW. The induction of macrophage gene expression by LPS predominantly utilizes Myd88-independent signaling cascades. Physiol Genomics. 2004;19:319–330. doi: 10.1152/physiolgenomics.00128.2004. [DOI] [PubMed] [Google Scholar]
- 20.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 21.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 22.Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med. 2003;198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- 24.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 25.Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophys Res Commun. 2008;368:94–99. doi: 10.1016/j.bbrc.2008.01.061. [DOI] [PubMed] [Google Scholar]
- 26.Halsey TA, Vazquez-Torres A, Gravdahl DJ, Fang FC, Libby SJ. The ferritin-like Dps protein is required for Salmonella enterica serovar Typhimurium oxidative stress resistance and virulence. Infect Immun. 2004;72:1155–1158. doi: 10.1128/IAI.72.2.1155-1158.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williamson R, Lee D, Hagaman J, Maeda N. Marked reduction of high density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein A-I; Proc Natl Acad Sci U S A; 1992. pp. 7134–7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirotani T, Yamamoto M, Kumagai Y, Uematsu S, Kawase I, Takeuchi O, Akira S. Regulation of lipopolysaccharide-inducible genes by MyD88 and Toll/IL-1 domain containing adaptor inducing IFN-beta. Biochem Biophys Res Commun. 2005;328:383–392. doi: 10.1016/j.bbrc.2004.12.184. [DOI] [PubMed] [Google Scholar]
- 29.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 30.Kenny EF, O’Neill LA. Signalling adaptors used by Toll-like receptors: an update. Cytokine. 2008;43:342–349. doi: 10.1016/j.cyto.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Parker TS, Levine DM, Chang JC, Laxer J, Coffin CC, Rubin AL. Reconstituted high-density lipoprotein neutralizes gram-negative bacterial lipopolysaccharides in human whole blood. Infect Immun. 1995;63:253–258. doi: 10.1128/iai.63.1.253-258.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grunfeld C, Feingold KR. HDL and innate immunity: a tale of two apolipoproteins. J Lipid Res. 2008;49:1605–1606. doi: 10.1194/jlr.E800011-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Atger VM, de la Llera Moya M, Stoudt GW, Rodrigueza WV, Phillips MC, Rothblat GH. Cyclodextrins as catalysts for the removal of cholesterol from macrophage foam cells. J Clin Invest. 1997;99:773–780. doi: 10.1172/JCI119223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, Beutler B. CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 35.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Harrington L, Trebicka E, Shi HN, Kagan JC, Hong CC, Lin HY, Babitt JL, Cherayil BJ. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J Clin Invest. 2009;119:3322–3328. doi: 10.1172/JCI39939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E; Proc Natl Acad Sci U S A; 2004. pp. 10679–10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wathelet MG, Szpirer J, Nols CB, Clauss IM, De Wit L, Islam MQ, Levan G, Horisberger MA, Content J, Szpirer C, Huezl GA. Cloning and chromosomal location of human genes inducible by type I interferon. Somat Cell Mol Genet. 1988;14:415–426. doi: 10.1007/BF01534709. [DOI] [PubMed] [Google Scholar]
- 40.Mach F, Sauty A, Iarossi AS, Sukhova GK, Neote K, Libby P, Luster AD. Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Invest. 1999;104:1041–1050. doi: 10.1172/JCI6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heller EA, Liu E, Tager AM, Yuan Q, Lin AY, Ahluwalia N, Jones K, Koehn SL, Lok VM, Aikawa E, Moore KJ, Luster AD, Gerszten RE. Chemokine CXCL 10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation. 2006;113:2301–2312. doi: 10.1161/CIRCULATIONAHA.105.605121. [DOI] [PubMed] [Google Scholar]
- 42.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hall JC, Rosen A. Type I interferons: crucial participants in disease amplification in autoimmunity. Nat Rev Rheumatol. 6:40–49. doi: 10.1038/nrrheum.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Lenten BJ, Wagner AC, Anantharamaiah GM, Garber DW, Fishbein MC, Adhikary L, Nayak DP, Hama S, Navab M, Fogelman AM. Influenza infection promotes macrophage traffic into arteries of mice that is prevented by D-4F, an apolipoprotein A-I mimetic peptide. Circulation. 2002;106:1127–1132. doi: 10.1161/01.cir.0000030182.35880.3e. [DOI] [PubMed] [Google Scholar]
- 45.Van Lenten BJ, Wagner AC, Navab M, Anantharamaiah GM, Hui EK, Nayak DP, Fogelman AM. D-4F, an apolipoprotein A-I mimetic peptide, inhibits the inflammatory response induced by influenza A infection of human type II pneumocytes. Circulation. 2004;110:3252–3258. doi: 10.1161/01.CIR.0000147232.75456.B3. [DOI] [PubMed] [Google Scholar]
- 46.Wilhelm AJ, Zabalawi M, Grayson JM, Weant AE, Major AS, Owen J, Bharadwaj M, Walzem R, Chan L, Oka K, Thomas MJ, Sorci-Thomas MG. Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler Thromb Vasc Biol. 2009;29:843–849. doi: 10.1161/ATVBAHA.108.183442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kane JP, Hardman DA, Dimpfl JC, Levy JA. Apolipoprotein is responsible for neutralization of xenotropic type C virus by mouse serum. Proc Natl Acad Sci U S A. 1979;76:5957–5961. doi: 10.1073/pnas.76.11.5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.